
Atypical Rho GTPases of the RhoBTB Subfamily: Roles in Vesicle Trafficking and Tumorigenesis



Abstract
:1. Introduction
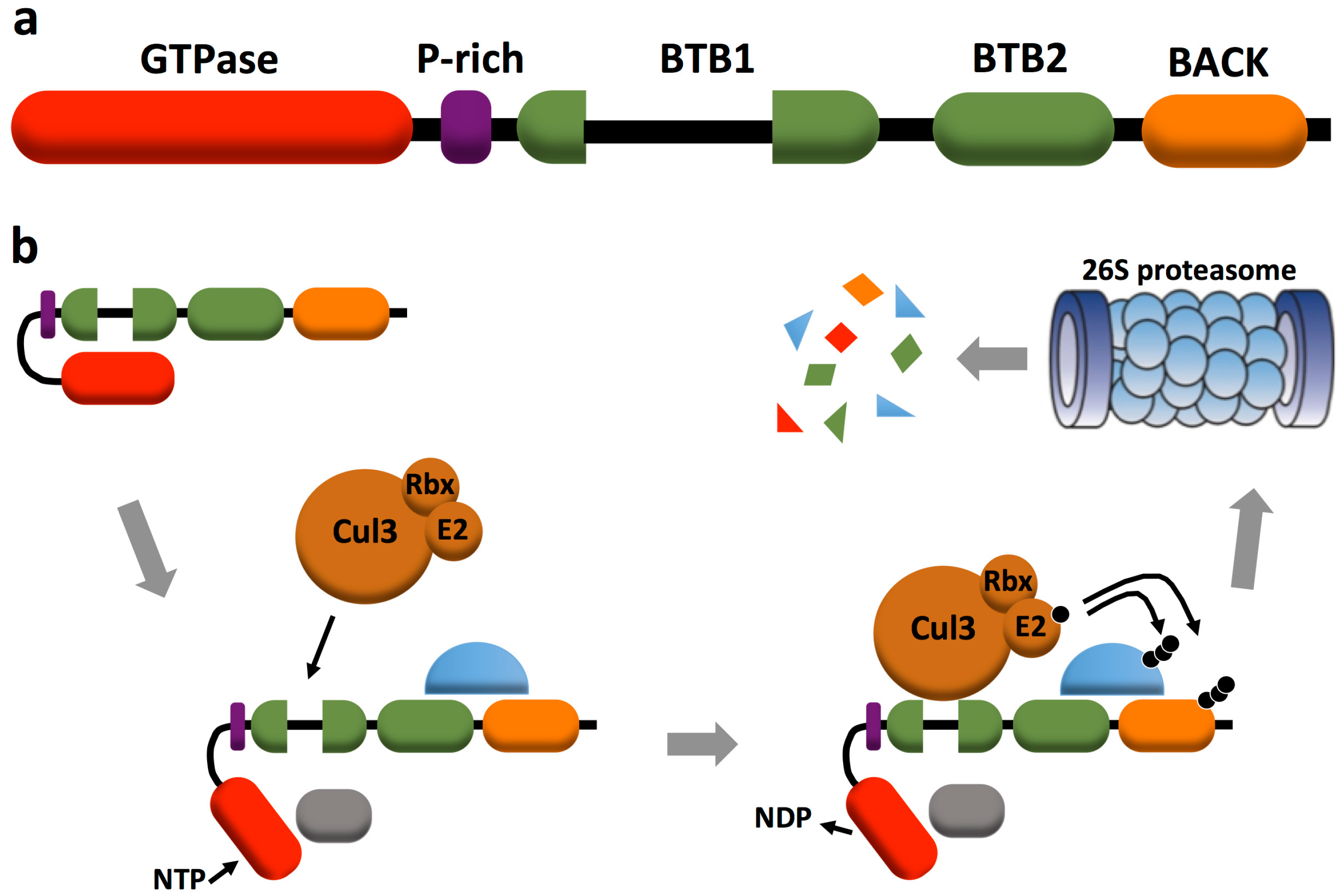
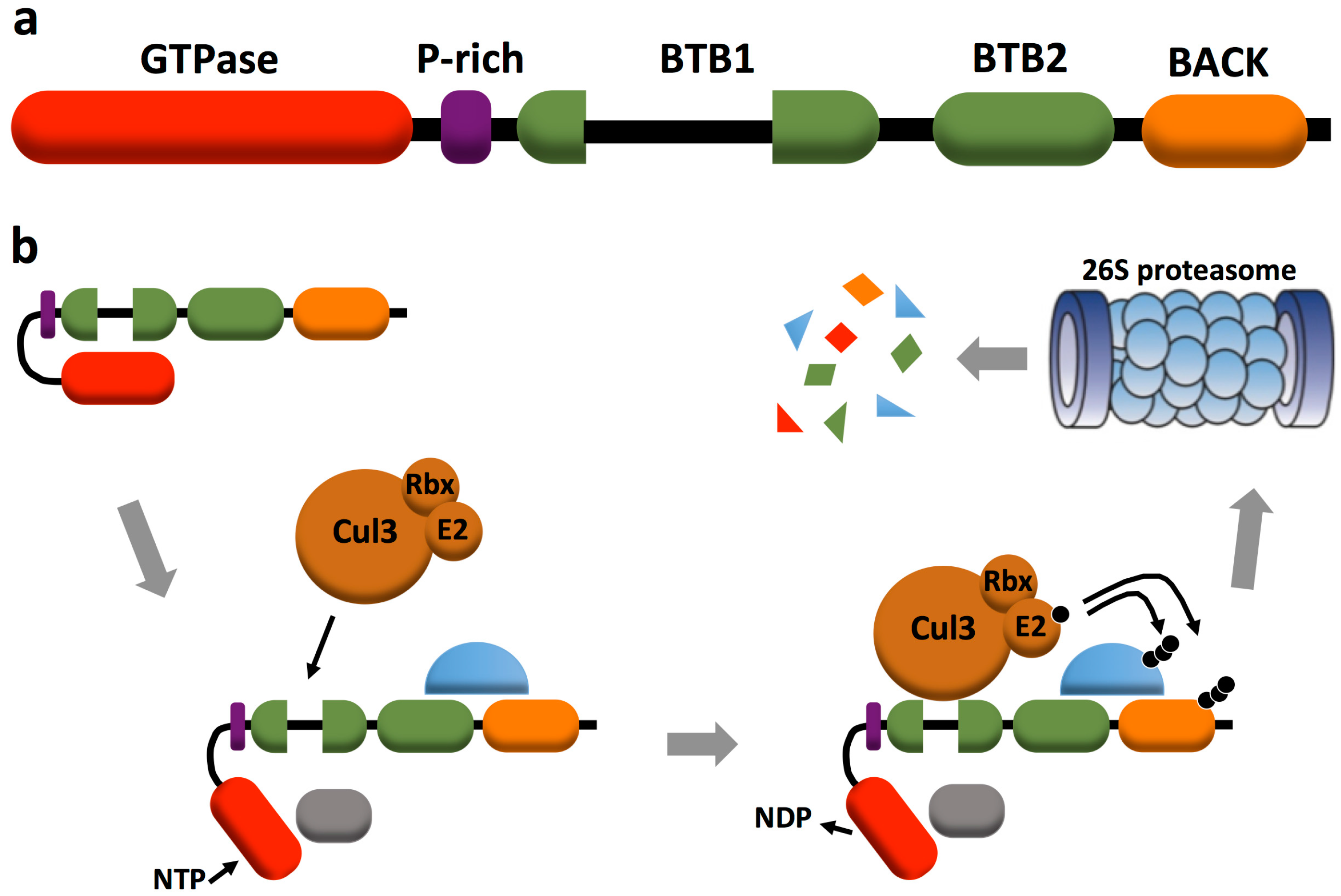
2. RhoBTB Architecture and Gene Expression
3. RhoBTB and Proteasomal Degradation
4. RhoBTB and Vesicle Trafficking
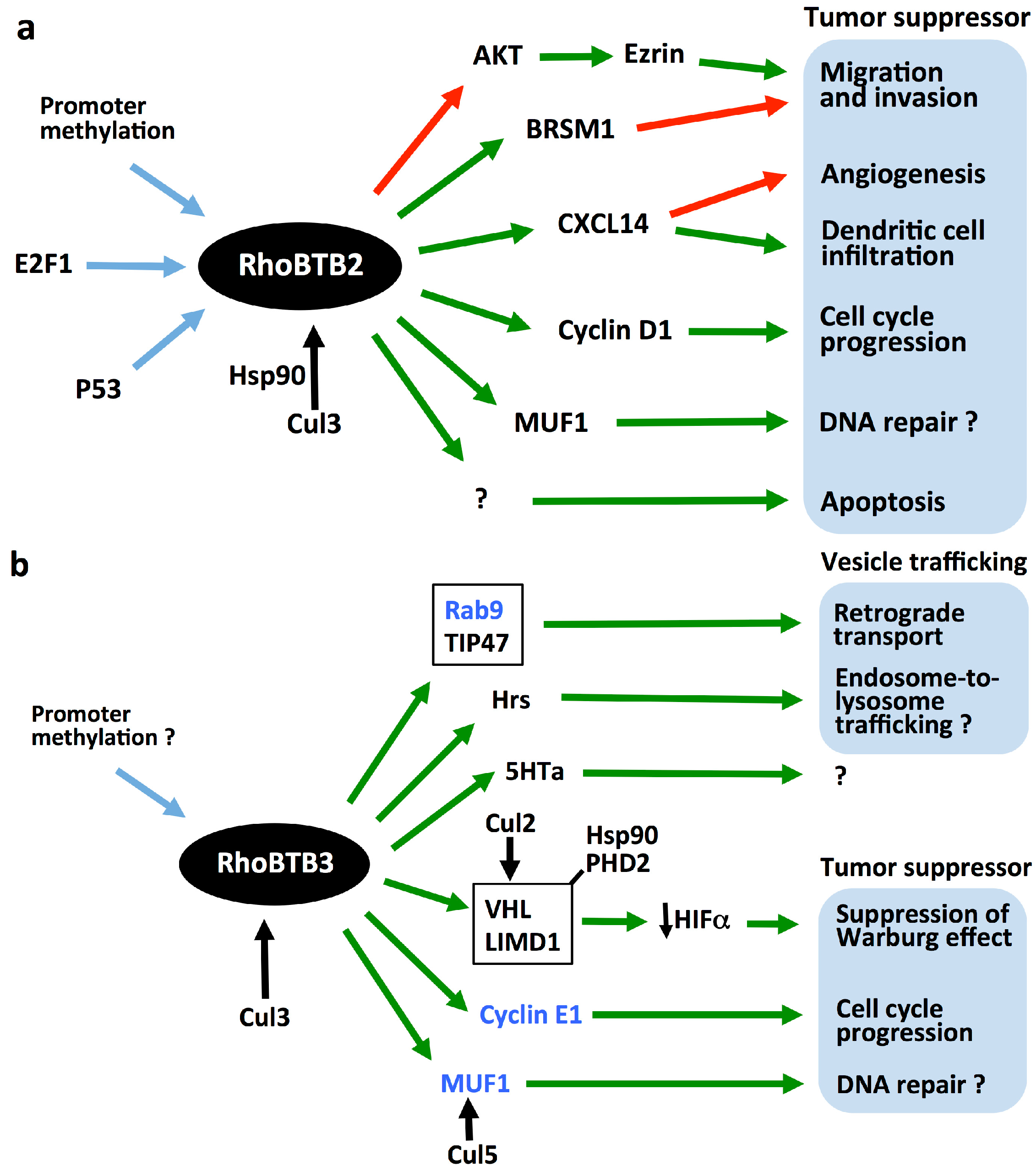
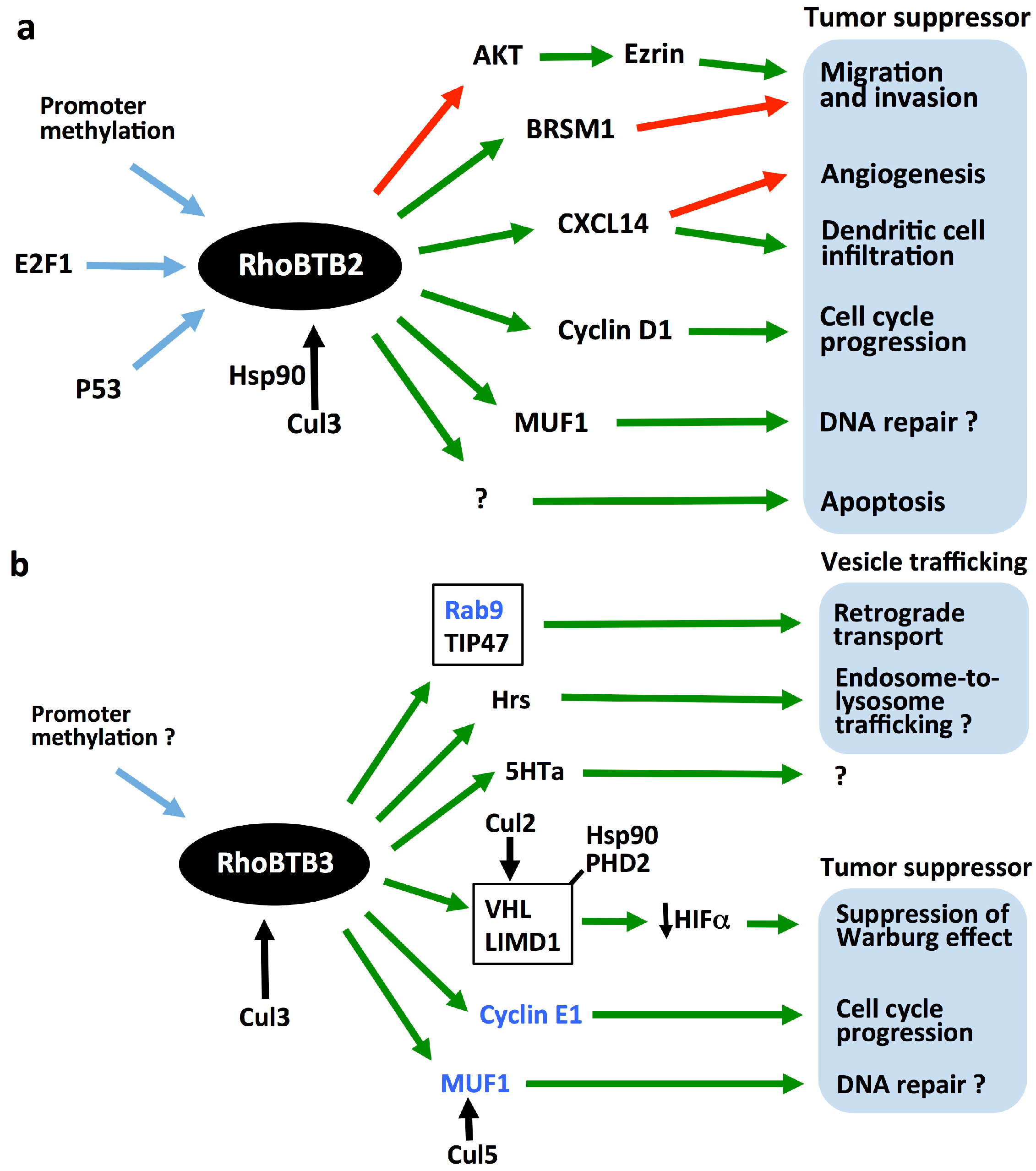
5. RhoBTBs as Tumor Suppressors
6. Tumorigenic Mechanisms of RhoBTB
6.1. Regulation of the Cell Cycle
6.2. Modulation of the Adaptive Response to Hypoxia
6.3. Other Potential Tumorigenic Mechanisms
7. Implications in Other Diseases and Animal Models of RhoBTB Function
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Goitre, L.; Trapani, E.; Trabalzini, L.; Retta, S. The Ras superfamily of small GTPases: The unlocked secrets. Methods Mol. Biol. 2014, 1120, 1–18. [Google Scholar] [PubMed]
- Ridley, A.J. Historical overview of Rho GTPases. Methods Mol. Biol. 2013, 827, 3–12. [Google Scholar]
- Sit, S.-T.; Manser, E. Rho GTPases and their role in organizing the actin cytoskeleton. J. Cell Sci. 2011, 124, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Eliás, M.; Klimes, V. Rho GTPases: Deciphering the evolutionary history of a complex protein family. Methods Mol. Biol. 2012, 827, 13–34. [Google Scholar] [PubMed]
- Rivero, F.; Dislich, H.; Glöckner, G.; Noegel, A.A. The Dictyostelium discoideum family of Rho-related proteins. Nucleic Acids Res. 2001, 29, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, M.; Meth, J.L.; von Klitzing, C.; Wei, W.; Esposito, D.; Rodgers, L.; Walsh, T.; Welcsh, P.; King, M.-C.; Wigler, M.H. DBC2, a candidate for a tumor suppressor gene involved in breast cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 13647–13652. [Google Scholar] [CrossRef] [PubMed]
- Berthold, J.; Schenkova, K.; Rivero, F. Rho GTPases of the RhoBTB subfamily and tumorigenesis. Acta Pharmacol. Sin. 2008, 29, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Manjarrez, J.R.; Sun, L.; Prince, T.; Matts, R.L. Hsp90-dependent assembly of the DBC2/RhoBTB2-cullin3 E3-ligase complex. PLoS ONE 2014, 9, e90054. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, E.J.; Calero, M.; Sridevi, K.; Pfeffer, S.R. RhoBTB3: A Rho GTPase-family ATPase required for endosome to Golgi transport. Cell 2009, 137, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Elias, M.; Rivero, F. The evolutionary history of RhoBTB proteins. Unpublished work. 2016. [Google Scholar]
- Berthold, J.; Schenková, K.; Ramos, S.; Miura, Y.; Furukawa, M.; Aspenström, P.; Rivero, F. Characterization of RhoBTB-dependent Cul3 ubiquitin ligase complexes--evidence for an autoregulatory mechanism. Exp. Cell Res. 2008, 314, 3453–3465. [Google Scholar] [CrossRef] [PubMed]
- Stogios, P.J.; Privé, G.G. The BACK domain in BTB-kelch proteins. Trends Biochem. Sci. 2004, 29, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Pfeffer, S.R. Golgi-associated RhoBTB3 targets cyclin E for ubiquitylation and promotes cell cycle progression. J. Cell Biol. 2013, 203, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.; Grimm-Gunter, E.-M.S.; Joshi, P.; Rivero, F. Expression analysis of mouse Rhobtb3 using a LacZ reporter and preliminary characterization of a knockout strain. Histochem. Cell Biol. 2014, 142, 511–548. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, A.; Ping, Q.; Carpenter, C.L. RhoBTB2 is a substrate of the mammalian Cul3 ubiquitin ligase complex. Genes Dev. 2004, 18, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Pridgeon, J.W.; Webber, E.A.; Sha, D.; Li, L.; Chin, L.-S. Proteomic analysis reveals Hrs ubiquitin-interacting motif-mediated ubiquitin signaling in multiple cellular processes. FEBS J. 2009, 276, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Matthys, A.; Van Craenenbroeck, K.; Lintermans, B.; Haegeman, G.; Vanhoenacker, P. RhoBTB3 interacts with the 5-HT7a receptor and inhibits its proteasomal degradation. Cell. Signal. 2012, 24, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-S.; Liu, Q.; Li, M.; Lin, S.-Y.; Peng, Y.; Wu, D.; Li, T.Y.; Fu, Q.; Jia, W.; Wang, X.; et al. RHOBTB3 promotes proteasomal degradation of HIFα through facilitating hydroxylation and suppresses the Warburg effect. Cell Res. 2015, 25, 1025–1042. [Google Scholar] [CrossRef] [PubMed]
- Schenková, K.; Lutz, J.; Kopp, M.; Ramos, S.; Rivero, F. MUF1/leucine-rich repeat containing 41 (LRRC41), a substrate of RhoBTB-dependent cullin 3 ubiquitin ligase complexes, is a predominantly nuclear dimeric protein. J. Mol. Biol. 2012, 422, 659–673. [Google Scholar] [CrossRef] [PubMed]
- Kato, J.; Yoneda-Kato, N. Mammalian COP9 signalosome. Genes Cells 2009, 14, 1209–1225. [Google Scholar] [CrossRef] [PubMed]
- Eckl, J.; Richter, K. Functions of the Hsp90 chaperone system: lifting client proteins to new heights. Int. J. Biochem. Mol. Biol. 2013, 4, 157–165. [Google Scholar] [PubMed]
- Chang, F.K.; Sato, N.; Kobayashi-Simorowski, N.; Yoshihara, T.; Meth, J.L.; Hamaguchi, M. DBC2 is essential for transporting vesicular stomatitis virus glycoprotein. J. Mol. Biol. 2006, 364, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.R. Multiple routes of protein transport from endosomes to the trans Golgi network. FEBS Lett. 2009, 583, 3811–3816. [Google Scholar] [CrossRef] [PubMed]
- Heselmeyer-Haddad, K.; Berroa Garcia, L.Y.; Bradley, A.; Ortiz-Melendez, C.; Lee, W.-J.; Christensen, R.; Prindiville, S.A.; Calzone, K.A.; Soballe, P.W.; Hu, Y.; et al. Single-cell genetic analysis of ductal carcinoma in situ and invasive breast cancer reveals enormous tumor heterogeneity yet conserved genomic imbalances and gain of MYC during progression. Am. J. Pathol. 2012, 181, 1807–1822. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Hou, L.; Song, J.; Lin, D.; Wu, L.; Ge, Y.; Ma, Z. Decreased expression of the DBC2 gene and its clinicopathological significance in breast cancer: correlation with aberrant DNA methylation. Biotechnol. Lett. 2013, 35, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Meng, L.; Shen, H.-C.; Du, J.-J. Loss of DBC2 expression is an early and progressive event in the development of lung adenocarcinoma. Asian Pac. J. Cancer Prev. 2012, 13, 2021–2023. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.A.; Aveyard, J.S.; Taylor, C.F.; Harnden, P.; Bass, S. Mutation analysis of the 8p candidate tumour suppressor genes DBC2 (RHOBTB2) and LZTS1 in bladder cancer. Cancer Lett. 2005, 225, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Chen, J.-Y.; Yang, J.; Li, B.; Chen, Z.-H.; Xiao, C.-G. DBC2 gene is silenced by promoter methylation in bladder cancer. Urol. Oncol. 2008, 26, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.G.; Choi, B.J.; Kim, C.J.; Song, J.H.; Zhang, C.; Nam, S.W.; Lee, J.Y.; Park, W.S. Genetic analysis of the DBC2 gene in gastric cancer. Acta Oncol. 2008, 47, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Han, Y.; Han, X. Downregulated RhoBTB2 expression contributes to poor outcome in osteosarcoma patients. Cancer Biother. Radiopharm. 2013, 28, 709–716. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, C.M.; Lygoe, K.A.; Skelton, L.; Mitter, R.; Mellor, H. The atypical Rho GTPase RhoBTB2 is required for expression of the chemokine CXCL14 in normal and cancerous epithelial cells. Oncogene 2008, 27, 6856–6865. [Google Scholar] [CrossRef] [PubMed]
- Beder, L.B.; Gunduz, M.; Ouchida, M.; Gunduz, E.; Sakai, A.; Fukushima, K.; Nagatsuka, H.; Ito, S.; Honjo, N.; Nishizaki, K.; et al. Identification of a candidate tumor suppressor gene RHOBTB1 located at a novel allelic loss region 10q21 in head and neck cancer. J. Cancer Res. Clin. Oncol. 2006, 132, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.-S.; Wu, X.-D.; Zhang, S.-Q.; Li, C.-F.; Yang, L.; Li, D.-D.; Zhang, B.-G.; Zhang, Y.; Jin, J.-P.; Zhang, B. The tumor suppressor gene RhoBTB1 is a novel target of miR-31 in human colon cancer. Int. J. Oncol. 2013, 42, 676–682. [Google Scholar] [PubMed]
- Bhattacharjee, D.; Shenoy, S.; Bairy, K. DNA methylation and chromatin remodeling: The blueprint of cancer epigenetics. Scientifica 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Hajikhan Mirzaei, M.; Noruzinia, M.; Karbassian, H.; Shafeghati, Y.; Keyhanee, M.; Bidmeshki-Pour, A. Evaluation of methylation status in the 5’UTR promoter region of the DBC2 gene as a biomarker in sporadic breast cancer. Cell J. 2012, 14, 19–24. [Google Scholar] [PubMed]
- Tang, W.; Wang, C.; Fu, F.; Chen, Q. RhoBTB2 gene in breast cancer is silenced by promoter methylation. Int. J. Mol. Med. 2014, 33, 722–728. [Google Scholar] [PubMed]
- Khakpour, G.; Pooladi, A.; Izadi, P.; Noruzinia, M.; Tavakkoly Bazzaz, J. DNA methylation as a promising landscape: A simple blood test for breast cancer prediction. Tumor Biol. 2015, 36, 4905–4912. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.; Croce, C. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Alder, H.; Taccioli, C.; Chen, H.; Jiang, Y.; Smalley, K.J.; Fadda, P.; Ozer, H.G.; Huebner, K.; Farber, J.L.; Croce, C.M.; et al. Dysregulation of miR-31 and miR-21 induced by zinc deficiency promotes esophageal cancer. Carcinogenesis 2012, 33, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Zhang, L.; Yang, Y.; Sun, J.; Deng, B.; Feng, J.; Shao, Q.; Feng, A.; Song, B.; Qu, X. RhoBTB2 (DBC2) functions as tumor suppressor via inhibiting proliferation, preventing colony formation and inducing apoptosis in breast cancer cells. Gene 2011, 486, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-J.; Yang, D.; Luo, Y.-W. RhoBTB2 (DBC2) functions as a multifunctional tumor suppressor in thyroid cancer cells via mitochondrial apoptotic pathway. Int. J. Clin. Exp. Med. 2015, 8, 5954–5958. [Google Scholar] [PubMed]
- Yoshihara, T.; Collado, D.; Hamaguchi, M. Cyclin D1 down-regulation is essential for DBC2’s tumor suppressor function. Biochem. Biophys. Res. Commun. 2007, 358, 1076–1079. [Google Scholar] [CrossRef] [PubMed]
- Collado, D.; Yoshihara, T.; Hamaguchi, M. DBC2 Resistance is Achieved by Enhancing 26S Proteasome-mediated Protein Degradation. Biochem. Biophys. Res. Commun. 2007, 360, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.C.; Ma, Y.; Cress, W.D. RhoBTB2 (DBC2) is a mitotic E2F1 target gene with a novel role in apoptosis. J. Biol. Chem. 2008, 283, 2353–2362. [Google Scholar] [CrossRef] [PubMed]
- Garritano, S.; Inga, A.; Gemignani, F.; Landi, S. More targets, more pathways and more clues for mutant p53. Oncogenesis 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.; Chu, C.; Harper, J. Culprits in the degradation of cyclin E apprehended. Genes Dev. 1999, 13, 2751–2757. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Fry, E. Novel molecular markers for breast cancer. Biomark. Cancer 2016, 8, 25–42. [Google Scholar] [CrossRef] [PubMed]
- LaGory, E.; Giaccia, A. The ever-expanding role of HIF in tumour and stromal biology. Nat. Cell Biol. 2016, 18, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Tanegashima, K. Pleiotropic functions of the CXC-type chemokine CXCL14 in mammals. J. Biochem. 2012, 151, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.-J.; Lu, C.; Zhou, G.-P.; Wang, S. Ectopic expression of RhoBTB2 inhibits migration and invasion of human breast cancer cells. Cancer Biol. Ther. 2010, 10, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.N.; Cress, W.D. RhoBTB2 (DBC2) comes of age as a multifunctional tumor suppressor. Cancer Biol. Ther. 2010, 10, 1123–1125. [Google Scholar] [CrossRef] [PubMed]
- Kurian, S.M.; Le-Niculescu, H.; Patel, S.D.; Bertram, D.; Davis, J.; Dike, C.; Yehyawi, N.; Lysaker, P.; Dustin, J.; Caligiuri, M.; et al. Identification of blood biomarkers for psychosis using convergent functional genomics. Mol. Psychiatry 2011, 16, 37–58. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Choi, K.-H.; Baykiz, A.F.; Gershenfeld, H.K. Suicide candidate genes associated with bipolar disorder and schizophrenia: An exploratory gene expression profiling analysis of post-mortem prefrontal cortex. BMC Genom. 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.A.; Woltjer, R.L.; Goodenbour, J.M.; Horvath, S.; Geschwind, D.H. Genes and pathways underlying regional and cell type changes in Alzheimer’s disease. Genome Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Pelham, C.J.; Ketsawatsomkron, P.; Groh, S.; Grobe, J.L.; de Lange, W.J.; Ibeawuchi, S.-R.C.; Keen, H.L.; Weatherford, E.T.; Faraci, F.M.; Sigmund, C.D. Cullin-3 regulates vascular smooth muscle function and arterial blood pressure via PPARγ and RhoA/Rho-kinase. Cell Metab. 2012, 16, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Stump, M.; Mukohda, M.; Hu, C.; Sigmund, C. PPARγ regulation in hypertension and metabolic syndrome. Curr. Hypertens. Rep. 2015, 17. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ullmann, R.; Langnick, C.; Menzel, C.; Wotschofsky, Z.; Hu, H.; Döring, A.; Hu, Y.; Kang, H.; Tzschach, A.; et al. Breakpoint analysis of balanced chromosome rearrangements by next-generation paired-end sequencing. Eur. J. Hum. Genet. 2010, 18, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Rivero, F. RhoBTB3 deficiency causes a fertility defect. Unpublished work. 2016. [Google Scholar]
- Rivero, F.; Naseem, K.M. Impaired platelet function in mice deficient in the novel rhoGTPase RhoBTB3. Manuscript in preparation.
- De la Vega, M.; Burrows, J.F.; Johnston, J.A. Added complexity in Ras and Rho family GTPase function Ubiquitination. Small GTPases 2011, 2, 192–201. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Protein | RhoBTB | Function | Reference |
|---|---|---|---|
| Cullin 3 | 1, 2, 3 (Y2H, IP) | E3 ubiquitin ligase component. | [8,11,15] |
| Cullin 5 | 2, 3 (Y2H) | E3 ubiquitin ligase component. | [11] |
| Cyclin E1, cyclin B1 | 3 (IP) | Regulation of the cell cycle. | [13] |
| Hrs | 3 (IVEC) | Protein sorting for lysosomal degradation. | [16] |
| 5-HT7a | 3 (Y2H) | Serotonin receptor. | [17] |
| Hsp90 chaperone | 2 (IP); 3 (IP) | Protein folding and stabilization. | [8,18] |
| LIMD1 | 3 (IP) | Multifunctional scaffold protein. | [18] |
| MUF1 | 1, 2, (IP); 3 (B2H) | Adaptor for cullin 5-dependent ubiquitin ligase complexes. | [19] |
| PHD2 | 3 (IP) | Prolyl hydroxylase. | [18] |
| Rab9A, Rab9B | 3 (Y2H) | Retrograde transport of membrane receptors. | [9] |
| TIP47 | 3 (CL) | Cargo packaging for endosomal transport. | [9] |
| VHL | 3 (Y2H, IP) | Adaptor for cullin 2-dependent ubiquitin ligase complexes. | [18] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, W.; Rivero, F. Atypical Rho GTPases of the RhoBTB Subfamily: Roles in Vesicle Trafficking and Tumorigenesis. Cells 2016, 5, 28. https://doi.org/10.3390/cells5020028
Ji W, Rivero F. Atypical Rho GTPases of the RhoBTB Subfamily: Roles in Vesicle Trafficking and Tumorigenesis. Cells. 2016; 5(2):28. https://doi.org/10.3390/cells5020028
Chicago/Turabian StyleJi, Wei, and Francisco Rivero. 2016. "Atypical Rho GTPases of the RhoBTB Subfamily: Roles in Vesicle Trafficking and Tumorigenesis" Cells 5, no. 2: 28. https://doi.org/10.3390/cells5020028
APA StyleJi, W., & Rivero, F. (2016). Atypical Rho GTPases of the RhoBTB Subfamily: Roles in Vesicle Trafficking and Tumorigenesis. Cells, 5(2), 28. https://doi.org/10.3390/cells5020028