
Photoreceptor Sensory Cilium: Traversing the Ciliary Gate

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
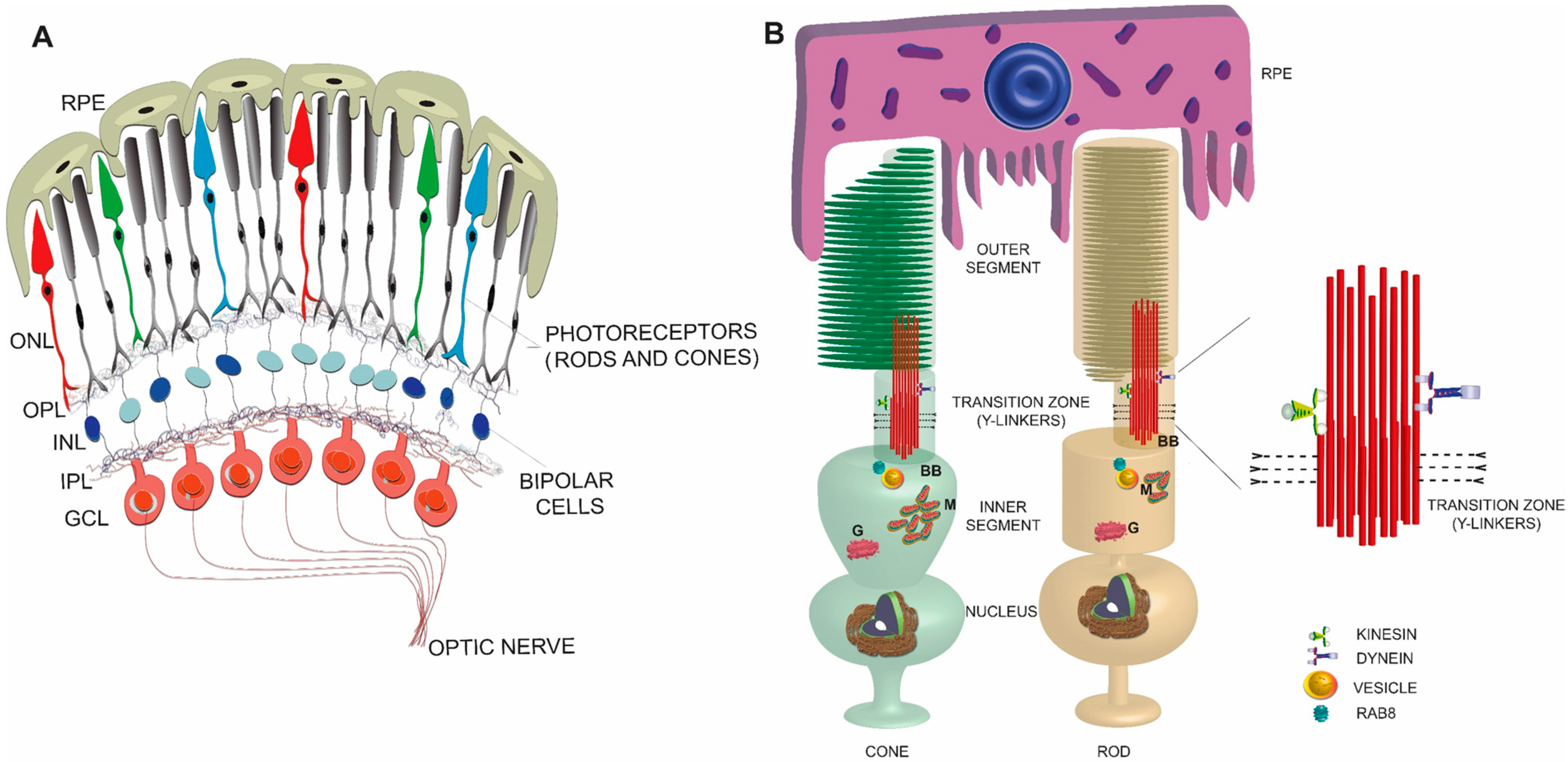
2. Retina
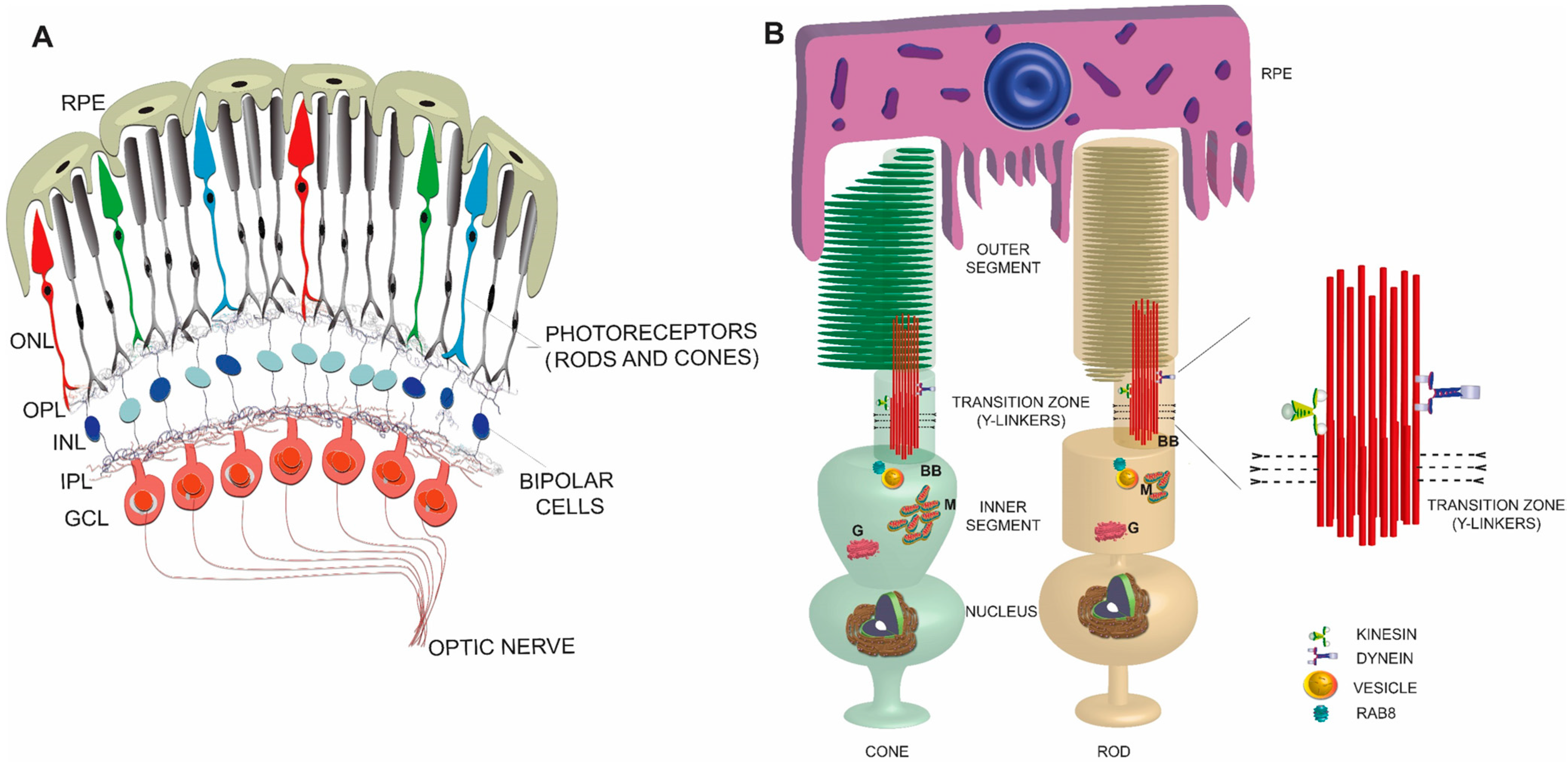
3. Photoreceptors
4. Photoreceptor Sensory (or Primary) Cilium
5. Photoreceptor Ciliogenesis
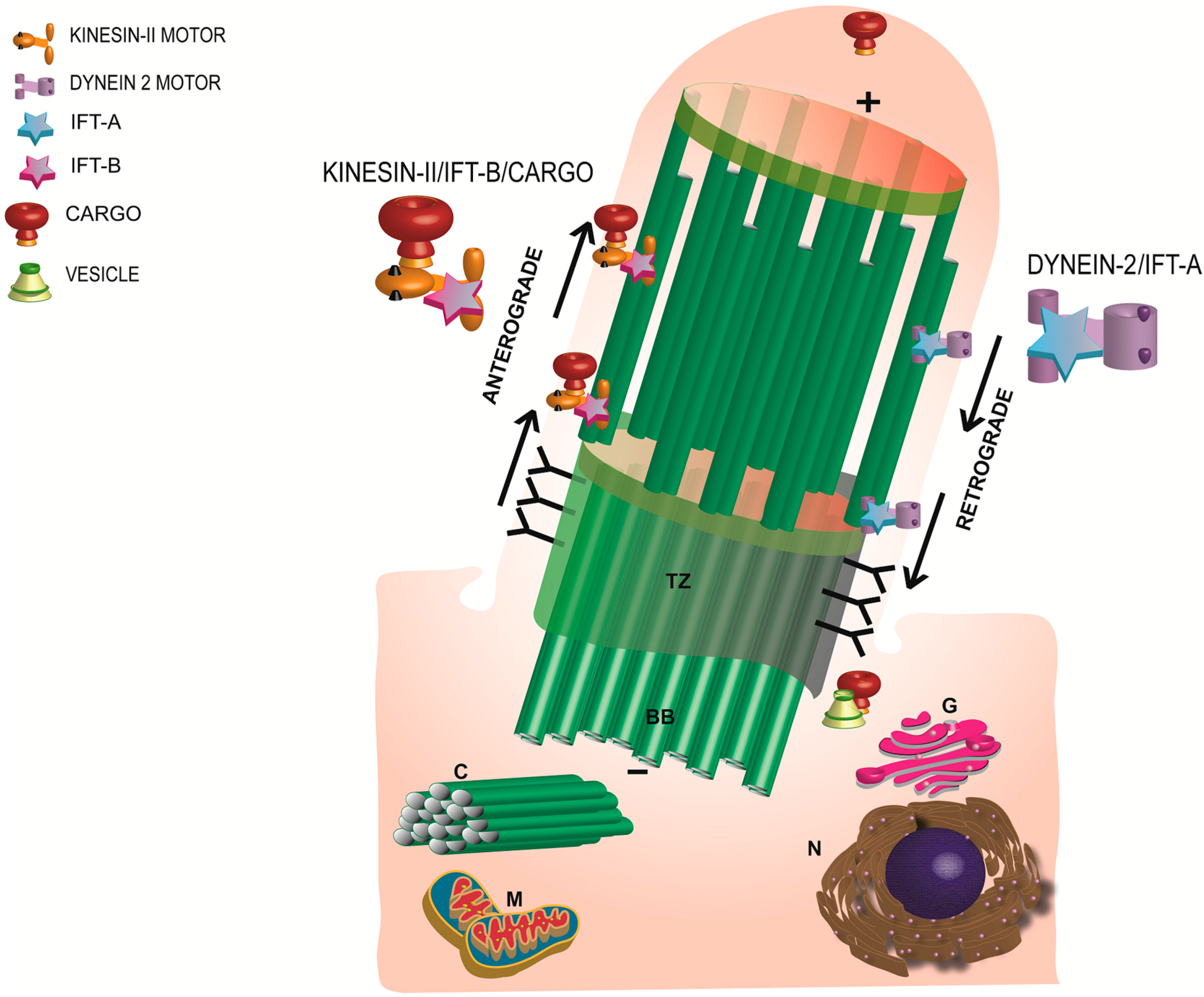
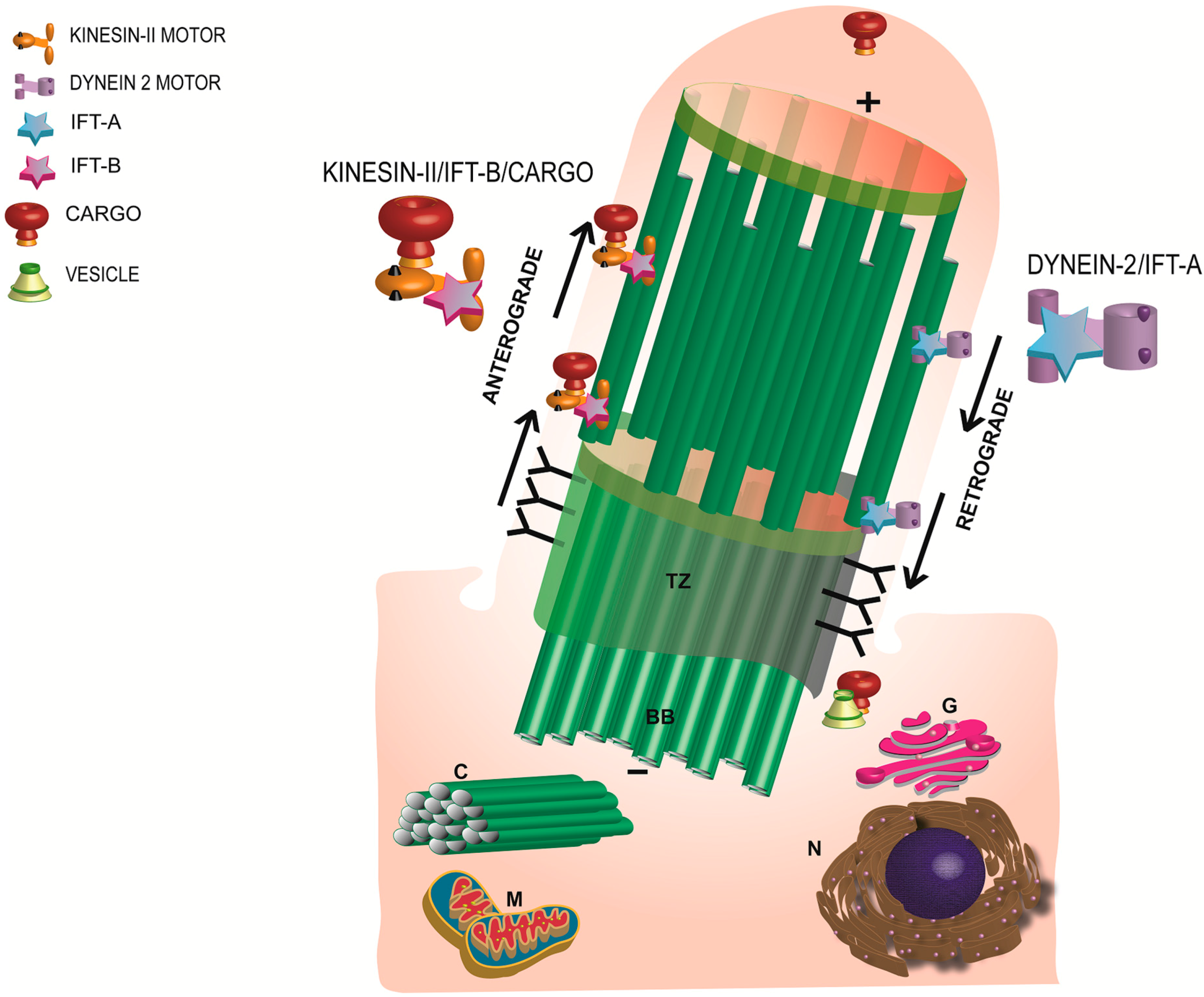
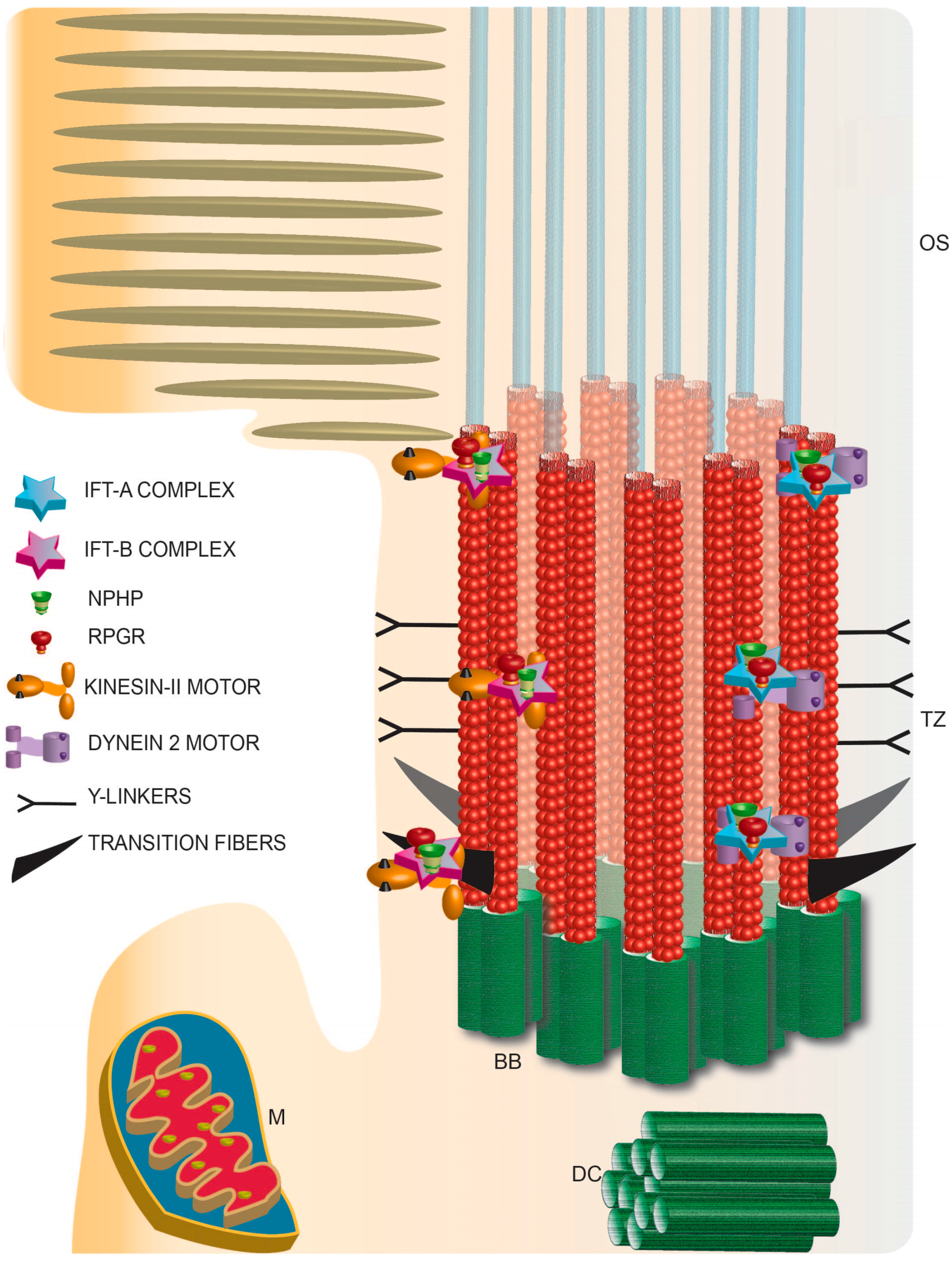
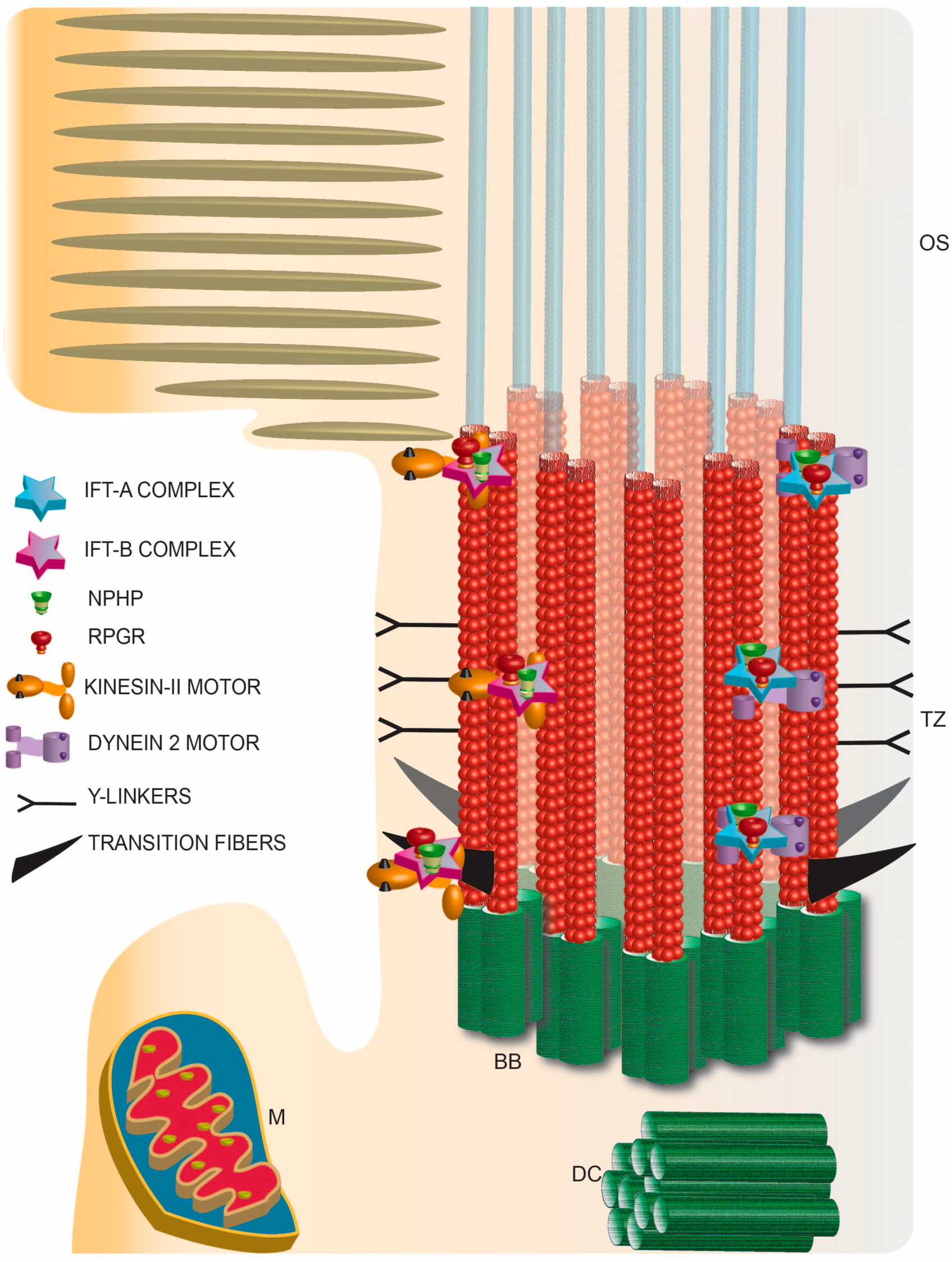
6. Photoreceptor Ciliary Trafficking
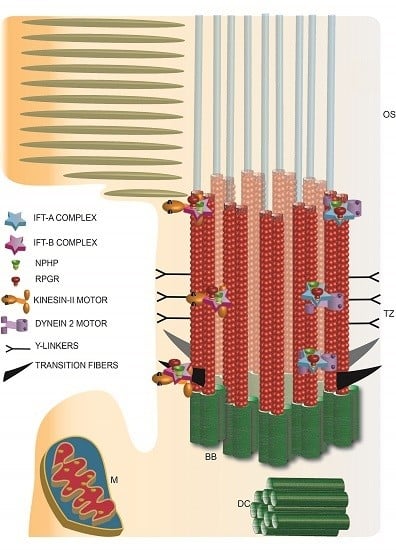
7. Photoreceptor Ciliary Gate
8. Traversing the Ciliary Gate
9. Concluding Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kandel, E.R. Principles of Neural Science, 5th ed.; McGraw-Hill: New York, NY, USA, 2013; p. 577. [Google Scholar]
- Gray, H.; Pick, T.P.; Howden, R. Anatomy, Descriptive and Surgical, Classic collector’s ed.; Fall River Press: New York, NY, USA, 1987; p. 1257. [Google Scholar]
- Lamb, T.D. Evolution of phototransduction, vertebrate photoreceptors and retina. Prog. Retin. Eye Res. 2013, 36, 52–119. [Google Scholar] [CrossRef] [PubMed]
- Pazour, G.J.; Witman, G.B. The vertebrate primary cilium is a sensory organelle. Curr. Opin. Cell Biol. 2003, 15, 105–110. [Google Scholar] [CrossRef]
- Berbari, N.F.; O’Connor, A.K.; Haycraft, C.J.; Yoder, B.K. The primary cilium as a complex signaling center. Curr. Biol. 2009, 19, R526–R535. [Google Scholar] [CrossRef] [PubMed]
- Eggenschwiler, J.T.; Anderson, K.V. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 2007, 23, 345–373. [Google Scholar] [CrossRef] [PubMed]
- Singla, V.; Reiter, J.F. The primary cilium as the cell's antenna: Signaling at a sensory organelle. Science 2006, 313, 629–633. [Google Scholar] [CrossRef] [PubMed]
- McEwen, D.P.; Koenekoop, R.K.; Khanna, H.; Jenkins, P.M.; Lopez, I.; Swaroop, A.; Martens, J.R. Hypomorphic cep290/nphp6 mutations result in anosmia caused by the selective loss of g proteins in cilia of olfactory sensory neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 15917–15922. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, O.; Khanna, H. Ciliary signaling cascades in photoreceptors. Vis. Res. 2012, 75, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.B.; Veland, I.R.; Schroder, J.M.; Christensen, S.T. Assembly of primary cilia. Dev. Dyn. 2008, 237, 1993–2006. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [PubMed]
- Hildebrandt, F.; Otto, E. Cilia and centrosomes: A unifying pathogenic concept for cystic kidney disease? Nat. Rev. Genet. 2005, 6, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Nachury, M.V. The bbsome. Curr. Biol. 2009, 19, R472–R473. [Google Scholar] [CrossRef] [PubMed]
- Fliegauf, M.; Benzing, T.; Omran, H. When cilia go bad: Cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007, 8, 880–893. [Google Scholar] [CrossRef] [PubMed]
- De Robertis, E. Morphogenesis of the retinal rods; an electron microscope study. J. Biophys. Biochem. Cytol. 1956, 2, 209–218. [Google Scholar] [CrossRef] [PubMed]
- De Robertis, E. Some observations on the ultrastructure and morphogenesis of photoreceptors. J. Gen. Physiol. 1960, 43, 1–13. [Google Scholar] [CrossRef]
- Sjostrand, F.S. The ultrastructure of the innersegments of the retinal rods of the guinea pig eye as revealed by electron microscopy. J. Cell. Physiol. 1953, 42, 45–70. [Google Scholar] [CrossRef] [PubMed]
- Sjostrand, F.S. The ultrastructure of the outer segments of rods and cones of the eye as revealed by the electron microscope. J. Cell. Physiol. 1953, 42, 15–44. [Google Scholar] [CrossRef] [PubMed]
- Tokuyasu, K.; Yamada, E. The fine structure of the retina studied with the electron microscope. Iv. Morphogenesis of outer segments of retinal rods. J. Biophys. Biochem. Cytol. 1959, 6, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Rajala, A.; Dighe, R.; Agbaga, M.P.; Anderson, R.E.; Rajala, R.V. Insulin receptor signaling in cones. J. Biol. Chem. 2013, 288, 19503–19515. [Google Scholar] [CrossRef] [PubMed]
- Besharse, J.C. The Retina: A Model for Cell Biological Studies Part I; Academic: New York, NY, USA, 1986; pp. 297–352. [Google Scholar]
- Wang, J.; Deretic, D. Molecular complexes that direct rhodopsin transport to primary cilia. Prog. Retin. Eye Res. 2014, 38, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Aveldano, M.I.; Bazan, N.G. Molecular species of phosphatidylcholine, -ethanolamine, -serine, and -inositol in microsomal and photoreceptor membranes of bovine retina. J. Lipid Res. 1983, 24, 620–627. [Google Scholar] [PubMed]
- Doxsey, S. Re-evaluating centrosome function. Nat. Rev. Mol. Cell Biol. 2001, 2, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Basto, R.; Lau, J.; Vinogradova, T.; Gardiol, A.; Woods, C.G.; Khodjakov, A.; Raff, J.W. Flies without centrioles. Cell 2006, 125, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt-Dias, M.; Rodrigues-Martins, A.; Carpenter, L.; Riparbelli, M.; Lehmann, L.; Gatt, M.K.; Carmo, N.; Balloux, F.; Callaini, G.; Glover, D.M. Sak/plk4 is required for centriole duplication and flagella development. Curr. Biol. 2005, 15, 2199–2207. [Google Scholar] [CrossRef] [PubMed]
- Marshall, W.F. Centriole evolution. Curr. Opin. Cell Biol. 2009, 21, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, J.M.; Davis, E.E.; Katsanis, N. The vertebrate primary cilium in development, homeostasis, and disease. Cell 2009, 137, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J. Intraflagellar transport. Curr. Biol. 2002, 12, R125. [Google Scholar] [CrossRef]
- Rosenbaum, J.L.; Witman, G.B. Intraflagellar transport. Nat. Rev. Mol. Cell Biol. 2002, 3, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Pazour, G.J.; Baker, S.A.; Deane, J.A.; Cole, D.G.; Dickert, B.L.; Rosenbaum, J.L.; Witman, G.B.; Besharse, J.C. The intraflagellar transport protein, ift88, is essential for vertebrate photoreceptor assembly and maintenance. J. Cell Biol. 2002, 157, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, M.; Malicki, J. Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron 2004, 42, 703–716. [Google Scholar] [CrossRef]
- Roepman, R.; Wolfrum, U. Protein networks and complexes in photoreceptor cilia. Subcell. Biochem. 2007, 43, 209–235. [Google Scholar] [PubMed]
- Sedmak, T.; Sehn, E.; Wolfrum, U. Immunoelectron microscopy of vesicle transport to the primary cilium of photoreceptor cells. Methods Cell Biol. 2009, 94, 259–272. [Google Scholar] [PubMed]
- Sedmak, T.; Wolfrum, U. Intraflagellar transport proteins in ciliogenesis of photoreceptor cells. Biol. Cell 2011, 103, 449–466. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Tan, G.; Levenkova, N.; Li, T.; Pugh, E.N., Jr.; Rux, J.J.; Speicher, D.W.; Pierce, E.A. The proteome of the mouse photoreceptor sensory cilium complex. Mol. Cell. Proteom. 2007, 6, 1299–1317. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.N.; Li, L.; Anand, M.; Khanna, H. Ablation of retinal ciliopathy protein rpgr results in altered photoreceptor ciliary composition. Sci. Rep. 2015, 5, 11137. [Google Scholar] [CrossRef]
- Follit, J.A.; Tuft, R.A.; Fogarty, K.E.; Pazour, G.J. The intraflagellar transport protein ift20 is associated with the golgi complex and is required for cilia assembly. Mol. Biol. Cell 2006, 17, 3781–3792. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.A.; Lopes, V.S.; Sanagustin, J.T.; Keady, B.T.; Williams, D.S.; Pazour, G.J. Distinct functions for ift140 and ift20 in opsin transport. Cytoskelet. (Hoboken) 2014, 71, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wei, Y.; Ronquillo, C.C.; Marc, R.E.; Yoder, B.K.; Frederick, J.M.; Baehr, W. Heterotrimeric kinesin-2 (kif3) mediates transition zone and axoneme formation of mouse photoreceptors. J. Biol. Chem. 2015, 290, 12765–12778. [Google Scholar] [CrossRef] [PubMed]
- Marszalek, J.R.; Liu, X.; Roberts, E.A.; Chui, D.; Marth, J.D.; Williams, D.S.; Goldstein, L.S. Genetic evidence for selective transport of opsin and arrestin by kinesin-ii in mammalian photoreceptors. Cell 2000, 102, 175–187. [Google Scholar] [CrossRef]
- Hollingsworth, T.J.; Gross, A.K. Defective trafficking of rhodopsin and its role in retinal degenerations. Int. Rev. Cell Mol. Biol. 2012, 293, 1–44. [Google Scholar]
- Mazelova, J.; Astuto-Gribble, L.; Inoue, H.; Tam, B.M.; Schonteich, E.; Prekeris, R.; Moritz, O.L.; Randazzo, P.A.; Deretic, D. Ciliary targeting motif vxpx directs assembly of a trafficking module through arf4. EMBO J. 2009, 28, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Papermaster, D.S.; Schneider, B.G.; Besharse, J.C. Vesicular transport of newly synthesized opsin from the golgi apparatus toward the rod outer segment. Ultrastructural immunocytochemical and autoradiographic evidence in xenopus retinas. Investig. Ophthalmol. Vis. Sci. 1985, 26, 1386–1404. [Google Scholar]
- Rao, K.N.; Khanna, H. Role of small gtpases in polarized vesicle transport to primary cilium. Res. Rep. Biol. 2015, 6, 17–24. [Google Scholar]
- Baker, S.A.; Haeri, M.; Yoo, P.; Gospe, S.M., 3rd; Skiba, N.P.; Knox, B.E.; Arshavsky, V.Y. The outer segment serves as a default destination for the trafficking of membrane proteins in photoreceptors. J. Cell Biol. 2008, 183, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Kizhatil, K.; Baker, S.A.; Arshavsky, V.Y.; Bennett, V. Ankyrin-g promotes cyclic nucleotide-gated channel transport to rod photoreceptor sensory cilia. Science 2009, 323, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Conley, S.M.; Stuck, M.W.; Naash, M.I. Differences in rds trafficking, assembly and function in cones versus rods: Insights from studies of c150s-rds. Hum. Mol. Genet. 2010, 19, 4799–4812. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Ropelewski, P.; Nemet, I.; Lee, R.; Lodowski, K.H.; Imanishi, Y. An unconventional secretory pathway mediates the cilia targeting of peripherin/rds. J. Neurosci. 2014, 34, 992–1006. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Li, M.; Sun, J.; Baker, S.A.; Insinna, C.; Besharse, J.C. Photoreceptor ift complexes containing chaperones, guanylyl cyclase 1 and rhodopsin. Traffic 2009, 10, 648–663. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; White, S.R.; Shida, T.; Schulz, S.; Aguiar, M.; Gygi, S.P.; Bazan, J.F.; Nachury, M.V. The conserved bardet-biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 2010, 141, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Nachury, M.V.; Loktev, A.V.; Zhang, Q.; Westlake, C.J.; Peranen, J.; Merdes, A.; Slusarski, D.C.; Scheller, R.H.; Bazan, J.F.; Sheffield, V.C.; et al. A core complex of bbs proteins cooperates with the gtpase rab8 to promote ciliary membrane biogenesis. Cell 2007, 129, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Calvert, P.D.; Strissel, K.J.; Schiesser, W.E.; Pugh, E.N., Jr.; Arshavsky, V.Y. Light-driven translocation of signaling proteins in vertebrate photoreceptors. Trends Cell Biol. 2006, 16, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, M.; Lyubarsky, A.L.; Strissel, K.J.; Savchenko, A.B.; Govardovskii, V.I.; Pugh, E.N., Jr.; Arshavsky, V.Y. Massive light-driven translocation of transducin between the two major compartments of rod cells: A novel mechanism of light adaptation. Neuron 2002, 34, 95–106. [Google Scholar] [CrossRef]
- Strissel, K.J.; Lishko, P.V.; Trieu, L.H.; Kennedy, M.J.; Hurley, J.B.; Arshavsky, V.Y. Recoverin undergoes light-dependent intracellular translocation in rod photoreceptors. J. Biol. Chem. 2005, 280, 29250–29255. [Google Scholar] [CrossRef] [PubMed]
- Nair, K.S.; Hanson, S.M.; Mendez, A.; Gurevich, E.V.; Kennedy, M.J.; Shestopalov, V.I.; Vishnivetskiy, S.A.; Chen, J.; Hurley, J.B.; Gurevich, V.V.; et al. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron 2005, 46, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Slepak, V.Z.; Hurley, J.B. Mechanism of light-induced translocation of arrestin and transducin in photoreceptors: Interaction-restricted diffusion. IUBMB Life 2008, 60, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.C.; Chuang, J.Z.; Sung, C.H. Light regulates the ciliary protein transport and outer segment disc renewal of mammalian photoreceptors. Dev. Cell 2015, 32, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Sang, L.; Miller, J.J.; Corbit, K.C.; Giles, R.H.; Brauer, M.J.; Otto, E.A.; Baye, L.M.; Wen, X.; Scales, S.J.; Kwong, M.; et al. Mapping the nphp-jbts-mks protein network reveals ciliopathy disease genes and pathways. Cell 2011, 145, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Fliegauf, M.; Horvath, J.; von Schnakenburg, C.; Olbrich, H.; Muller, D.; Thumfart, J.; Schermer, B.; Pazour, G.J.; Neumann, H.P.; Zentgraf, H.; et al. Nephrocystin specifically localizes to the transition zone of renal and respiratory cilia and photoreceptor connecting cilia. J. Am. Soc. Nephrol. 2006, 17, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Murga-Zamalloa, C.A.; Desai, N.J.; Hildebrandt, F.; Khanna, H. Interaction of ciliary disease protein retinitis pigmentosa gtpase regulator with nephronophthisis-associated proteins in mammalian retinas. Mol. Vis. 2010, 16, 1373–1381. [Google Scholar] [PubMed]
- Anand, M.; Khanna, H. Ciliary transition zone (tz) proteins rpgr and cep290: Role in photoreceptor cilia and degenerative diseases. Expert Opin. Ther. Targets 2012, 16, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Benzing, T.; Schermer, B. Transition zone proteins and cilia dynamics. Nat. Genet. 2011, 43, 723–724. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.F.; Blacque, O.E.; Leroux, M.R. The base of the cilium: Roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 2012, 13, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Craige, B.; Tsao, C.C.; Diener, D.R.; Hou, Y.; Lechtreck, K.F.; Rosenbaum, J.L.; Witman, G.B. Cep290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J. Cell Biol. 2010, 190, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Besharse, J.C.; Bok, D. The Retina and its Disorders; Academic Press: Amsterdam, The Netherlands; Boston, MA, USA, 2011; p. xvi, 912. [Google Scholar]
- Peters, K.R.; Palade, G.E.; Schneider, B.G.; Papermaster, D.S. Fine structure of a periciliary ridge complex of frog retinal rod cells revealed by ultrahigh resolution scanning electron microscopy. J. Cell Biol. 1983, 96, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.; Detwiler, P.B.; Bunt-Milam, A.H. Distribution of membrane proteins in mechanically dissociated retinal rods. Investig. Ophthalmol. Vis. Sci. 1988, 29, 1012–1020. [Google Scholar]
- Den Hollander, A.I.; Koenekoop, R.K.; Yzer, S.; Lopez, I.; Arends, M.L.; Voesenek, K.E.; Zonneveld, M.N.; Strom, T.M.; Meitinger, T.; Brunner, H.G.; et al. Mutations in the cep290 (nphp6) gene are a frequent cause of leber congenital amaurosis. Am. J. Hum. Genet. 2006, 79, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Koenekoop, R.K. An overview of leber congenital amaurosis: A model to understand human retinal development. Surv. Ophthalmol. 2004, 49, 379–398. [Google Scholar] [CrossRef] [PubMed]
- Murga-Zamalloa, C.A.; Ghosh, A.K.; Patil, S.B.; Reed, N.A.; Chan, L.S.; Davuluri, S.; Peranen, J.; Hurd, T.W.; Rachel, R.A.; Khanna, H. Accumulation of the raf-1 kinase inhibitory protein (rkip) is associated with cep290-mediated photoreceptor degeneration in ciliopathies. J. Biol. Chem. 2011, 286, 28276–28286. [Google Scholar] [CrossRef]
- Sayer, J.A.; Otto, E.A.; O’Toole, J.F.; Nurnberg, G.; Kennedy, M.A.; Becker, C.; Hennies, H.C.; Helou, J.; Attanasio, M.; Fausett, B.V.; et al. The centrosomal protein nephrocystin-6 is mutated in joubert syndrome and activates transcription factor atf4. Nat. Genet. 2006, 38, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Chih, B.; Liu, P.; Chinn, Y.; Chalouni, C.; Komuves, L.G.; Hass, P.E.; Sandoval, W.; Peterson, A.S. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat. Cell Biol. 2012, 14, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalo, F.R.; Corbit, K.C.; Sirerol-Piquer, M.S.; Ramaswami, G.; Otto, E.A.; Noriega, T.R.; Seol, A.D.; Robinson, J.F.; Bennett, C.L.; Josifova, D.J.; et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet. 2011, 43, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.L.; Li, C.; Kida, K.; Inglis, P.N.; Mohan, S.; Semenec, L.; Bialas, N.J.; Stupay, R.M.; Chen, N.; Blacque, O.E.; et al. Mks and nphp modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J. Cell Biol. 2011, 192, 1023–1041. [Google Scholar] [CrossRef] [PubMed]
- Boldt, K.; Mans, D.A.; Won, J.; van Reeuwijk, J.; Vogt, A.; Kinkl, N.; Letteboer, S.J.; Hicks, W.L.; Hurd, R.E.; Naggert, J.K.; et al. Disruption of intraflagellar protein transport in photoreceptor cilia causes leber congenital amaurosis in humans and mice. J. Clin. Investig. 2011, 121, 2169–2180. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Malicki, J. Nephrocystins and mks proteins interact with ift particle and facilitate transport of selected ciliary cargos. EMBO J. 2011, 30, 2532–2544. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.T.; Chiou, Y.Y.; Wang, E.; Chien, Y.L.; He, H.H.; Tsai, F.J.; Lin, C.Y.; Tsai, S.P.; Li, H. Essential role of nephrocystin in photoreceptor intraflagellar transport in mouse. Hum. Mol. Genet. 2009, 18, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Calvert, P.D.; Schiesser, W.E.; Pugh, E.N., Jr. Diffusion of a soluble protein, photoactivatable gfp, through a sensory cilium. J. Gen. Physiol. 2010, 135, 173–196. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Calvert, P.D. Transport and localization of signaling proteins in ciliated cells. Vis. Res. 2012, 75, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Kee, H.L.; Verhey, K.J. Molecular connections between nuclear and ciliary import processes. Cilia 2013, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Niewiadomski, P.; Lin, B.; Nakamura, H.; Phua, S.C.; Jiao, J.; Levchenko, A.; Inoue, T.; Rohatgi, R.; Inoue, T. Chemically inducible diffusion trap at cilia reveals molecular sieve-like barrier. Nat. Chem. Biol. 2013, 9, 437–443. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khanna, H. Photoreceptor Sensory Cilium: Traversing the Ciliary Gate. Cells 2015, 4, 674-686. https://doi.org/10.3390/cells4040674
Khanna H. Photoreceptor Sensory Cilium: Traversing the Ciliary Gate. Cells. 2015; 4(4):674-686. https://doi.org/10.3390/cells4040674
Chicago/Turabian StyleKhanna, Hemant. 2015. "Photoreceptor Sensory Cilium: Traversing the Ciliary Gate" Cells 4, no. 4: 674-686. https://doi.org/10.3390/cells4040674
APA StyleKhanna, H. (2015). Photoreceptor Sensory Cilium: Traversing the Ciliary Gate. Cells, 4(4), 674-686. https://doi.org/10.3390/cells4040674