
NAD+-Metabolizing Ectoenzymes in Remodeling Tumor–Host Interactions: The Human Myeloma Model

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Premises
Biogenesis of NAD+
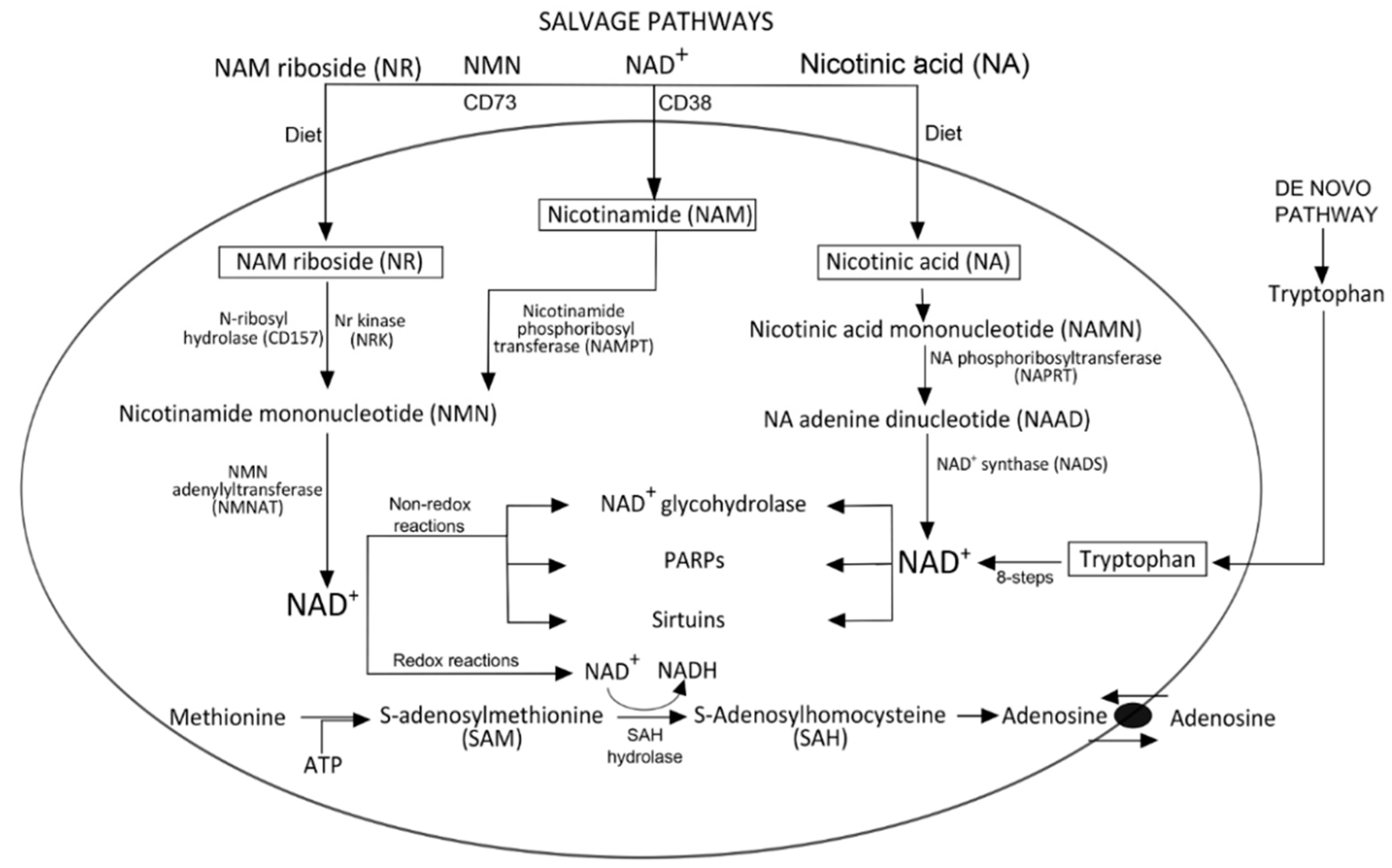
3. NAD+ Degradation
3.1. Role of NAD+ as Cofactor
3.2. Role of NAD+ as a Precursor for Additional Reactions
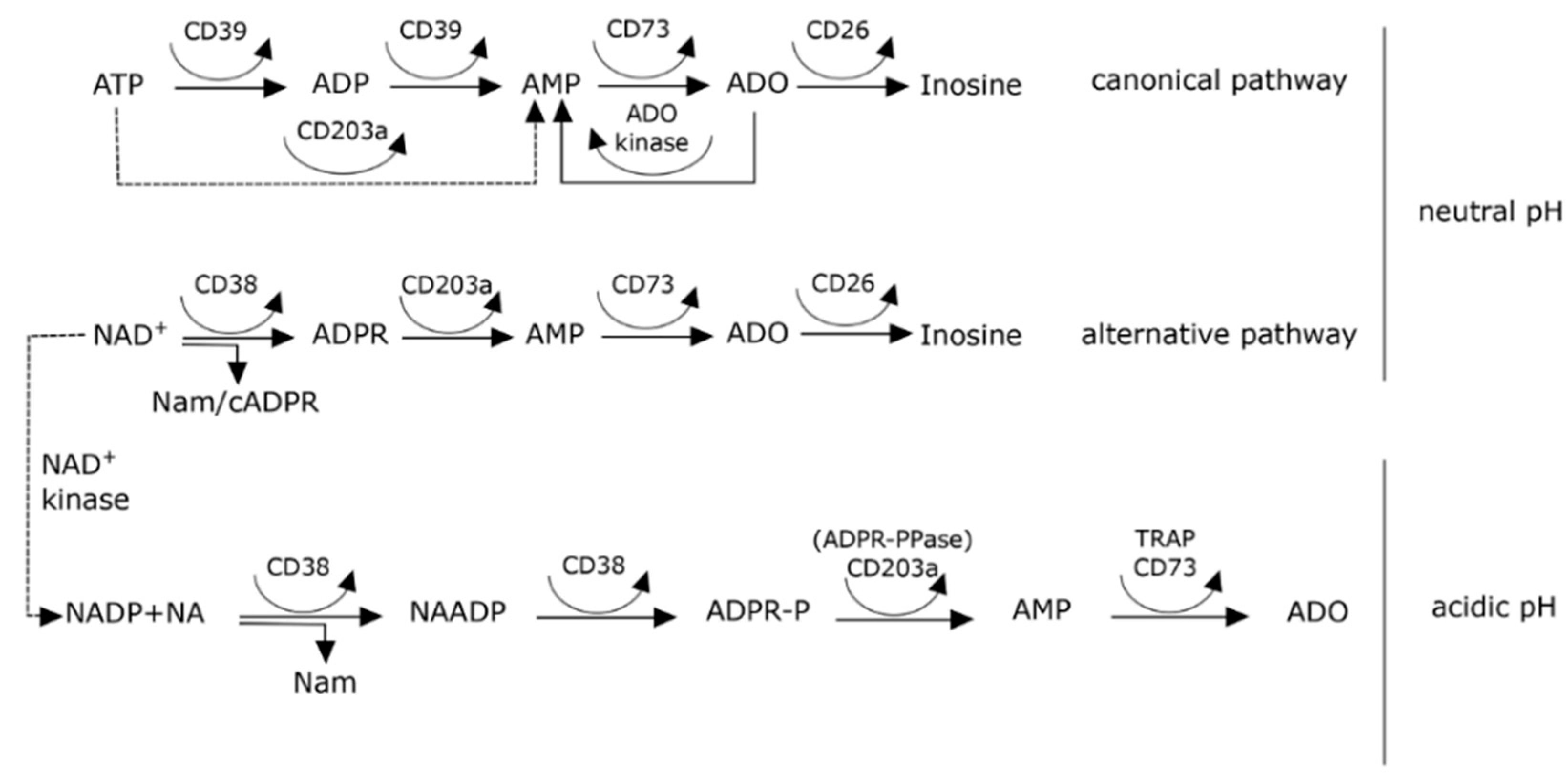
4. Extracellular NAD+ Metabolism
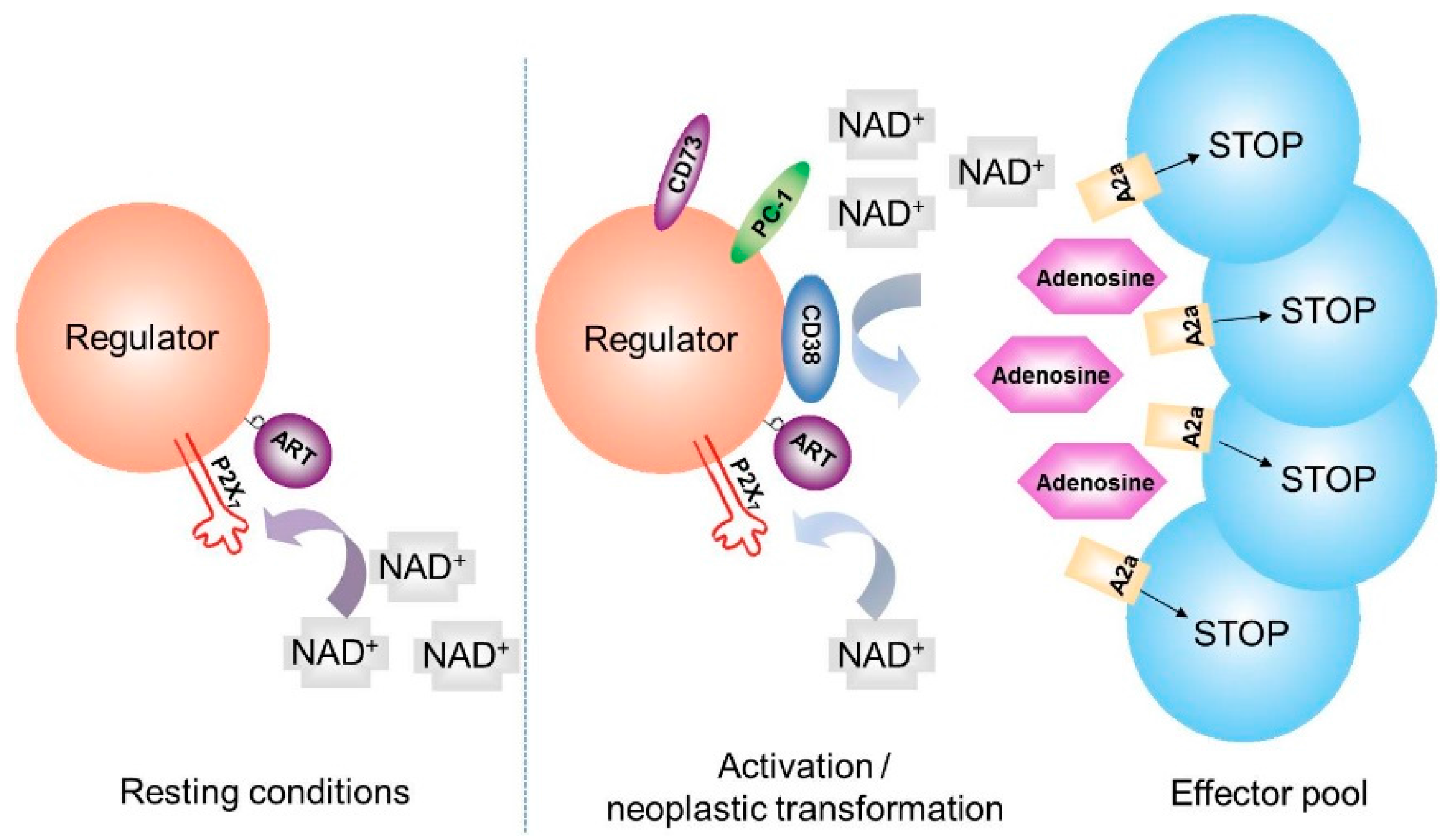
5. CD38 and Generation of Adenosine
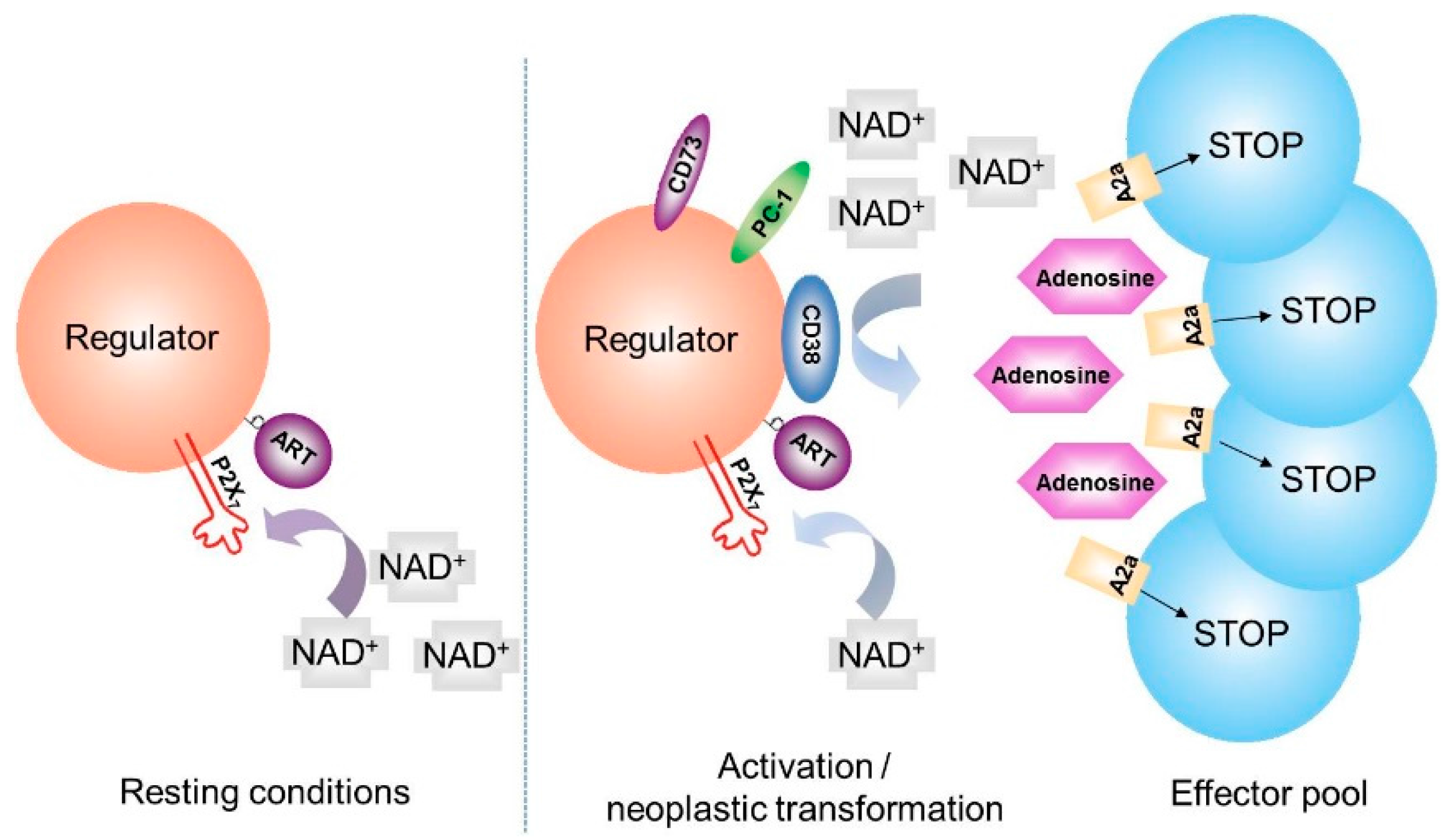
6. NAD+ Balance in Cancer: A Working Hypothesis Confirmed in a Human Myeloma Model
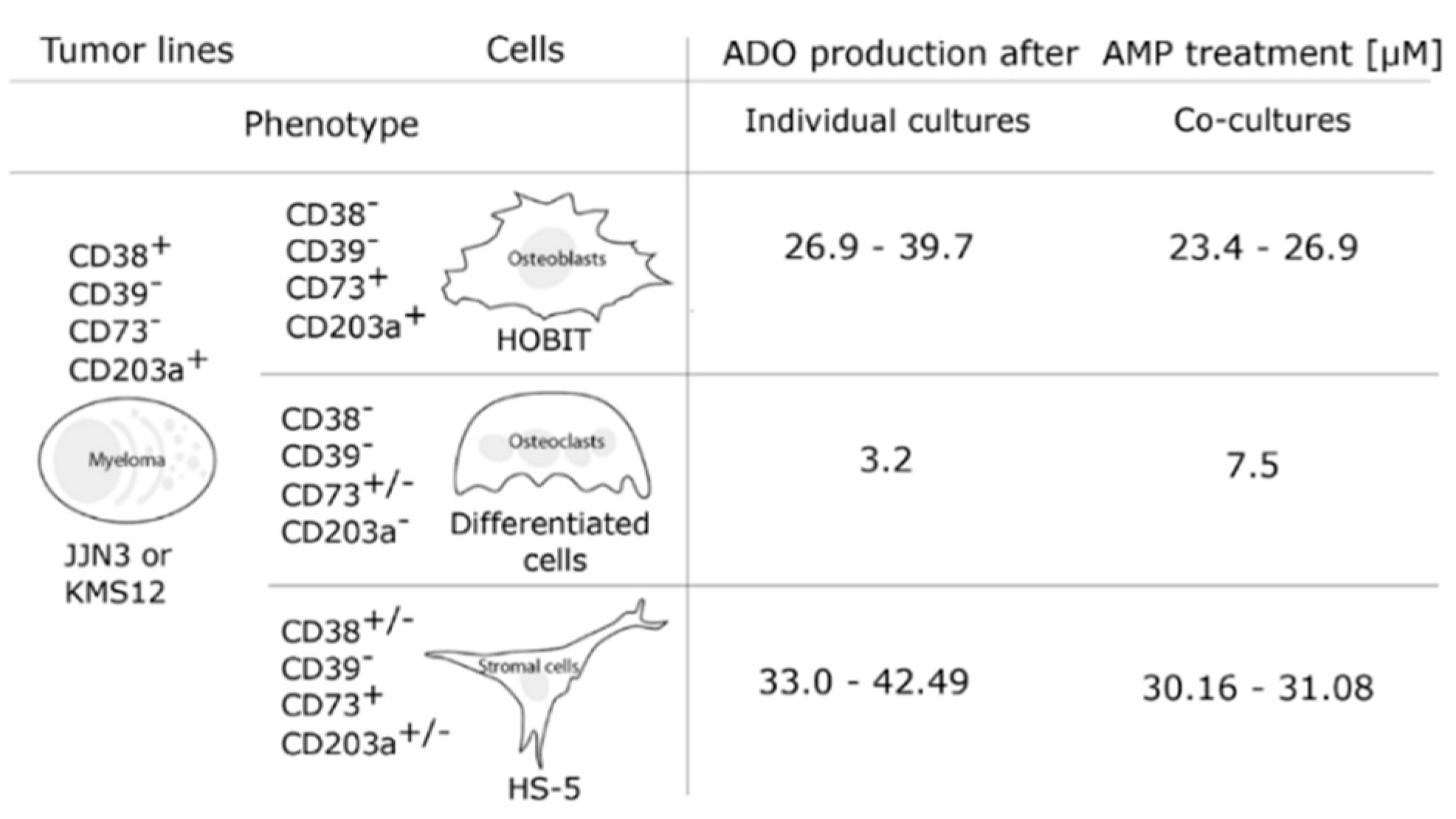
7. Ectoenzymes in the Myeloma Niche

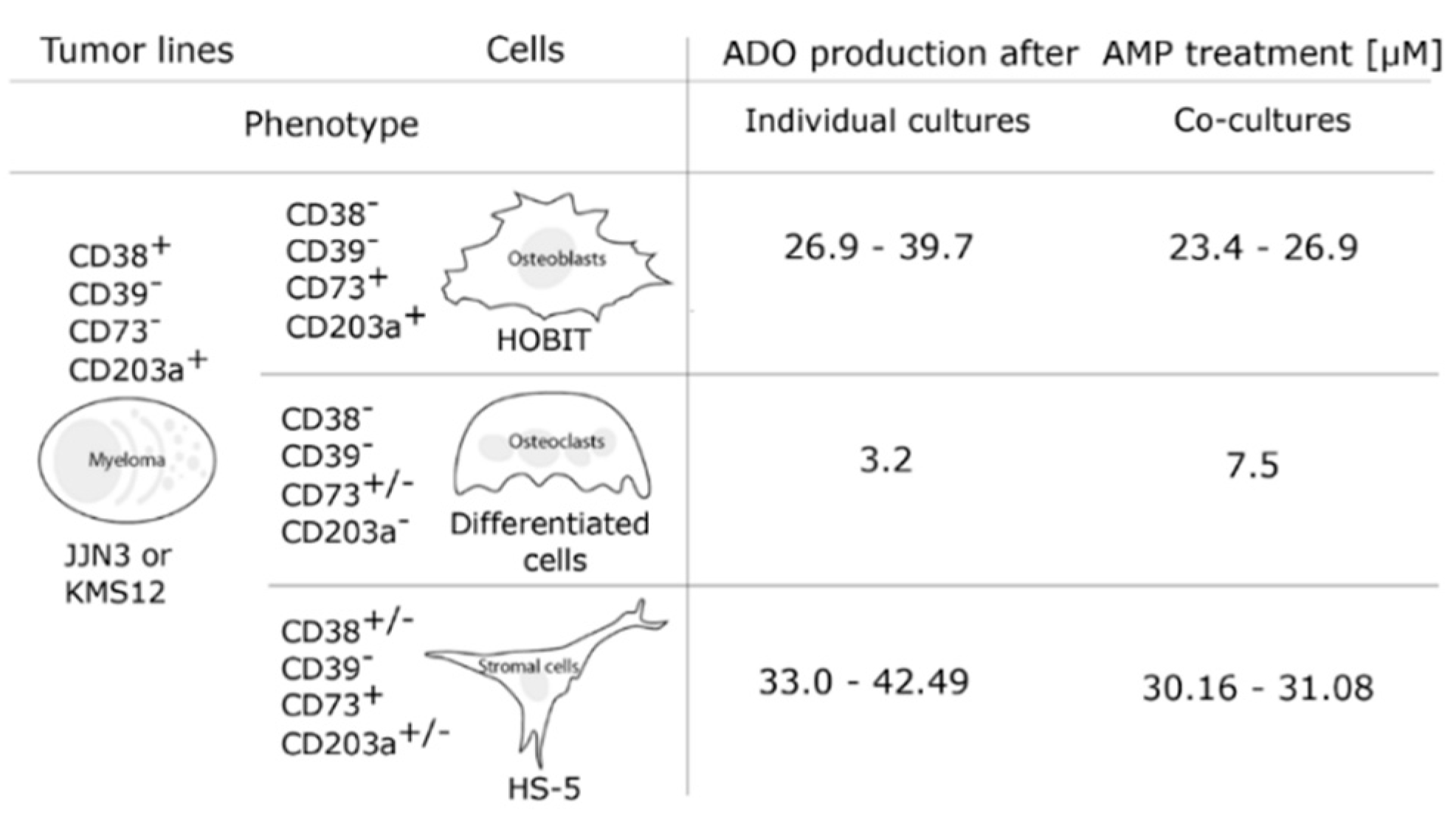
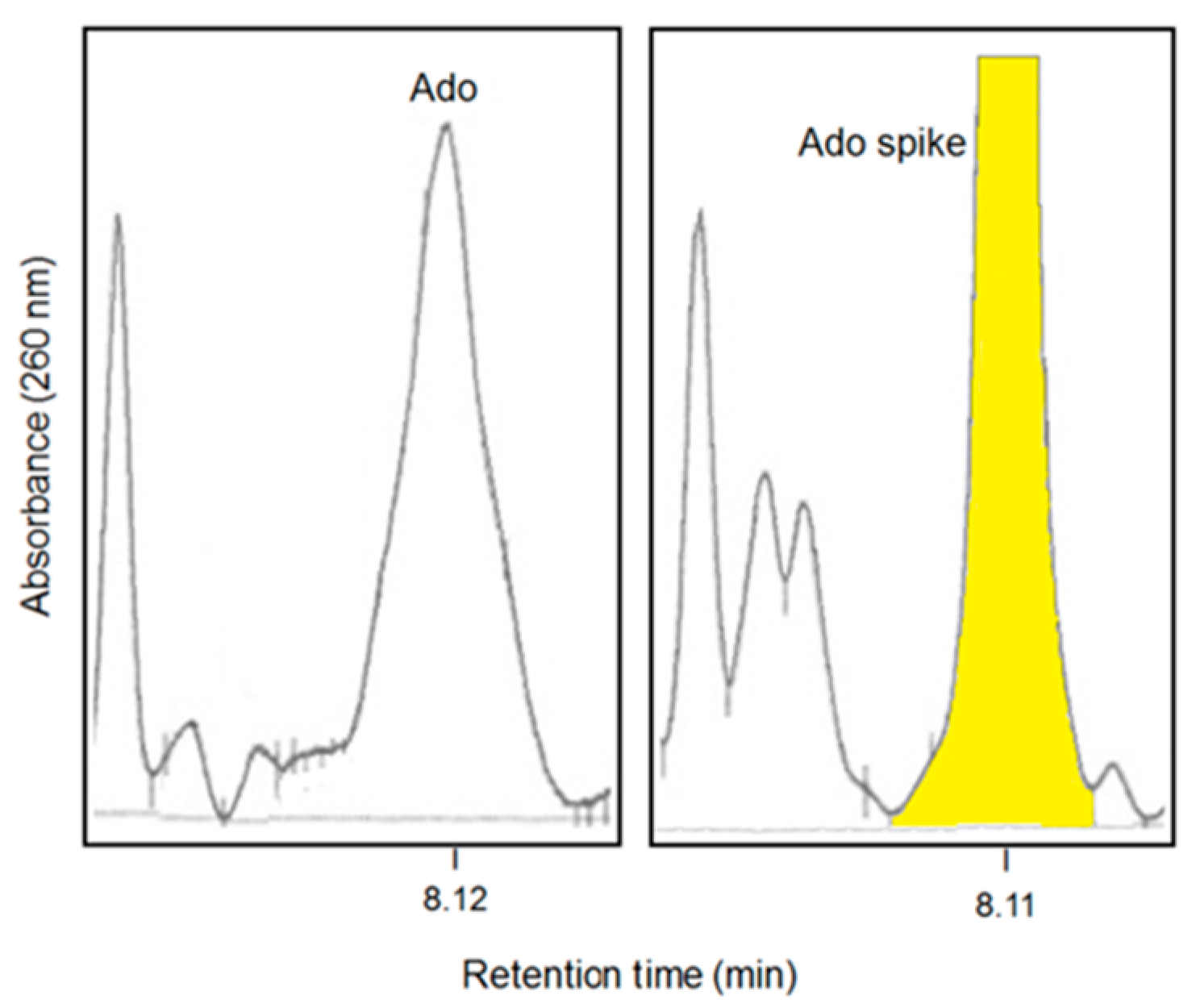
8. Effects of pH on Adenosine Production in the Myeloma Niche
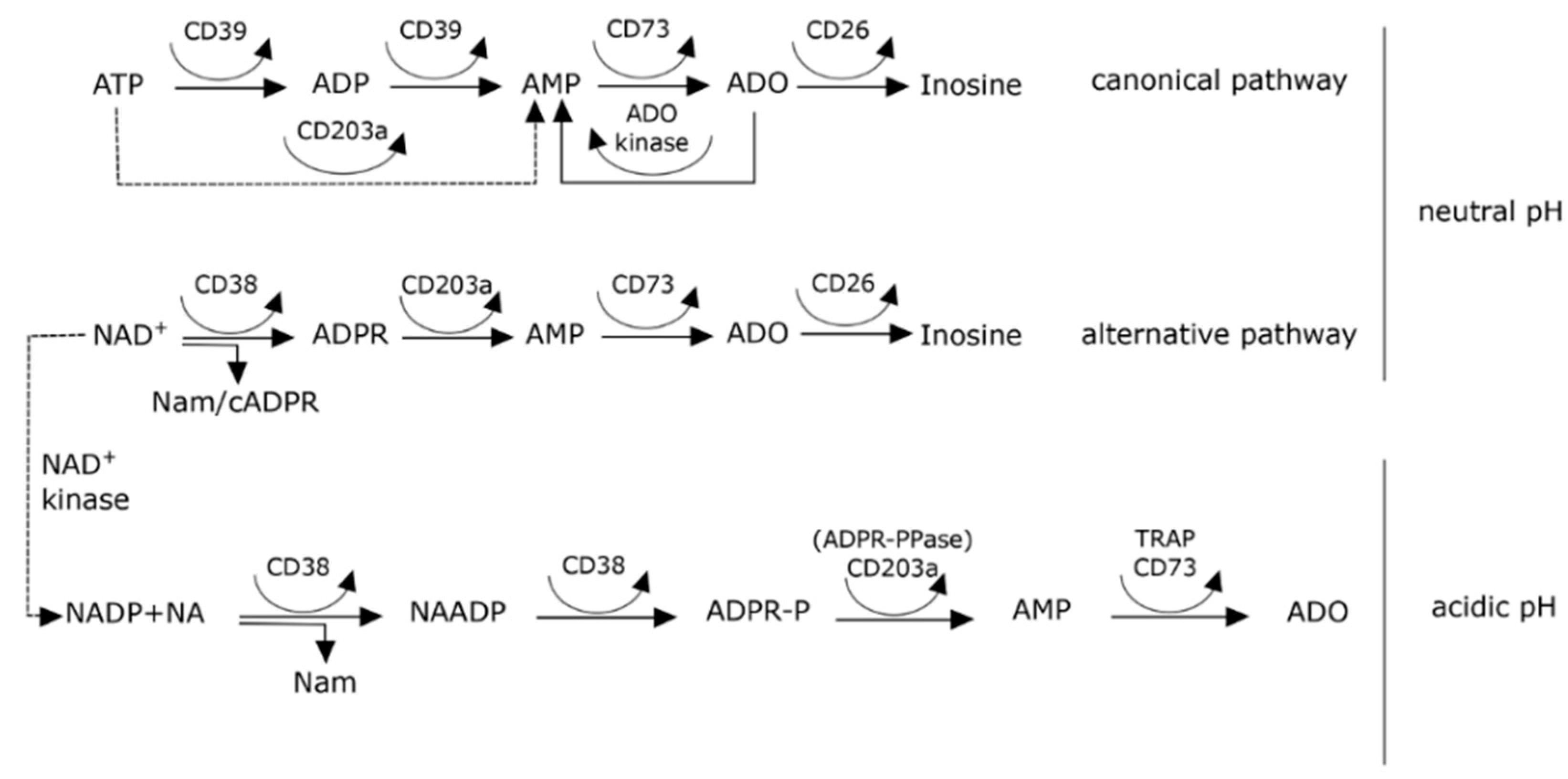
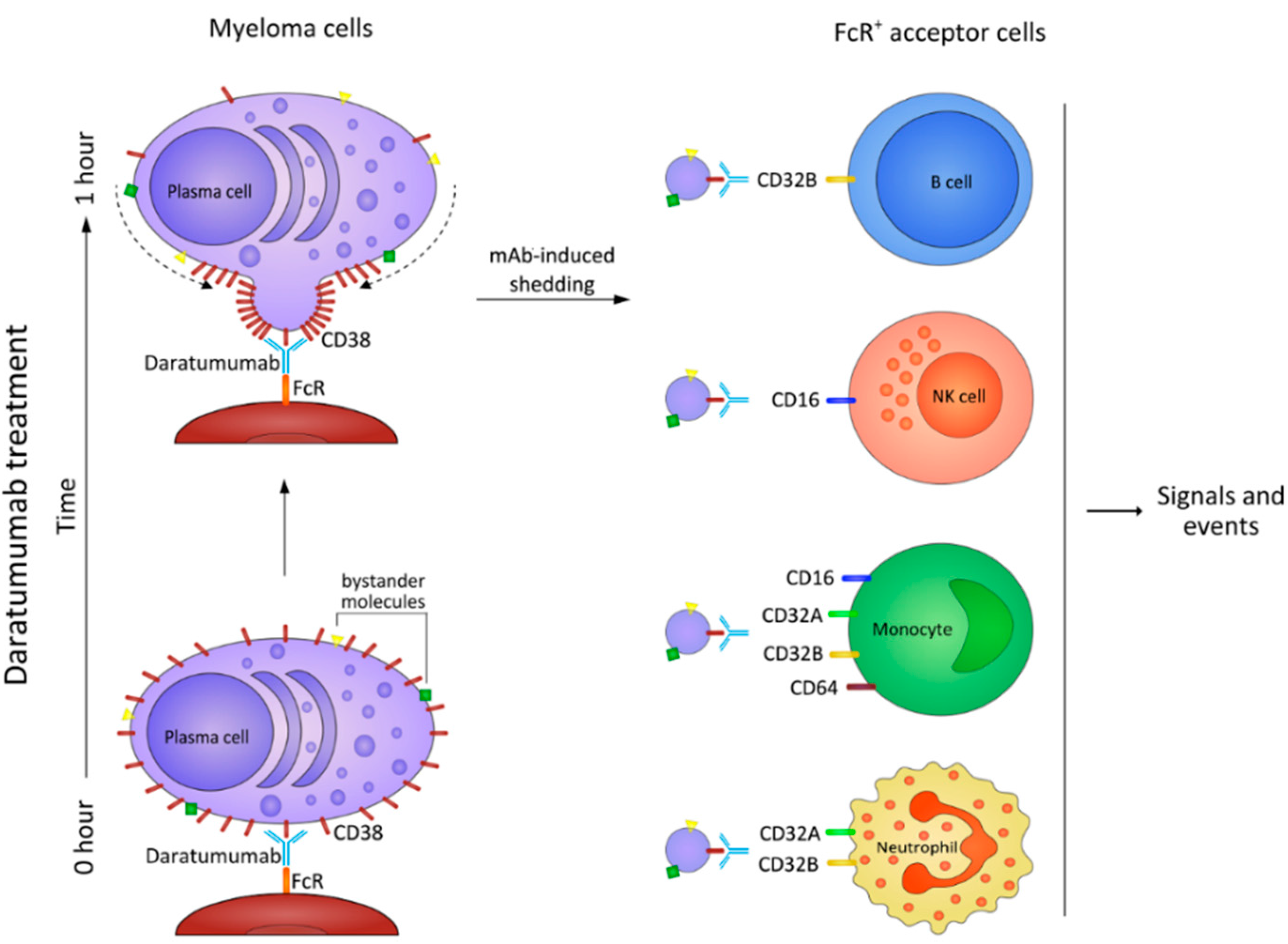
9. The NAD+-Consuming CD38 Enzyme as a Local Modulator in the BM Myeloma Niche: Can Therapeutic mAb Confirm the Hypothesis?
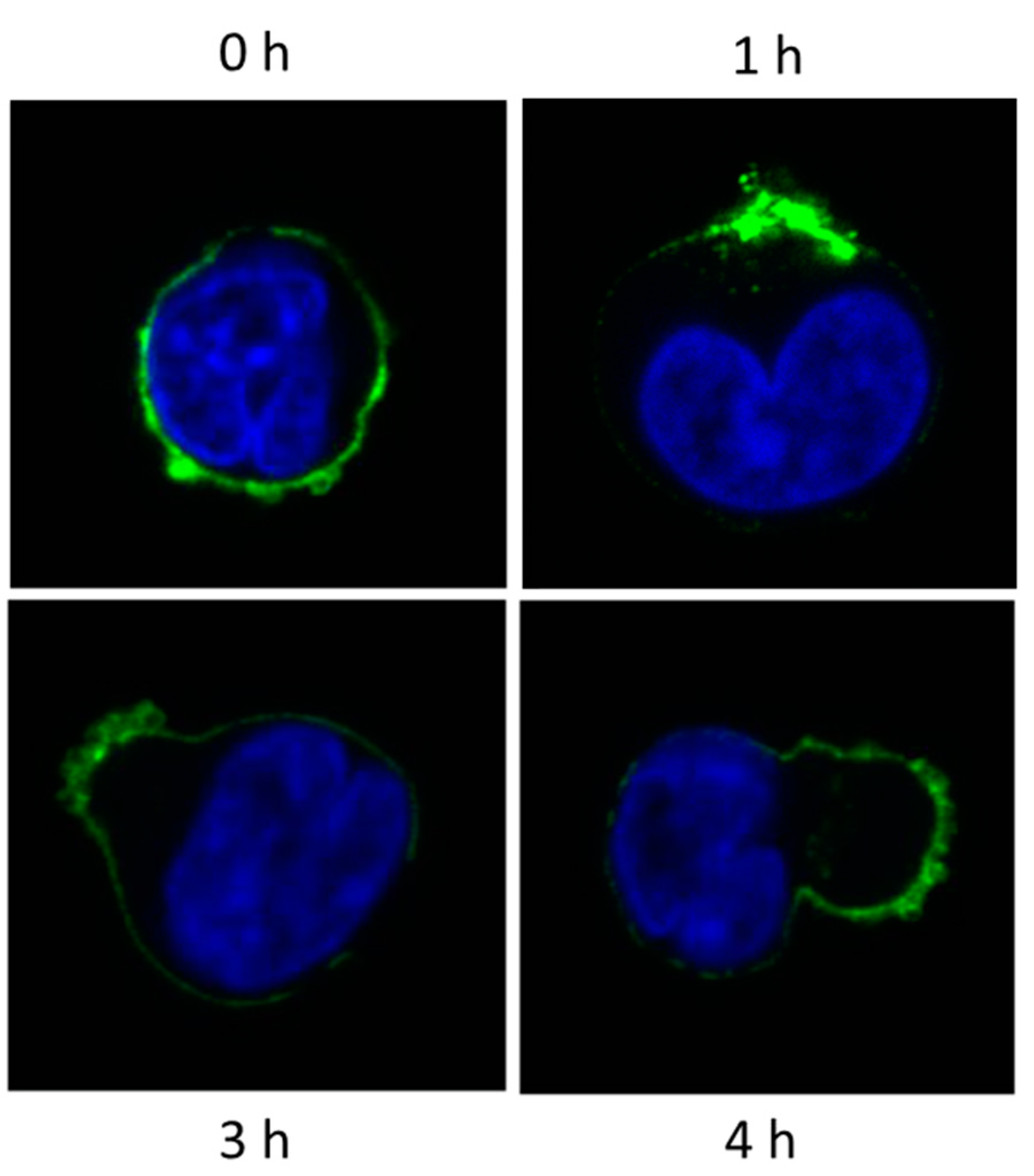
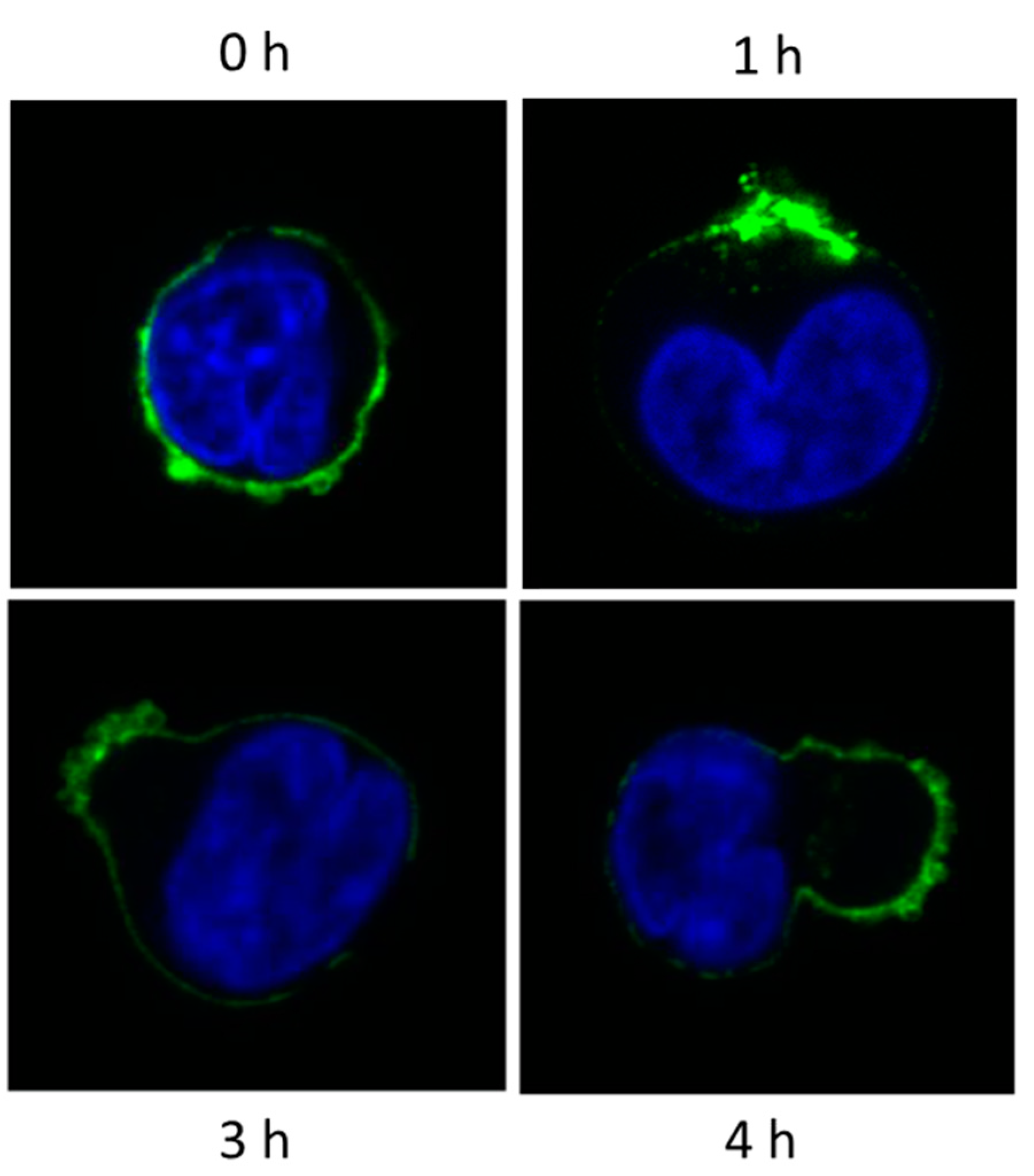
10. Myeloma Cells Release Microvesicles upon DARA mAb Ligation
Fate of MV
11. Conclusions and Open Issues
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Berger, F.; Ramirez-Hernandez, M.H.; Ziegler, M. The new life of a centenarian: Signalling functions of NAD(P). Trends Biochem. Sci. 2004, 29, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, A.; Dolle, C.; Felici, R.; Ziegler, M. The NAD metabolome—A key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Chan, N.Y.; Sauve, A.A. Syntheses of nicotinamide riboside and derivatives: Effective agents for increasing nicotinamide adenine dinucleotide concentrations in mammalian cells. J. Med. Chem. 2007, 50, 6458–6461. [Google Scholar] [CrossRef] [PubMed]
- Bender, D.A. Biochemistry of tryptophan in health and disease. Mol. Aspects Med. 1983, 6, 101–197. [Google Scholar] [CrossRef]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Burgos, E.S. NAMPT in regulated NAD biosynthesis and its pivotal role in human metabolism. Curr. Med. Chem. 2011, 18, 1947–1961. [Google Scholar] [CrossRef] [PubMed]
- Travelli, C.; Drago, V.; Maldi, E.; Kaludercic, N.; Galli, U.; Boldorini, R.; Di Lisa, F.; Tron, G.C.; Canonico, P.L.; Genazzani, A.A. Reciprocal potentiation of the antitumoral activities of FK866, an inhibitor of nicotinamide phosphoribosyltransferase, and etoposide or cisplatin in neuroblastoma cells. J. Pharmacol. Exp. Ther. 2011, 338, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Sauve, A.A. Nicotinamide riboside, a trace nutrient in foods, is a vitamin B3 with effects on energy metabolism and neuroprotection. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Preugschat, F.; Carter, L.H.; Boros, E.E.; Porter, D.J.; Stewart, E.L.; Shewchuk, L.M. A pre-steady state and steady state kinetic analysis of the N-ribosyl hydrolase activity of hCD157. Arch. Biochem. Biophys. 2014, 564, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Christian, W.; Griese, A. Wasserstoffübertragendes Co-Ferment, seine Zusammensetzung und Wirkungsweise. Biochem. Z. 1935, 282, 157–205. [Google Scholar]
- Malavasi, F.; Deaglio, S.; Zaccarello, G.; Horenstein, A.L.; Chillemi, A.; Audrito, V.; Serra, S.; Gandione, M.; Zitella, A.; Tizzani, A. The hidden life of NAD+-consuming ectoenzymes in the endocrine system. J. Mol. Endocrinol. 2010, 45, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Corda, D.; di Girolamo, M. Functional aspects of protein mono-ADP-ribosylation. EMBO J. 2003, 22, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 2008, 13, 3046–3082. [Google Scholar] [CrossRef] [PubMed]
- Sauve, A.A. Sirtuin chemical mechanisms. Biochim. Biophys. Acta 2010, 1804, 1591–1603. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Ferri, V.; Bolzoni, M.; Quarona, V.; Chillemi, A.; Zaccarello, G.; Giuliani, N.; Malavasi, F. Ectoenzymes and Their Products as Communicators in the Human Myeloma Niche. In Proceedings of the 56th ASH Annual Meeting and Exposition, San Fancisco, CA, USA, 6–9 December 2014; p. 2064.
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [PubMed]
- Guida, L.; Bruzzone, S.; Sturla, L.; Franco, L.; Zocchi, E.; de Flora, A. Equilibrative and concentrative nucleoside transporters mediate influx of extracellular cyclic ADP-ribose into 3T3 murine fibroblasts. J. Biol. Chem. 2002, 277, 47097–47105. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [PubMed]
- Quarona, V.; Ferri, V.; Chillemi, A.; Bolzoni, M.; Mancini, C.; Zaccarello, G.; Roato, I.; Morandi, F.; Marimpietri, D.; Faccani, G.; et al. Unraveling the contribution of ectoenzymes to myeloma life and survival in the bone marrow niche. Ann. N. Y. Acad. Sci. 2015, 1335, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, F.; Deaglio, S.; Damle, R.; Cutrona, G.; Ferrarini, M.; Chiorazzi, N. CD38 and chronic lymphocytic leukemia: A decade later. Blood 2011, 118, 3470–3478. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Liu, H.X.; Hirai, H.; Torashima, T.; Nagai, T.; Lopatina, O.; Shnayder, N.A.; Yamada, K.; Noda, M.; Seike, T.; et al. CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 2007, 446, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Cakir-Kiefer, C.; Muller-Steffner, H.; Oppenheimer, N.; Schuber, F. Kinetic competence of the cADP-ribose-CD38 complex as an intermediate in the CD38/NAD+ glycohydrolase-catalysed reactions: Implication for CD38 signalling. Biochem. J. 2001, 358, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Dousa, T.P.; Chini, E.N.; Beers, K.W. Adenine nucleotide diphosphates: Emerging second messengers acting via intracellular Ca2+ release. Am. J. Physiol. 1996, 271, C1007–C1024. [Google Scholar] [PubMed]
- Zhao, Y.J.; Lam, C.M.; Lee, H.C. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci. Signal. 2012, 5, ra67. [Google Scholar] [CrossRef] [PubMed]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Chillemi, A.; Zaccarello, G.; Bruzzone, S.; Quarona, V.; Zito, A.; Serra, S.; Malavasi, F. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology 2013, 2, e26246. [Google Scholar] [CrossRef] [PubMed]
- Grahnert, A.; Grahnert, A.; Klein, C.; Schilling, E.; Wehrhahn, J.; Hauschildt, S. Review: NAD+: A modulator of immune functions. Innate Immun. 2011, 17, 212–233. [Google Scholar] [CrossRef] [PubMed]
- Hubert, S.; Rissiek, B.; Klages, K.; Huehn, J.; Sparwasser, T.; Haag, F.; Koch-Nolte, F.; Boyer, O.; Seman, M.; Adriouch, S. Extracellular NAD+ shapes the Foxp3+ regulatory T cell compartment through the ART2-P2X7 pathway. J. Exp. Med. 2010, 207, 2561–2568. [Google Scholar] [CrossRef] [PubMed]
- Seman, M.; Adriouch, S.; Scheuplein, F.; Krebs, C.; Freese, D.; Glowacki, G.; Deterre, P.; Haag, F.; Koch-Nolte, F. NAD-induced T cell death: ADP-ribosylation of cell surface proteins by ART2 activates the cytolytic P2X7 purinoceptor. Immunity 2003, 19, 571–582. [Google Scholar] [CrossRef]
- Rissiek, B.; Haag, F.; Boyer, O.; Koch-Nolte, F.; Adriouch, S. ADP-ribosylation of P2X7: A matter of life and death for regulatory T cells and natural killer T cells. Curr. Top. Microbiol. Immunol. 2015, 384, 107–126. [Google Scholar] [PubMed]
- Karakasheva, T.A.; Waldron, T.J.; Eruslanov, E.; Lee, J.; O’Brien, S.; Hinck, P.D.; Basu, D.; Singhal, D.; Malavasi, F.; Rustgi, A.K. CD38-expressing Myeloid Derived Suppressor Cells promote tumor growth in a murine model of esophageal cancer. Cancer Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chillemi, A.; Zaccarello, G.; Quarona, V.; Lazzaretti, M.; Martella, E.; Giuliani, N.; Ferracini, R.; Pistoia, V.; Horenstein, A.L.; Malavasi, F. CD38 and bone marrow microenvironment. Front. Biosci. (Landmark Ed.) 2014, 19, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Pradas, G.S.; Levitt, D.G.; Lee, H.C.; Stout, C.D. Crystallization of ADP-ribosyl cyclase from Aplysia californica. Proteins 1996, 24, 138–140. [Google Scholar] [PubMed]
- Morandi, F.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Chiesa, S.; Imperatori, A.; Zanellato, S.; Mortara, L.; Gattorno, M.; Pistoia, V.; et al. CD56brightCD16- NK Cells Produce Adenosine through a CD38-Mediated Pathway and Act as Regulatory Cells Inhibiting Autologous CD4+ T Cell Proliferation. J. Immunol. 2015, 195, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Morandi, B.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Zaccarello, G.; Carrega, P.; Ferlazzo, G.; Mingari, M.C.; Moretta, L.; et al. A non-canonical adenosinergic pathway lead by CD38 in human melanoma cells induces suppression of T cell proliferation. Oncotarget 2015. [Google Scholar]
- Storti, P.; Bolzoni, M.; Donofrio, G.; Airoldi, I.; Guasco, D.; Toscani, D.; Martella, E.; Lazzaretti, M.; Mancini, C.; Agnelli, L.; et al. Hypoxia-inducible factor (HIF)-1alpha suppression in myeloma cells blocks tumoral growth in vivo inhibiting angiogenesis and bone destruction. Leukemia 2013, 27, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, D.E.; Smith, P.T.; Caldwell, C.C.; Ohta, A.; Apasov, S.G.; Sitkovsky, M.V. Analysis of A2a receptor-deficient mice reveals no significant compensatory increases in the expression of A2b, A1, and A3 adenosine receptors in lymphoid organs. Biochem. Pharmacol. 2003, 65, 2081–2090. [Google Scholar] [CrossRef]
- Zito, A.; Chillemi, A.; Quarona, V.; Mandili, G.; Cassone, A.; Horenstein, A.L.; Malavasi, F. Bispecific murine CD73xCD38 monoclonal antibody as a potential tool to overcome adenosine-mediated immunosuppression. In Proceedings of the EACR-AACR-SIC 2015 Special Conference, Anticancer Drug Action and Drug Resistance: From Cancer Biology to the Clinic, Florence, Italy, 20–23 June 2015.
- Sitkovsky, M.; Ohta, A. Targeting the hypoxia-adenosinergic signaling pathway to improve the adoptive immunotherapy of cancer. J. Mol. Med. (Berl.) 2013, 91, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Belikoff, B.; Schreiber, T.H.; Sethumadhavan, S.; Abbott, R.; Philbrook, P.; Thayer, M.; Shujia, D.; et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1alpha-dependent and extracellular adenosine-mediated tumor protection. J. Mol. Med. (Berl.) 2014, 92, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Yegutkin, G.G. Enzymes involved in metabolism of extracellular nucleotides and nucleosides: Functional implications and measurement of activities. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 473–497. [Google Scholar] [CrossRef] [PubMed]
- Chillemi, A.; Zaccarello, G.; Quarona, V.; Ferracin, M.; Ghimenti, C.; Massaia, M.; Horenstein, A.L.; Malavasi, F. Anti-CD38 antibody therapy: Windows of opportunity yielded by the functional characteristics of the target molecule. Mol. Med. 2013, 19, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Roghanian, A.; Teige, I.; Martensson, L.; Cox, K.L.; Kovacek, M.; Ljungars, A.; Mattson, J.; Sundberg, A.; Vaughan, A.T.; Shah, V.; et al. Antagonistic human FcgammaRIIB (CD32B) antibodies have anti-tumor activity and overcome resistance to antibody therapy in vivo. Cancer Cell 2015, 27, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- D’Souza-Schorey, C.; Clancy, J.W. Tumor-derived microvesicles: Shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012, 26, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Arendt, B.K.; Walters, D.K.; Wu, X.; Tschumper, R.C.; Jelinek, D.F. Multiple myeloma dell-derived microvesicles are enriched in CD147 expression and enhance tumor cell proliferation. Oncotarget 2014, 5, 5686–5699. [Google Scholar] [CrossRef] [PubMed]
- Zumaquero, E.; Munoz, P.; Cobo, M.; Lucena, G.; Pavon, E.J.; Martin, A.; Navarro, P.; Garcia-Perez, A.; Ariza-Veguillas, A.; Malavasi, F.; et al. Exosomes from human lymphoblastoid B cells express enzymatically active CD38 that is associated with signaling complexes containing CD81, Hsc-70 and Lyn. Exp. Cell Res. 2010, 316, 2692–2706. [Google Scholar] [CrossRef] [PubMed]
- Zomer, A.; Maynard, C.; Verweij, F.J.; Kamermans, A.; Schafer, R.; Beerling, E.; Schiffelers, R.M.; de Wit, E.; Berenguer, J.; Ellenbroek, S.I.; et al. In Vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell 2015, 161, 1046–1057. [Google Scholar] [CrossRef] [PubMed]
- DiLillo, D.J.; Ravetch, J.V. Differential Fc-Receptor Engagement Drives an Anti-tumor Vaccinal Effect. Cell 2015, 161, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Cagnetta, A.; Cea, M.; Calimeri, T.; Acharya, C.; Fulciniti, M.; Tai, Y.T.; Hideshima, T.; Chauhan, D.; Zhong, M.Y.; Patrone, F.; et al. Intracellular NAD(+) depletion enhances bortezomib-induced anti-myeloma activity. Blood 2013, 122, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Galli, U.; Travelli, C.; Massarotti, A.; Fakhfouri, G.; Rahimian, R.; Tron, G.C.; Genazzani, A.A. Medicinal chemistry of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors. J. Med. Chem. 2013, 56, 6279–6296. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horenstein, A.L.; Chillemi, A.; Quarona, V.; Zito, A.; Roato, I.; Morandi, F.; Marimpietri, D.; Bolzoni, M.; Toscani, D.; Oldham, R.J.; et al. NAD+-Metabolizing Ectoenzymes in Remodeling Tumor–Host Interactions: The Human Myeloma Model. Cells 2015, 4, 520-537. https://doi.org/10.3390/cells4030520
Horenstein AL, Chillemi A, Quarona V, Zito A, Roato I, Morandi F, Marimpietri D, Bolzoni M, Toscani D, Oldham RJ, et al. NAD+-Metabolizing Ectoenzymes in Remodeling Tumor–Host Interactions: The Human Myeloma Model. Cells. 2015; 4(3):520-537. https://doi.org/10.3390/cells4030520
Chicago/Turabian StyleHorenstein, Alberto L., Antonella Chillemi, Valeria Quarona, Andrea Zito, Ilaria Roato, Fabio Morandi, Danilo Marimpietri, Marina Bolzoni, Denise Toscani, Robert J. Oldham, and et al. 2015. "NAD+-Metabolizing Ectoenzymes in Remodeling Tumor–Host Interactions: The Human Myeloma Model" Cells 4, no. 3: 520-537. https://doi.org/10.3390/cells4030520
APA StyleHorenstein, A. L., Chillemi, A., Quarona, V., Zito, A., Roato, I., Morandi, F., Marimpietri, D., Bolzoni, M., Toscani, D., Oldham, R. J., Cuccioloni, M., Sasser, A. K., Pistoia, V., Giuliani, N., & Malavasi, F. (2015). NAD+-Metabolizing Ectoenzymes in Remodeling Tumor–Host Interactions: The Human Myeloma Model. Cells, 4(3), 520-537. https://doi.org/10.3390/cells4030520