
Cellular Mechanisms of Ciliary Length Control

Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Cilium: Types and Structure
3. Cilia and the Cell Cycle
4. Programs of Ciliogenesis
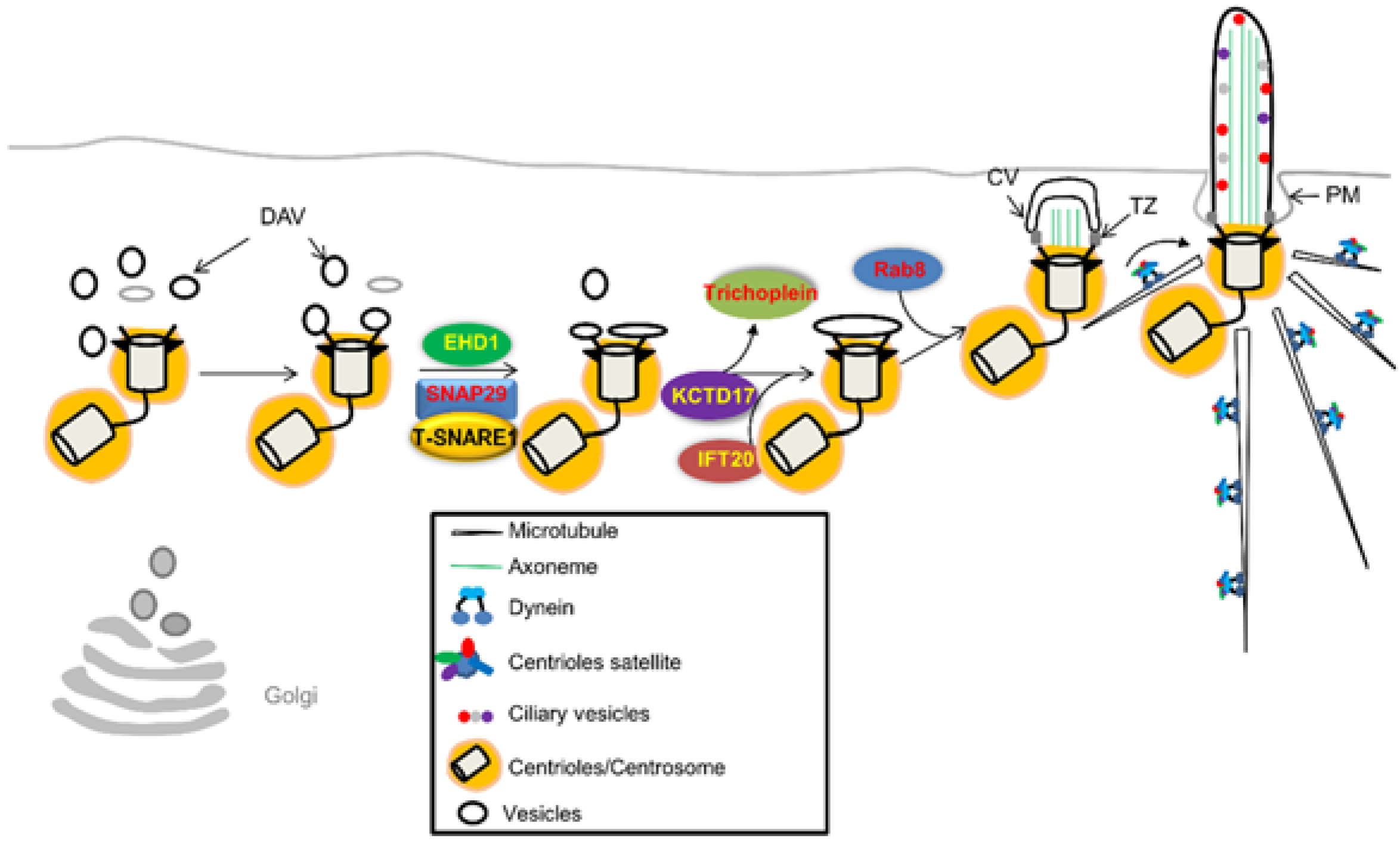
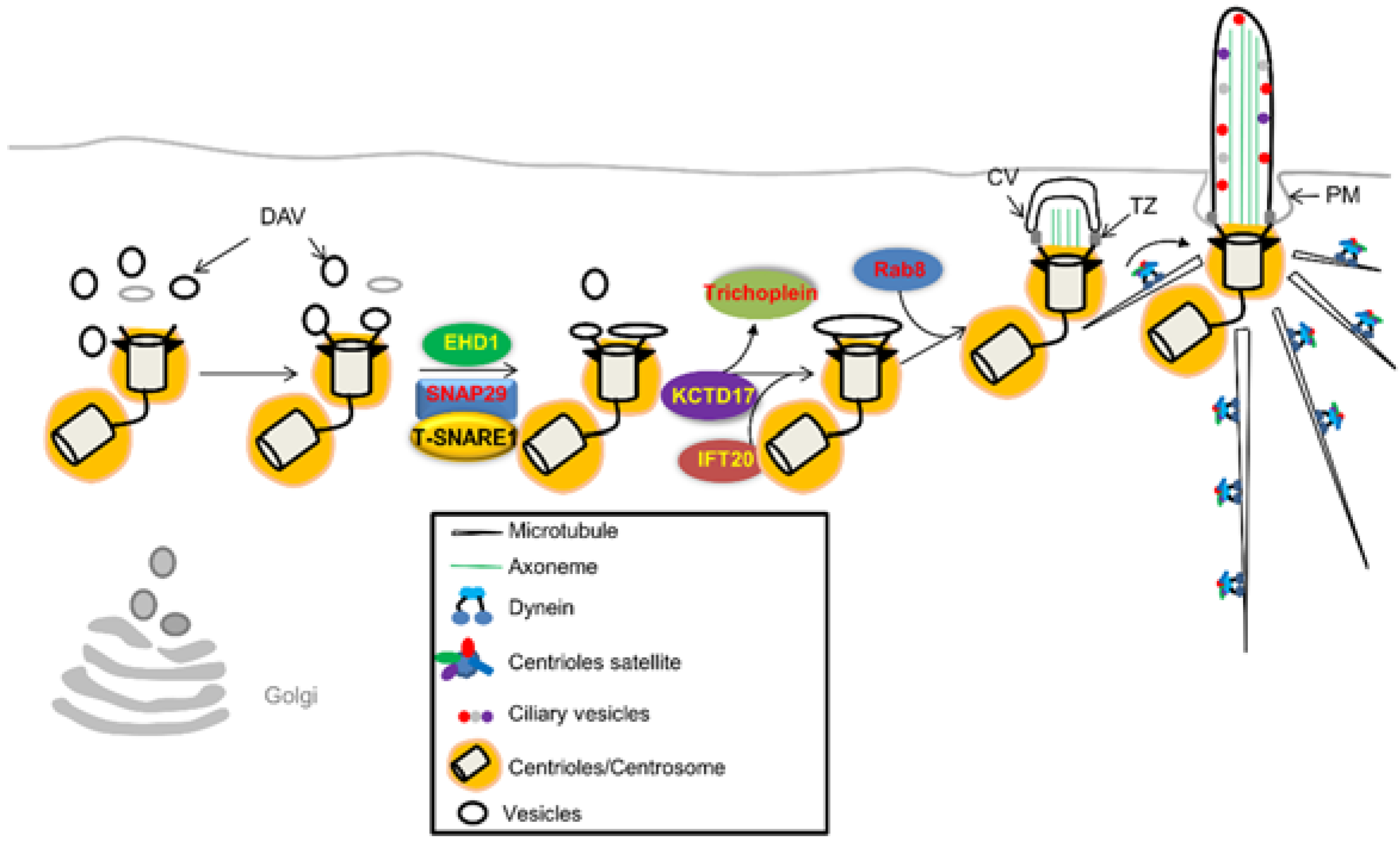
4.1. Initiation of Ciliary Assembly
4.2. Maintenance of Ciliary Length
4.3. Regulators of Ciliogenesis
4.3.1. CP110 Destruction/Dislocation by TTBK2 and MARK4
4.3.2. Trichoplein is Degraded by CRL3KCTD17
4.3.3. Ofd1 is Removed by Autophagy
5. Ciliary Length Control Mechanisms
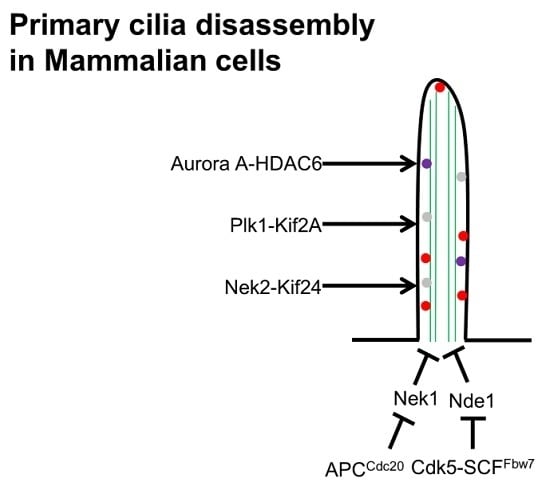
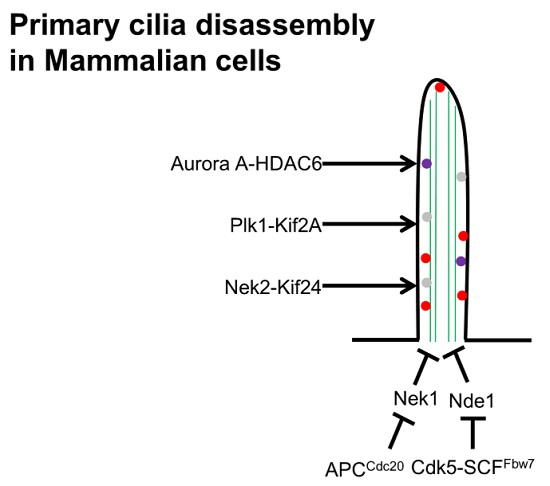
5.1. Nde1 is Regulated by CDK5-SCFfbw7
5.2. APC in Ciliogenesis
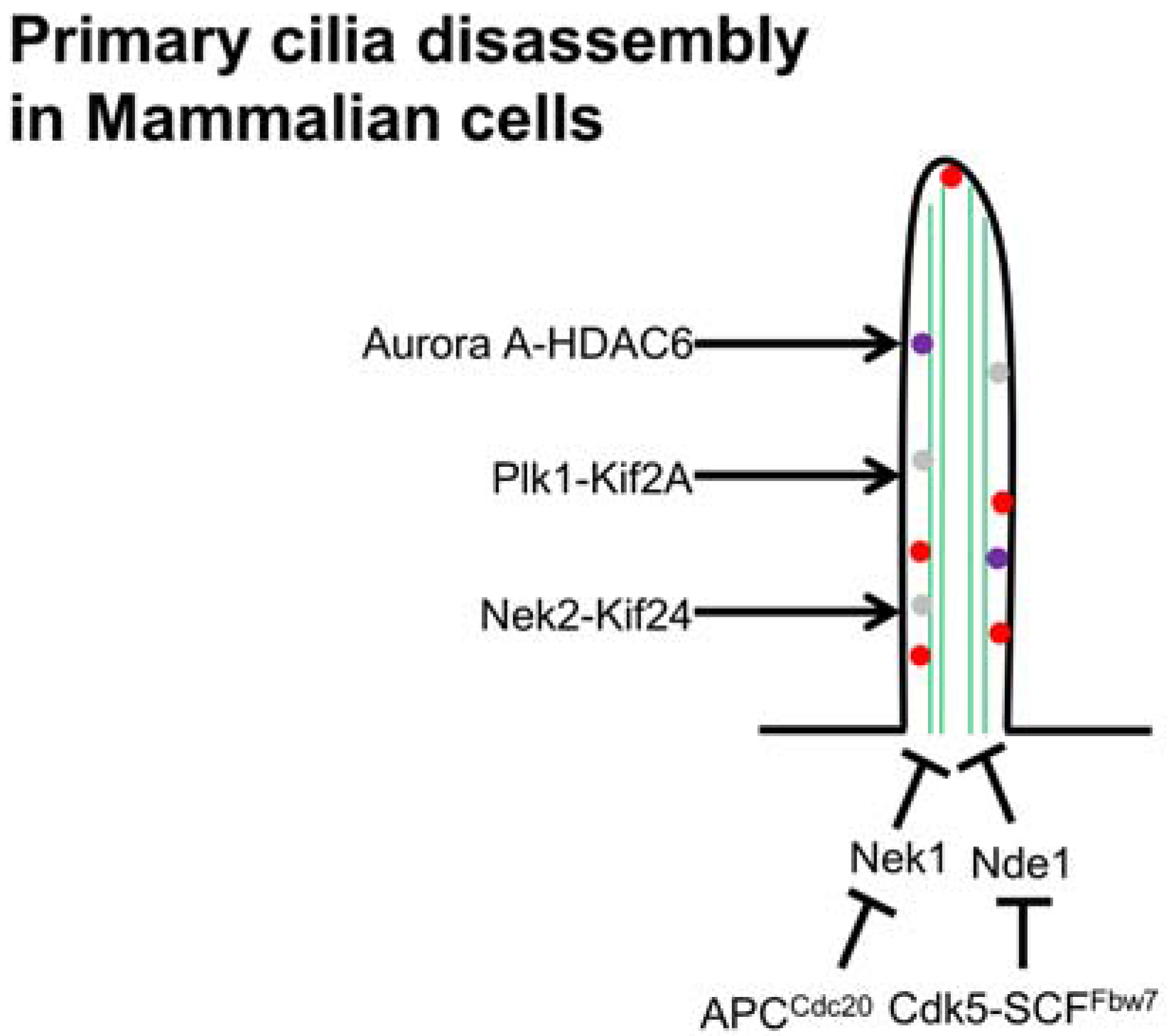
5.3. Arf and Arl Members in Length Control
6. Cilia Disassembly
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Pedersen, L.B.; Veland, I.R.; Schrøder, J.M.; Christensen, S.T. Assembly of primary cilia. Dev. Dynam. 2008, 237, 1993–2006. [Google Scholar] [CrossRef] [PubMed]
- Silverman, M.A.; Leroux, M.R. Intraflagellar transport and the generation of dynamic, structurally and functionally diverse cilia. Trends Cell Biol. 2009, 19, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. 2010, 11, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Badano, J.L.; Mitsuma, N.; Beales, P.L.; Katsanis, N. The ciliopathies: An emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 2006, 7, 125–148. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, D.N.; Wang, A.M.; Strugnell, G.E. Expression of primary cilia in mammalian cells. Cell Biol. Int. 1996, 20, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A.; Raff, J.W. Centrioles, centrosomes, and cilia in health and disease. Cell 2009, 139, 663–678. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.S.; Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. New Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Anderson, K.V. Cilia and hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 11325–11330. [Google Scholar] [CrossRef] [PubMed]
- Plotnikova, O.V.; Golemis, E.A.; Pugacheva, E.N. Cell cycle-dependent ciliogenesis and cancer. Cancer Res. 2008, 68, 2058–2061. [Google Scholar] [CrossRef] [PubMed]
- Avasthi, P.; Marshall, W.F. Stages of ciliogenesis and regulation of ciliary length. Differentiation 2012, 83, S30–S42. [Google Scholar] [CrossRef] [PubMed]
- McMurray, R.; Wann, A.; Thompson, C.; Connelly, J.; Knight, M. Surface topography regulates wnt signaling through control of primary cilia structure in mesenchymal stem cells. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Subramanian, R.; Bangs, F.; Omelchenko, T.; Liem, K.F., Jr.; Kapoor, T.M.; Anderson, K.V. The kinesin-4 protein kif7 regulates mammalian hedgehog signalling by organizing the cilium tip compartment. Nat. Cell Biol. 2014, 16, 663–672. [Google Scholar] [PubMed]
- Satir, P.; Christensen, S.T. Overview of structure and function of mammalian cilia. Annu. Rev. Physiol. 2007, 69, 377–400. [Google Scholar] [PubMed]
- Christensen, S.T.; Pedersen, L.B.; Schneider, L.; Satir, P. Sensory cilia and integration of signal transduction in human health and disease. Traffic 2007, 8, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Takeda, S.; Tanaka, Y.; Belmonte, J.-C.I.; Hirokawa, N. Mechanism of nodal flow: A conserved symmetry breaking event in left-right axis determination. Cell 2005, 121, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, S.; Tanaka, Y.; Okada, Y.; Takeda, S.; Harada, A.; Kanai, Y.; Kido, M.; Hirokawa, N. Randomization of left–right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking kif3b motor protein. Cell 1998, 95, 829–837. [Google Scholar] [CrossRef]
- Kim, S.; Tsiokas, L. Cilia and cell cycle re-entry: More than a coincidence. Cell Cycle 2011, 10, 2683–2690. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Dynlacht, B.D. Regulating the transition from centriole to basal body. J. Cell Biol. 2011, 193, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, S. Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J. Cell Biol. 1962, 15, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Doxsey, S.; Zimmerman, W.; Mikule, K. Centrosome control of the cell cycle. Trends Cell Biol. 2005, 15, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Snell, W. The primary cilium: Keeper of the key to cell division. Cell 2007, 129, 1255–1257. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A.; Stearns, T. The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat. Cell Biol. 2011, 13, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Berbari, N.F.; Sharma, N.; Malarkey, E.B.; Pieczynski, J.N.; Boddu, R.; Gaertig, J.; Guay-Woodford, L.; Yoder, B.K. Microtubule modifications and stability are altered by cilia perturbation and in cystic kidney disease. Cytoskeleton 2013, 70, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Wloga, D.; Gaertig, J. Post-translational modifications of microtubules. J. Cell Sci. 2010, 123, 3447–3455. [Google Scholar] [CrossRef] [PubMed]
- Westermann, S.; Weber, K. Post-translational modifications regulate microtubule function. Nat. Rev. Mol. Cell Biol. 2003, 4, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.W.; Cai, D.; Verhey, K.J. Tubulin modifications and their cellular functions. Curr. Opin. Cell Biol. 2008, 20, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.; Ackerman, B.; Gundersen, G.G. Cell biology (communication arising): Tubulin acetylation and cell motility. Nature 2003, 421, 230. [Google Scholar] [CrossRef] [PubMed]
- Konno, A.; Setou, M.; Ikegami, K. 3. Ciliary and flagellar structure and function—their regulations by posttranslational modifications of axonemal tubulin. Int. Rev. Cell Mol. Biol. 2012, 294, 133. [Google Scholar] [PubMed]
- Tucker, R.W.; Pardee, A.B.; Fujiwara, K. Centriole ciliation is related to quiescence and DNA synthesis in 3t3 cells. Cell 1979, 17, 527–535. [Google Scholar] [CrossRef]
- Tucker, R.W.; Scher, C.D.; Stiles, C.D. Centriole deciliation associated with the early response of 3t3 cells to growth factors but not to sv40. Cell 1979, 18, 1065–1072. [Google Scholar] [CrossRef]
- Paridaen, J.T.; Wilsch-Brauninger, M.; Huttner, W.B. Asymmetric inheritance of centrosome-associated primary cilium membrane directs ciliogenesis after cell division. Cell 2013, 155, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.B. Protoplasm. It’s composition and structure. In The Cell in Development and Heredity, 3rd ed.; Macmillan: New York, NY, USA, 1928. [Google Scholar]
- Sung, C.H.; Leroux, M.R. The roles of evolutionarily conserved functional modules in cilia-related trafficking. Nat. Cell Biol. 2013, 15, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Bornens, M. The centrosome in cells and organisms. Science 2012, 335, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Marshall, W.F. Ciliogenesis: Building the cell's antenna. Nat. Rev. Mol. Cell Biol. 2011, 12, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Ghossoub, R.; Molla-Herman, A.; Bastin, P.; Benmerah, A. The ciliary pocket: A once-forgotten membrane domain at the base of cilia. Biol. Cell 2011, 103, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Molla-Herman, A.; Ghossoub, R.; Blisnick, T.; Meunier, A.; Serres, C.; Silbermann, F.; Emmerson, C.; Romeo, K.; Bourdoncle, P.; Schmitt, A. The ciliary pocket: An endocytic membrane domain at the base of primary and motile cilia. J. Cell Sci. 2010, 123, 1785–1795. [Google Scholar] [CrossRef] [PubMed]
- Latta, H.; Maunsbach, A.B.; Madden, S.C. Cilia in different segments of the rat nephron. J. Biophys. Biochem. Cytol. 1961, 11, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Dalen, H. An ultrastructural study of the tracheal epithelium of the guinea-pig with special reference to the ciliary structure. J. Anat. 1983, 136, 47. [Google Scholar] [PubMed]
- Baudoin, J.-P.; Viou, L.; Launay, P.-S.; Luccardini, C.; Gil, S.E.; Kiyasova, V.; Irinopoulou, T.; Alvarez, C.; Rio, J.-P.; Boudier, T. Tangentially migrating neurons assemble a primary cilium that promotes their reorientation to the cortical plate. Neuron 2012, 76, 1108–1122. [Google Scholar] [CrossRef] [PubMed]
- Rieder, C.L.; Jensen, C.G.; Jensen, L.C. The resorption of primary cilia during mitosis in a vertebrate (ptk1) cell line. J. Ultrastruct. Res. 1979, 68, 173–185. [Google Scholar] [CrossRef]
- Kiprilov, E.N.; Awan, A.; Desprat, R.; Velho, M.; Clement, C.A.; Byskov, A.G.; Andersen, C.Y.; Satir, P.; Bouhassira, E.E.; Christensen, S.T. Human embryonic stem cells in culture possess primary cilia with hedgehog signaling machinery. J. Cell Biol. 2008, 180, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Iomini, C.; Tejada, K.; Mo, W.; Vaananen, H.; Piperno, G. Primary cilia of human endothelial cells disassemble under laminar shear stress. J. Cell Biol. 2004, 164, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Pitaval, A.; Tseng, Q.; Bornens, M.; Théry, M. Cell shape and contractility regulate ciliogenesis in cell cycle–arrested cells. J. Cell Biol. 2010, 191, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Boisvieux-Ulrich, E.; Lainé, M.-C.; Sandoz, D. Cytochalasin d inhibits basal body migration and ciliary elongation in quail oviduct epithelium. Cell Tissue Res. 1990, 259, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Bennett, F.C.; Harvey, K.F. Fat cadherin modulates organ size in drosophila via the salvador/warts/hippo signaling pathway. Curr. Biol. 2006, 16, 2101–2110. [Google Scholar] [CrossRef] [PubMed]
- Sansores-Garcia, L.; Bossuyt, W.; Wada, K.I.; Yonemura, S.; Tao, C.; Sasaki, H.; Halder, G. Modulating f-actin organization induces organ growth by affecting the hippo pathway. EMBO J. 2011, 30, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, T.; Hoff, S.; Schell, C.; Helmstädter, M.; Kretz, O.; Kuechlin, S.; Yakulov, T.A.; Engel, C.; Müller, B.; Bensch, R. The polarity protein inturned links nphp4 to daam1 to control the subapical actin network in multiciliated cells. J. Cell Biol. 2015, 211, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Habbig, S.; Bartram, M.P.; Müller, R.U.; Schwarz, R.; Andriopoulos, N.; Chen, S.; Sägmüller, J.G.; Hoehne, M.; Burst, V.; Liebau, M.C. Nphp4, a cilia-associated protein, negatively regulates the hippo pathway. J. Cell Biol. 2011, 193, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Graser, S.; Stierhof, Y.D.; Lavoie, S.B.; Gassner, O.S.; Lamla, S.; Le Clech, M.; Nigg, E.A. Cep164, a novel centriole appendage protein required for primary cilium formation. J. Cell Biol. 2007, 179, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamed, Z.A.; Natarajan, S.; Wheway, G.; Inglehearn, C.F.; Toomes, C.; Johnson, C.A.; Jagger, D.J. The meckel-gruber syndrome protein tmem67 controls basal body positioning and epithelial branching morphogenesis via the non-canonical wnt pathway. Dis. Model. Mech. 2015. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.N.; Kuhns, S.; Neuner, A.; Hub, B.; Zentgraf, H.; Pereira, G. Cep164 mediates vesicular docking to the mother centriole during early steps of ciliogenesis. J. Cell Biol. 2012, 199, 1083–1101. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Insinna, C.; Ott, C.; Stauffer, J.; Pintado, P.A.; Rahajeng, J.; Baxa, U.; Walia, V.; Cuenca, A.; Hwang, Y.S.; et al. Early steps in primary cilium assembly require ehd1/ehd3-dependent ciliary vesicle formation. Nat. Cell Biol. 2015, 17, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Slaats, G.G.; Ghosh, A.K.; Falke, L.L.; Le Corre, S.; Shaltiel, I.A.; van de Hoek, G.; Klasson, T.D.; Stokman, M.F.; Logister, I.; Verhaar, M.C. Nephronophthisis-associated cep164 regulates cell cycle progression, apoptosis and epithelial-to-mesenchymal transition. PLoS. Genet. 2014, e1004594. [Google Scholar] [CrossRef] [PubMed]
- Tanos, B.E.; Yang, H.-J.; Soni, R.; Wang, W.-J.; Macaluso, F.P.; Asara, J.M.; Tsou, M.-F.B. Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev. 2013, 27, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Bangs, F.; Paton, I.R.; Prescott, A.; James, J.; Davey, M.G.; Whitley, P.; Genikhovich, G.; Technau, U.; Burt, D.W.; et al. The talpid3 gene (kiaa0586) encodes a centrosomal protein that is essential for primary cilia formation. Development 2009, 136, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Kim, S.; Lin, Y.C.; Inoue, T.; Dynlacht, B.D. The cp110-interacting proteins talpid3 and cep290 play overlapping and distinct roles in cilia assembly. J. Cell Biol. 2014, 204, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Nachury, M.V.; Loktev, A.V.; Zhang, Q.; Westlake, C.J.; Peranen, J.; Merdes, A.; Slusarski, D.C.; Scheller, R.H.; Bazan, J.F.; Sheffield, V.C.; et al. A core complex of bbs proteins cooperates with the gtpase rab8 to promote ciliary membrane biogenesis. Cell 2007, 129, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Westlake, C.J.; Baye, L.M.; Nachury, M.V.; Wright, K.J.; Ervin, K.E.; Phu, L.; Chalouni, C.; Beck, J.S.; Kirkpatrick, D.S.; Slusarski, D.C. Primary cilia membrane assembly is initiated by rab11 and transport protein particle ii (trappii) complex-dependent trafficking of rabin8 to the centrosome. Proc. Natl. Acad. Sci. USA 2011, 108, 2759–2764. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.-C.; Tong, Z.J.; Westfall, J.E.; Ault, J.G.; Page-McCaw, P.S.; Ferland, R.J. Ahi1, whose human ortholog is mutated in joubert syndrome, is required for rab8a localization, ciliogenesis and vesicle trafficking. Hum. Mol. Genet. 2009, 18, 3926–3941. [Google Scholar] [CrossRef] [PubMed]
- Dixon-Salazar, T.; Silhavy, J.L.; Marsh, S.E.; Louie, C.M.; Scott, L.C.; Gururaj, A.; Al-Gazali, L.; Al-Tawari, A.A.; Kayserili, H.; Sztriha, L. Mutations in the ahi1 gene, encoding jouberin, cause joubert syndrome with cortical polymicrogyria. Am. J. Hum. Genet. 2004, 75, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Guo, W. Rabs and the exocyst in ciliogenesis, tubulogenesis and beyond. Trends Cell Biol. 2011, 21, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Hammer, J.A.; Wu, X.S. Rabs grab motors: Defining the connections between rab gtpases and motor proteins. Curr. Opin. Cell Biol. 2002, 14, 69–75. [Google Scholar] [CrossRef]
- Sahlender, D.A.; Roberts, R.C.; Arden, S.D.; Spudich, G.; Taylor, M.J.; Luzio, J.P.; Kendrick-Jones, J.; Buss, F. Optineurin links myosin vi to the golgi complex and is involved in golgi organization and exocytosis. J. Cell Biol. 2005, 169, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Mazelova, J.; Ransom, N.; Astuto-Gribble, L.; Wilson, M.C.; Deretic, D. Syntaxin 3 and SNAP-25 pairing, regulated by omega-3 docosahexaenoic acid, controls the delivery of rhodopsin for the biogenesis of cilia-derived sensory organelles, the rod outer segments. J. Cell Sci. 2009, 122, 2003–2013. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gao, J.; Adamian, M.; Wen, X.-H.; Pawlyk, B.; Zhang, L.; Sanderson, M.J.; Zuo, J.; Makino, C.L.; Li, T. The ciliary rootlet maintains long-term stability of sensory cilia. Mol. Cell. Boil. 2005, 25, 4129–4137. [Google Scholar] [CrossRef] [PubMed]
- Kee, H.L.; Dishinger, J.F.; Blasius, T.L.; Liu, C.-J.; Margolis, B.; Verhey, K.J. A size-exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nat. Cell Biol. 2012, 14, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.L.; Child, F. Flagellar regeneration in protozoan flagellates. J. Cell Biol. 1967, 34, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.-G.; Kwok, B.H.; Kernan, M.J. Intraflagellar transport is required in drosophila to differentiate sensory cilia but not sperm. Curr. Biol. 2003, 13, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Kozminski, K.G.; Johnson, K.A.; Forscher, P.; Rosenbaum, J.L. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc. Natl. Acad. Sci. USA 1993, 90, 5519–5523. [Google Scholar] [CrossRef] [PubMed]
- Snow, J.J.; Ou, G.; Gunnarson, A.L.; Walker, M.R.S.; Zhou, H.M.; Brust-Mascher, I.; Scholey, J.M. Two anterograde intraflagellar transport motors cooperate to build sensory cilia on C. elegans neurons. Nat. Cell Biol. 2004, 6, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Ou, G.; Civelekoglu-Scholey, G.; Blacque, O.E.; Endres, N.F.; Tao, L.; Mogilner, A.; Leroux, M.R.; Vale, R.D.; Scholey, J.M. Mechanism of transport of ift particles in C. elegans cilia by the concerted action of kinesin-II and OSM-3 motors. J. Cell Biol. 2006, 174, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Jurczyk, A.; Gromley, A.; Redick, S.; San Agustin, J.; Witman, G.; Pazour, G.J.; Peters, D.J.; Doxsey, S. Pericentrin forms a complex with intraflagellar transport proteins and polycystin-2 and is required for primary cilia assembly. J. Cell Biol. 2004, 166, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.B.; Geimer, S.; Rosenbaum, J.L. Dissecting the molecular mechanisms of intraflagellar transport in chlamydomonas. Curr. Biol. 2006, 16, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.B.; Rosenbaum, J.L. Chapter two intraflagellar transport (IFT): Role in ciliary assembly, resorption and signalling. Curr. Topics Dev. Biol. 2008, 85, 23–61. [Google Scholar]
- Piperno, G.; Siuda, E.; Henderson, S.; Segil, M.; Vaananen, H.; Sassaroli, M. Distinct mutants of retrograde intraflagellar transport (ift) share similar morphological and molecular defects. J. Cell Biol. 1998, 143, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Ou, G.; Blacque, O.E.; Snow, J.J.; Leroux, M.R.; Scholey, J.M. Functional coordination of intraflagellar transport motors. Nature 2005, 436, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Liem, K.F., Jr.; Ashe, A.; He, M.; Satir, P.; Moran, J.; Beier, D.; Wicking, C.; Anderson, K.V. The ift-a complex regulates shh signaling through cilia structure and membrane protein trafficking. J. Cell Biol. 2012, 197, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Keady, B.T.; Samtani, R.; Tobita, K.; Tsuchya, M.; San Agustin, J.T.; Follit, J.A.; Jonassen, J.A.; Subramanian, R.; Lo, C.W.; Pazour, G.J. Ift25 links the signal-dependent movement of hedgehog components to intraflagellar transport. Dev. Cell 2012, 22, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Eguether, T.; San Agustin, J.T.; Keady, B.T.; Jonassen, J.A.; Liang, Y.; Francis, R.; Tobita, K.; Johnson, C.A.; Abdelhamed, Z.A.; Lo, C.W. Ift27 links the bbsome to ift for maintenance of the ciliary signaling compartment. Dev. Cell 2014, 31, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Bhogaraju, S.; Cajanek, L.; Fort, C.; Blisnick, T.; Weber, K.; Taschner, M.; Mizuno, N.; Lamla, S.; Bastin, P.; Nigg, E.A. Molecular basis of tubulin transport within the cilium by ift74 and ift81. Science 2013, 341, 1009–1012. [Google Scholar] [CrossRef] [PubMed]
- Craft, J.M.; Harris, J.A.; Hyman, S.; Kner, P.; Lechtreck, K.F. Tubulin transport by ift is upregulated during ciliary growth by a cilium-autonomous mechanism. J. Cell Biol. 2015, 208, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Breslow, D.K.; Koslover, E.F.; Spakowitz, A.J.; Nelson, W.J.; Nachury, M.V. Single molecule imaging reveals a major role for diffusion in the exploration of ciliary space by signaling receptors. eLife 2013, 2, e00654. [Google Scholar] [CrossRef] [PubMed]
- Cole, D.G.; Diener, D.R.; Himelblau, A.L.; Beech, P.L.; Fuster, J.C.; Rosenbaum, J.L. Chlamydomonas kinesin-ii–dependent intraflagellar transport (ift): Ift particles contain proteins required for ciliary assembly in caenorhabditis elegans sensory neurons. J. Cell Biol. 1998, 141, 993–1008. [Google Scholar] [CrossRef] [PubMed]
- Blacque, O.E.; Reardon, M.J.; Li, C.; McCarthy, J.; Mahjoub, M.R.; Ansley, S.J.; Badano, J.L.; Mah, A.K.; Beales, P.L.; Davidson, W.S. Loss of C. elegans Bbs-7 and bbs-8 protein function results in cilia defects and compromised intraflagellar transport. Genes Dev. 2004, 18, 1630–1642. [Google Scholar] [CrossRef] [PubMed]
- Pazour, G.J.; Wilkerson, C.G.; Witman, G.B. A dynein light chain is essential for the retrograde particle movement of intraflagellar transport (ift). J. Cell Biol. 1998, 141, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Blaineau, C.; Tessier, M.; Dubessay, P.; Tasse, L.; Crobu, L.; Pagès, M.; Bastien, P. A novel microtubule-depolymerizing kinesin involved in length control of a eukaryotic flagellum. Curr. Biol. 2007, 17, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Tsang, W.Y.; Li, J.; Lane, W.; Dynlacht, B.D. Centriolar kinesin kif24 interacts with cp110 to remodel microtubules and regulate ciliogenesis. Cell 2011, 145, 914–925. [Google Scholar] [CrossRef] [PubMed]
- San Agustin, J.T.; Pazour, G.J.; Witman, G.B. Intraflagellar transport is essential for mammalian spermiogenesis but is absent in mature sperm. Mol. Boil. Cell 2015, 26, 4358–4372. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.I.; Kleylein-Sohn, J.; Westendorf, J.; Le Clech, M.; Lavoie, S.B.; Stierhof, Y.D.; Nigg, E.A. Control of centriole length by cpap and cp110. Curr. Biol. 2009, 19, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Spektor, A.; Tsang, W.Y.; Khoo, D.; Dynlacht, B.D. Cep97 and cp110 suppress a cilia assembly program. Cell 2007, 130, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Tsang, W.Y.; Dynlacht, B.D. Cp110 and its network of partners coordinately regulate cilia assembly. Cilia 2013, 2, 9. [Google Scholar] [PubMed]
- Tsang, W.Y.; Bossard, C.; Khanna, H.; Peranen, J.; Swaroop, A.; Malhotra, V.; Dynlacht, B.D. Cp110 suppresses primary cilia formation through its interaction with cep290, a protein deficient in human ciliary disease. Dev. Cell 2008, 15, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Toedt, G.; Gouveia, S.M.; Davey, N.E.; Hua, S.; van der Vaart, B.; Grigoriev, I.; Larsen, J.; Pedersen, L.B.; Bezstarosti, K. A proteome-wide screen for mammalian sxip motif-containing microtubule plus-end tracking proteins. Curr. Biol. 2012, 22, 1800–1807. [Google Scholar] [CrossRef] [PubMed]
- Kuhns, S.; Schmidt, K.N.; Reymann, J.; Gilbert, D.F.; Neuner, A.; Hub, B.; Carvalho, R.; Wiedemann, P.; Zentgraf, H.; Erfle, H. The microtubule affinity regulating kinase mark4 promotes axoneme extension during early ciliogenesis. J. Cell Biol. 2013, 200, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Goetz, S.C.; Liem, K.F., Jr.; Anderson, K.V. The spinocerebellar ataxia-associated gene tau tubulin kinase 2 controls the initiation of ciliogenesis. Cell 2012, 151, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Kubo, A.; Tsukita, S. Odf2-deficient mother centrioles lack distal/subdistal appendages and the ability to generate primary cilia. Nat. Cell Biol. 2005, 7, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Inoko, A.; Matsuyama, M.; Goto, H.; Ohmuro-Matsuyama, Y.; Hayashi, Y.; Enomoto, M.; Ibi, M.; Urano, T.; Yonemura, S.; Kiyono, T.; et al. Trichoplein and aurora a block aberrant primary cilia assembly in proliferating cells. J. Cell Biol. 2012, 197, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, K.; Kawakami, Y.; Kiyono, T.; Yonemura, S.; Kawamura, Y.; Era, S.; Matsuzaki, F.; Goshima, N.; Inagaki, M. Ubiquitin-proteasome system controls ciliogenesis at the initial step of axoneme extension. Nat. Commun. 2014, 5, 5081. [Google Scholar] [CrossRef]
- Singla, V.; Romaguera-Ros, M.; Garcia-Verdugo, J.M.; Reiter, J.F. Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev. Cell 2010, 18, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, M.I.; Zullo, A.; Barra, A.; Bimonte, S.; Messaddeq, N.; Studer, M.; Dollé, P.; Franco, B. Oral-facial-digital type i protein is required for primary cilia formation and left-right axis specification. Nat. Genet. 2006, 38, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Lopes, C.A.; Prosser, S.L.; Romio, L.; Hirst, R.A.; O'Callaghan, C.; Woolf, A.S.; Fry, A.M. Centriolar satellites are assembly points for proteins implicated in human ciliopathies, including oral-facial-digital syndrome 1. J. Cell Sci. 2011, 124, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Lin, M.G.; Stowe, T.R.; Chen, S.; Zhu, M.; Stearns, T.; Franco, B.; Zhong, Q. Autophagy promotes primary ciliogenesis by removing ofd1 from centriolar satellites. Nature 2013, 502, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Stephens, R.E. Synthesis and turnover of embryonic sea urchin ciliary proteins during selective inhibition of tubulin synthesis and assembly. Mol. Biol. Cell 1997, 8, 2187–2198. [Google Scholar] [CrossRef] [PubMed]
- Marshall, W.F.; Rosenbaum, J.L. Intraflagellar transport balances continuous turnover of outer doublet microtubules implications for flagellar length control. J. Cell Biol. 2001, 155, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, B.; Asai, D.; Tang, W.; Hays, T.; Gibbons, I. Phylogeny and expression of axonemal and cytoplasmic dynein genes in sea urchins. Mol. Biol. Cell 1994, 5, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Marshall, W.F.; Qin, H.; Rodrigo Brenni, M.; Rosenbaum, J.L. Flagellar length control system: Testing a simple model based on intraflagellar transport and turnover. Mol. Biol. Cell 2005, 16, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Besschetnova, T.Y.; Kolpakova-Hart, E.; Guan, Y.; Zhou, J.; Olsen, B.R.; Shah, J.V. Identification of signaling pathways regulating primary cilium length and flow-mediated adaptation. Curr. Biol. 2010, 20, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Palmer, K.J.; MacCarthy-Morrogh, L.; Smyllie, N.; Stephens, D.J. A role for tctex-1 (dynlt1) in controlling primary cilium length. Eur. J. Cell Boil. 2011, 90, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Kosan, Z.A.; Stallworth, J.E.; Berbari, N.F.; Yoder, B.K. Soluble levels of cytosolic tubulin regulate ciliary length control. Mol. Boil. Cell 2011, 22, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Piao, T.; Cao, M.; Qin, T.; Huang, L.; Deng, H.; Mao, T.; Pan, J. Flagellar regeneration requires cytoplasmic microtubule depolymerization and kinesin-13. J. Cell Sci. 2013, 126, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.L.; Witman, G.B. Intraflagellar transport. Nat. Rev. Mol. Cell Biol. 2002, 3, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Ou, G.; Koga, M.; Blacque, O.E.; Murayama, T.; Ohshima, Y.; Schafer, J.C.; Li, C.; Yoder, B.K.; Leroux, M.R.; Scholey, J.M. Sensory ciliogenesis in caenorhabditis elegans: Assignment of ift components into distinct modules based on transport and phenotypic profiles. Mol. Biol. Cell 2007, 18, 1554–1569. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.E.; Swiderski, R.E.; Rahmouni, K.; Nishimura, D.Y.; Mullins, R.F.; Agassandian, K.; Philp, A.R.; Searby, C.C.; Andrews, M.P.; Thompson, S. A knockin mouse model of the bardet–biedl syndrome 1 m390r mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc. Natl. Acad. Sci. USA 2007, 104, 19422–19427. [Google Scholar] [CrossRef] [PubMed]
- Berbari, N.F.; Lewis, J.S.; Bishop, G.A.; Askwith, C.C.; Mykytyn, K. Bardet–biedl syndrome proteins are required for the localization of g protein-coupled receptors to primary cilia. Proc. Natl. Acad. Sci. USA 2008, 105, 4242–4246. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; White, S.R.; Shida, T.; Schulz, S.; Aguiar, M.; Gygi, S.P.; Bazan, J.F.; Nachury, M.V. The conserved bardet-biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 2010, 141, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Mykytyn, K.; Mullins, R.F.; Andrews, M.; Chiang, A.P.; Swiderski, R.E.; Yang, B.; Braun, T.; Casavant, T.; Stone, E.M.; Sheffield, V.C. Bardet-biedl syndrome type 4 (bbs4)-null mice implicate bbs4 in flagella formation but not global cilia assembly. Proc. Natl. Acad. Sci. USA 2004, 101, 8664–8669. [Google Scholar] [CrossRef] [PubMed]
- Berman, S.A.; Wilson, N.F.; Haas, N.A.; Lefebvre, P.A. A novel map kinase regulates flagellar length in chlamydomonas. Curr. Biol. 2003, 13, 1145–1149. [Google Scholar] [CrossRef]
- Burghoorn, J.; Dekkers, M.P.; Rademakers, S.; de Jong, T.; Willemsen, R.; Jansen, G. Mutation of the map kinase dyf-5 affects docking and undocking of kinesin-2 motors and reduces their speed in the cilia of caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2007, 104, 7157–7162. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.F.; Lefebvre, P.A. Regulation of flagellar assembly by glycogen synthase kinase 3 in chlamydomonas reinhardtii. Eukaryot. Cell 2004, 3, 1307–1319. [Google Scholar] [CrossRef] [PubMed]
- Sirajuddin, M.; Rice, L.M.; Vale, R.D. Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat. Cell Biol. 2014, 16, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Pathak, N.; Obara, T.; Mangos, S.; Liu, Y.; Drummond, I.A. The zebrafish fleer gene encodes an essential regulator of cilia tubulin polyglutamylation. Mol. Biol. Cell 2007, 18, 4353–4364. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Zaghloul, N.A.; Bubenshchikova, E.; Oh, E.C.; Rankin, S.; Katsanis, N.; Obara, T.; Tsiokas, L. Nde1-mediated suppression of ciliogenesis affects cell cycle re-entry. Nat. Cell Biol. 2011, 13, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Maskey, D.; Marlin, M.C.; Kim, S.; Kim, S.; Ong, E.C.; Li, G.; Tsiokas, L. Cell cycle-dependent ubiquitylation and destruction of nde1 by cdk5-fbw7 regulates ciliary length. EMBO J. 2015, 34, 2424–2440. [Google Scholar] [CrossRef] [PubMed]
- Takeishi, S.; Nakayama, K.I. Role of fbxw7 in the maintenance of normal stem cells and cancer-initiating cells. Br. J. Cancer 2014, 111, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Yada, M.; Nishiyama, M.; Kamura, T.; Takahashi, H.; Tsunematsu, R.; Susaki, E.; Nakagawa, T.; Matsumoto, A.; Nakayama, K.I. Fbxw7 contributes to tumor suppression by targeting multiple proteins for ubiquitin-dependent degradation. Cancer Sci. 2006, 97, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, T.; Kirschner, M.W. The master cell cycle regulator apc-cdc20 regulates ciliary length and disassembly of the primary cilium. eLife 2014, 3, e03083. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Porazinski, S.; Wang, H.; Borovina, A.; Ciruna, B.; Shimizu, A.; Kajii, T.; Kikuchi, A.; Furutani-Seiki, M.; Matsuura, S. Insufficiency of bubr1, a mitotic spindle checkpoint regulator, causes impaired ciliogenesis in vertebrates. Hum. Mol. Genet. 2011. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, J.G.; Jackson, C.L. Arf family g proteins and their regulators: Roles in membrane transport, development and disease. Nat. Rev. Mol. Cell Boil. 2011, 12, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.S.; Chua, C.E.L.; Tang, B.L. Rabs and other small gtpases in ciliary transport. Biol. Cell 2011, 103, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ling, K.; Hu, J. The emerging role of arf/arl small gtpases in cilia and ciliopathies. J. Cell. Biochem. 2012, 113, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wei, Q.; Zhang, Y.; Ling, K.; Hu, J. The small gtpases arl-13 and arl-3 coordinate intraflagellar transport and ciliogenesis. J. Cell Biol. 2010, 189, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Esmail, M.A.; Ansley, S.J.; Blacque, O.E.; Boroevich, K.; Ross, A.J.; Moore, S.J.; Badano, J.L.; May-Simera, H.; Compton, D.S. Mutations in a member of the ras superfamily of small gtp-binding proteins causes bardet-biedl syndrome. Nat. Genet. 2004, 36, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Larkins, C.E.; Aviles, G.D.G.; East, M.P.; Kahn, R.A.; Caspary, T. Arl13b regulates ciliogenesis and the dynamic localization of shh signaling proteins. Mol. Biol. Cell 2011, 22, 4694–4703. [Google Scholar] [CrossRef] [PubMed]
- Wiens, C.J.; Tong, Y.; Esmail, M.A.; Oh, E.; Gerdes, J.M.; Wang, J.; Tempel, W.; Rattner, J.B.; Katsanis, N.; Park, H.-W. Bardet-biedl syndrome-associated small gtpase arl6 (bbs3) functions at or near the ciliary gate and modulates wnt signaling. J. Biol. Chem. 2010, 285, 16218–16230. [Google Scholar] [CrossRef] [PubMed]
- Pigino, G.; Geimer, S.; Lanzavecchia, S.; Paccagnini, E.; Cantele, F.; Diener, D.R.; Rosenbaum, J.L.; Lupetti, P. Electron-tomographic analysis of intraflagellar transport particle trains in situ. J. Cell Biol. 2009, 187, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Cevik, S.; Hori, Y.; Kaplan, O.I.; Kida, K.; Toivenon, T.; Foley-Fisher, C.; Cottell, D.; Katada, T.; Kontani, K.; Blacque, O.E. Joubert syndrome arl13b functions at ciliary membranes and stabilizes protein transport in caenorhabditis elegans. J. Cell Biol. 2010, 188, 953–969. [Google Scholar] [CrossRef] [PubMed]
- Quarmby, L.M.; Parker, J.D. Cilia and the cell cycle? J. Cell Biol. 2005, 169, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Pugacheva, E.N.; Jablonski, S.A.; Hartman, T.R.; Henske, E.P.; Golemis, E.A. Hef1-dependent aurora a activation induces disassembly of the primary cilium. Cell 2007, 129, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Kinzel, D.; Boldt, K.; Davis, E.E.; Burtscher, I.; Trumbach, D.; Diplas, B.; Attie-Bitach, T.; Wurst, W.; Katsanis, N.; Ueffing, M.; et al. Pitchfork regulates primary cilia disassembly and left-right asymmetry. Dev. Cell 2010, 19, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Saito, M.; Chuang, J.Z.; Tseng, Y.Y.; Dedesma, C.; Tomizawa, K.; Kaitsuka, T.; Sung, C.H. Ciliary transition zone activation of phosphorylated tctex-1 controls ciliary resorption, s-phase entry and fate of neural progenitors. Nat. Cell Biol. 2011, 13, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Johmura, Y.; Yu, L.R.; Park, J.E.; Gao, Y.; Bang, J.K.; Zhou, M.; Veenstra, T.D.; Kim, B.Y.; Lee, K.S. Identification of a novel wnt5a–ck1ε–dvl2–plk1-mediated primary cilia disassembly pathway. EMBO J. 2012, 31, 3104–3117. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Li, G.; Pan, J. Regulation of cilia assembly, disassembly, and length by protein phosphorylation. Methods Cell Biol. 2009, 94, 333–346. [Google Scholar] [PubMed]
- Jacoby, M.; Cox, J.J.; Gayral, S.; Hampshire, D.J.; Ayub, M.; Blockmans, M.; Pernot, E.; Kisseleva, M.V.; Compere, P.; Schiffmann, S.N.; et al. Inpp5e mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat. Genet. 2009, 41, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Bielas, S.L.; Silhavy, J.L.; Brancati, F.; Kisseleva, M.V.; Al-Gazali, L.; Sztriha, L.; Bayoumi, R.A.; Zaki, M.S.; Abdel-Aleem, A.; Rosti, R.O.; et al. Mutations in inpp5e, encoding inositol polyphosphate-5-phosphatase e, link phosphatidyl inositol signaling to the ciliopathies. Nat. Genet. 2009, 41, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Rasi, M.Q.; Parker, J.D.; Feldman, J.L.; Marshall, W.F.; Quarmby, L.M. Katanin knockdown supports a role for microtubule severing in release of basal bodies before mitosis in chlamydomonas. Mol. Biol. Cell 2009, 20, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.D.; Hilton, L.K.; Diener, D.R.; Qasim Rasi, M.; Mahjoub, M.R.; Rosenbaum, J.L.; Quarmby, L.M. Centrioles are freed from cilia by severing prior to mitosis. Cytoskeleton 2010, 67, 425–430. [Google Scholar] [CrossRef] [PubMed]
- McNally, F.J.; Vale, R.D. Identification of katanin, an atpase that severs and disassembles stable microtubules. Cell 1993, 75, 419–429. [Google Scholar] [CrossRef]
- Sudo, H.; Baas, P.W. Acetylation of microtubules influences their sensitivity to severing by katanin in neurons and fibroblasts. J. Neurosci. 2010, 30, 7215–7226. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.F.; Pomp, O.; Ben-Omran, T.; Kodani, A.; Henke, K.; Mochida, G.H.; Timothy, W.Y.; Woodworth, M.B.; Bonnard, C.; Raj, G.S. Katanin p80 regulates human cortical development by limiting centriole and cilia number. Neuron 2014, 84, 1240–1257. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keeling, J.; Tsiokas, L.; Maskey, D. Cellular Mechanisms of Ciliary Length Control. Cells 2016, 5, 6. https://doi.org/10.3390/cells5010006
Keeling J, Tsiokas L, Maskey D. Cellular Mechanisms of Ciliary Length Control. Cells. 2016; 5(1):6. https://doi.org/10.3390/cells5010006
Chicago/Turabian StyleKeeling, Jacob, Leonidas Tsiokas, and Dipak Maskey. 2016. "Cellular Mechanisms of Ciliary Length Control" Cells 5, no. 1: 6. https://doi.org/10.3390/cells5010006
APA StyleKeeling, J., Tsiokas, L., & Maskey, D. (2016). Cellular Mechanisms of Ciliary Length Control. Cells, 5(1), 6. https://doi.org/10.3390/cells5010006