MicroRNA-421 Dysregulation is Associated with Tetralogy of Fallot

Abstract
:1. Introduction
2. Experimental Section
2.1. Subjects

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | Gender | Mean Age (Range) | Analyzed by Array | Analyzed by qRT-PCR | |
|---|---|---|---|---|---|
| TOF | 16 | 11M/5F | 276 days (98–510) | 16 | 16 |
| control | 8 | 3M/5F | 142 days (28–382) | 8 | 8 |
| additional TOF subjects used for validation | 8 | 4M/4F | 292 days (167–425) | 0 | 8 |
2.2. Cell Cultures and Transfection

2.3. RNA Preparation
2.4. Microarray Analysis
2.5. Real-Time Quantitative Polymerase Chain Reaction
3. Results and Discussion
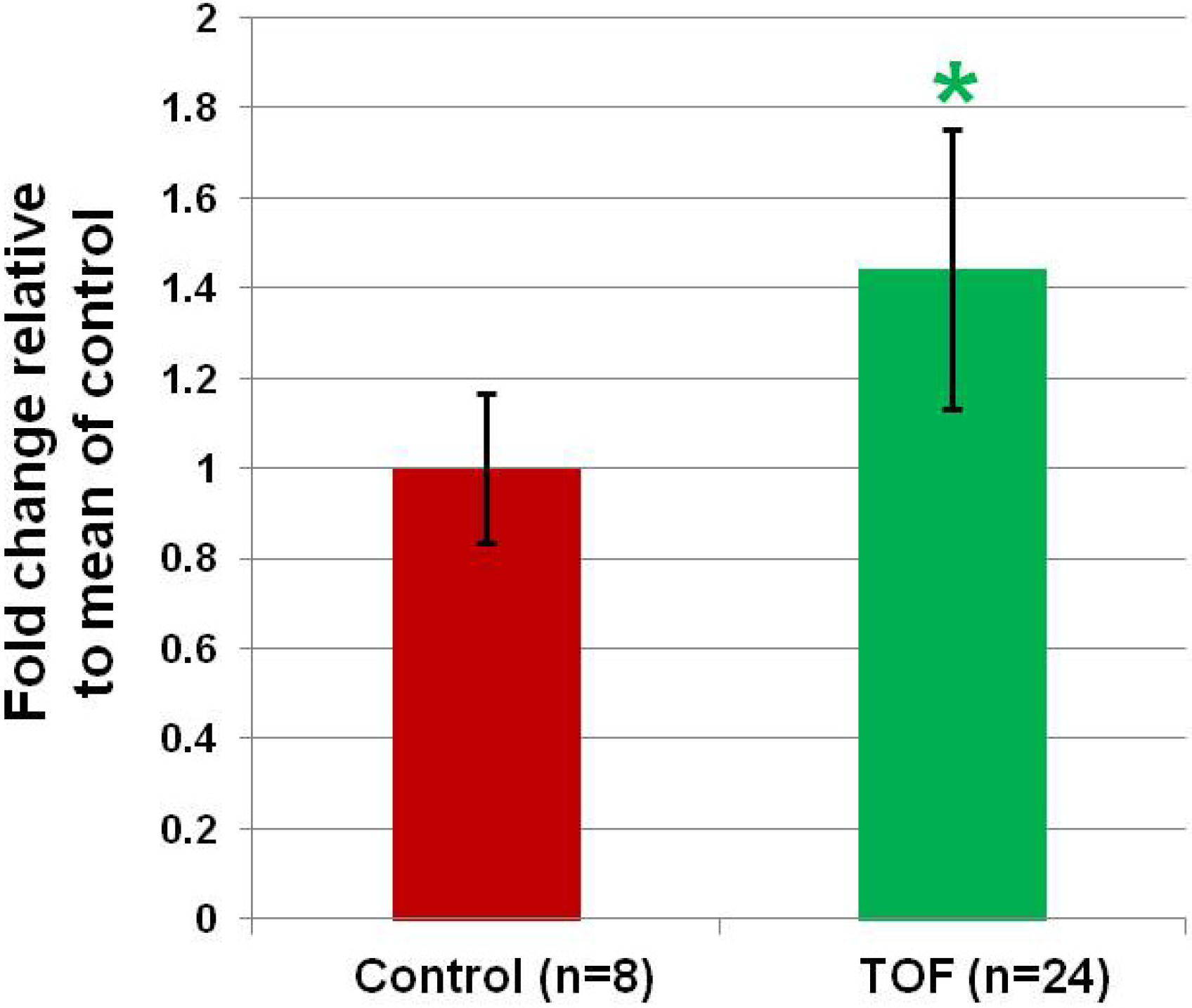
3.1. MiR-421 Expression is Increased in the RV of Infants with TOF.
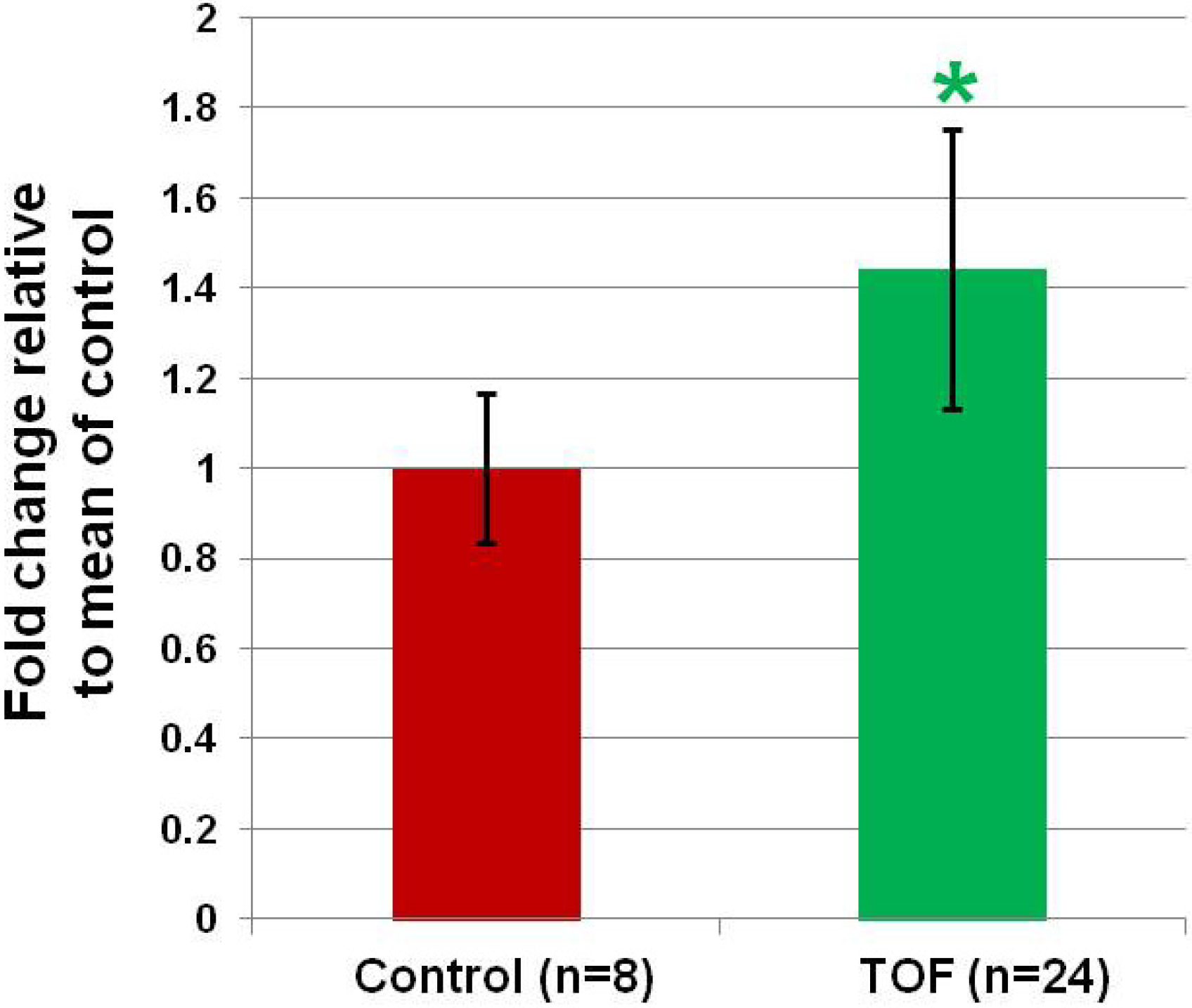
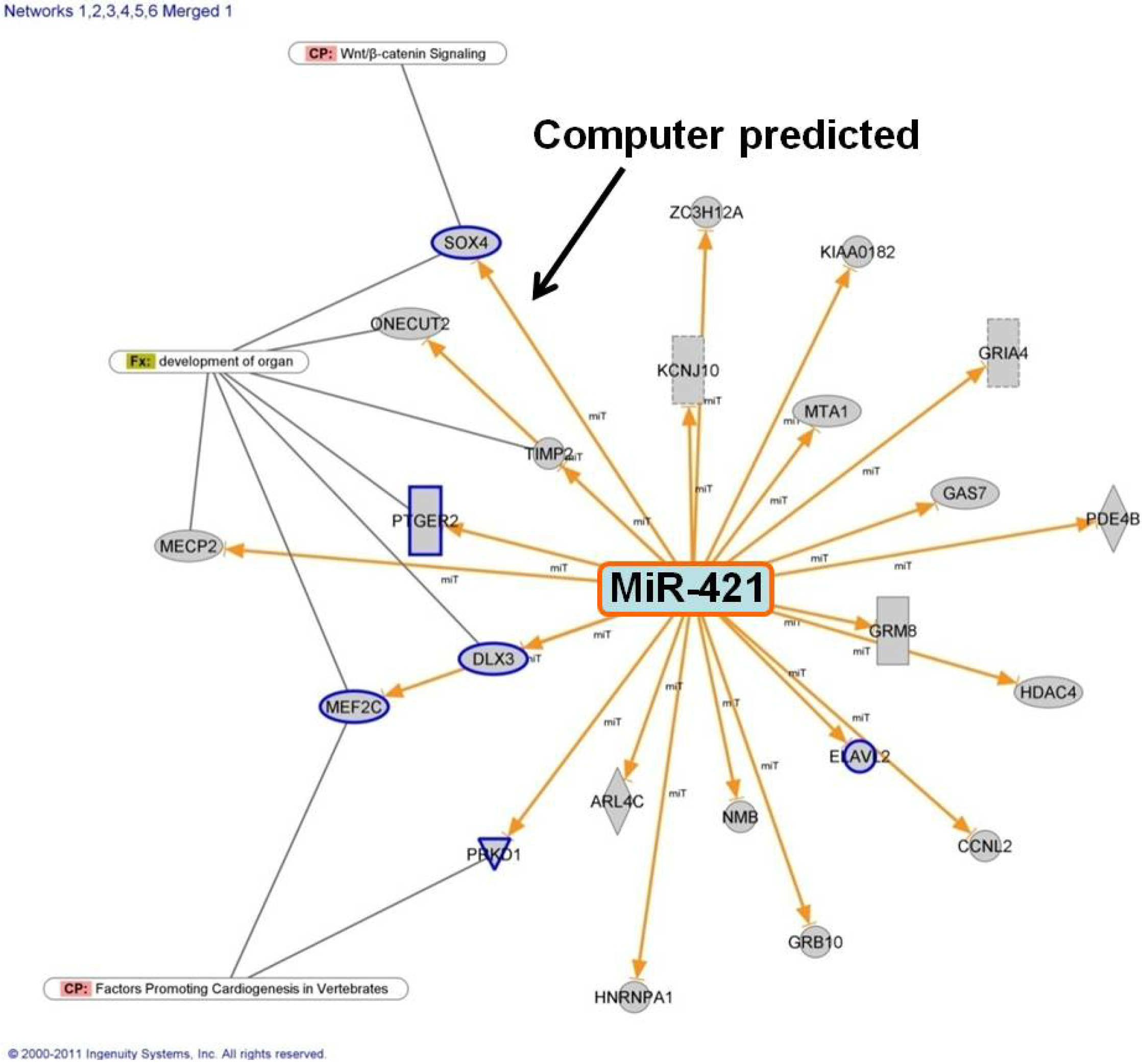
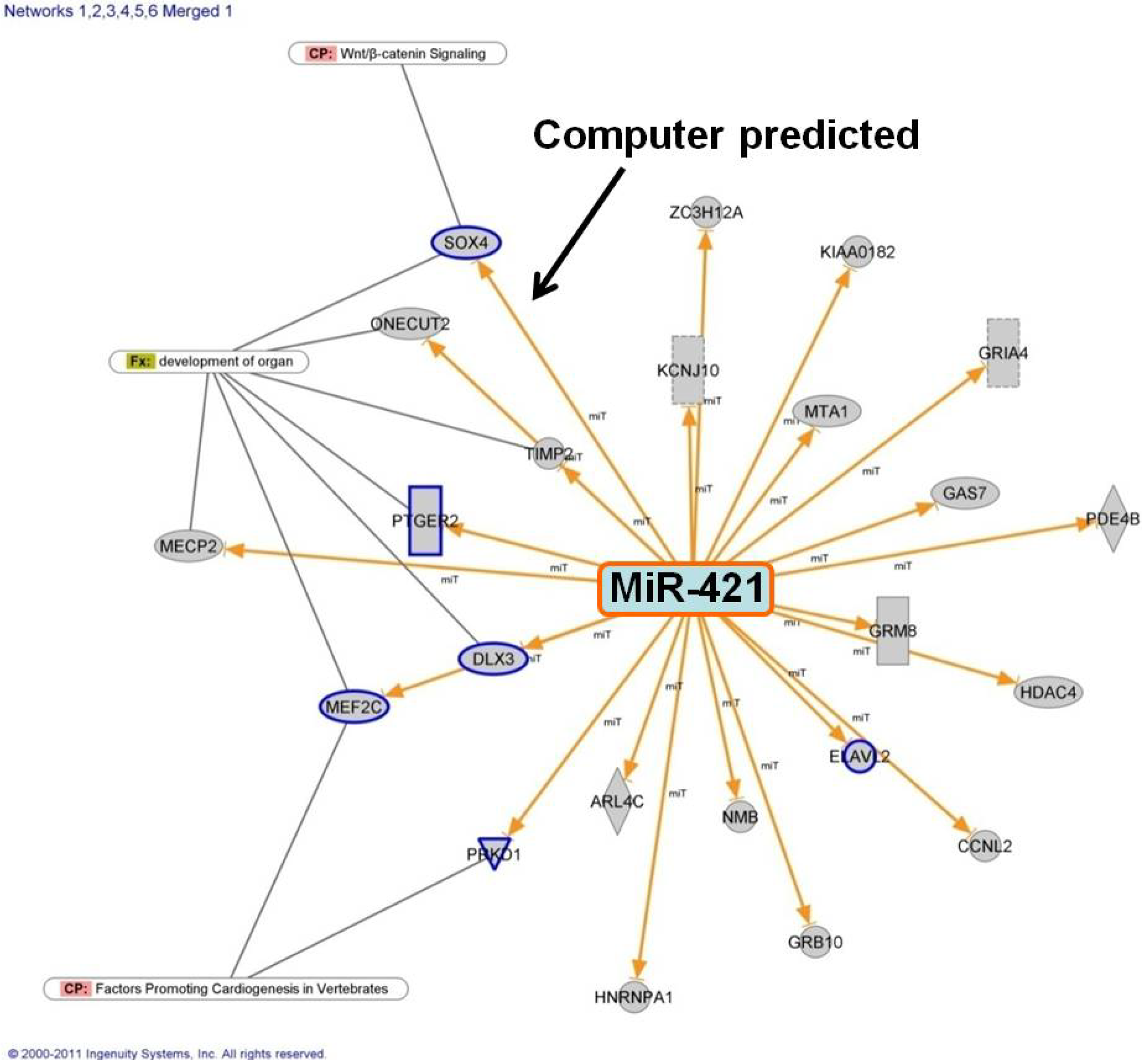
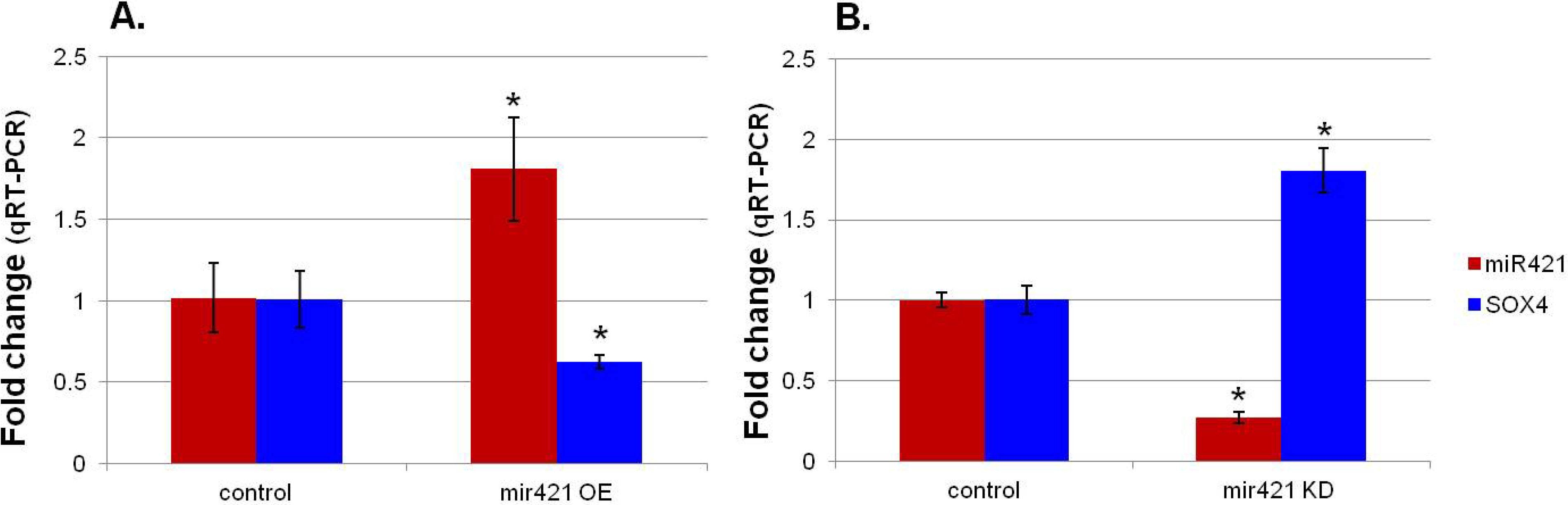
3.2. MiR-421 Expression is Inversely Correlated with SOX4 Expression
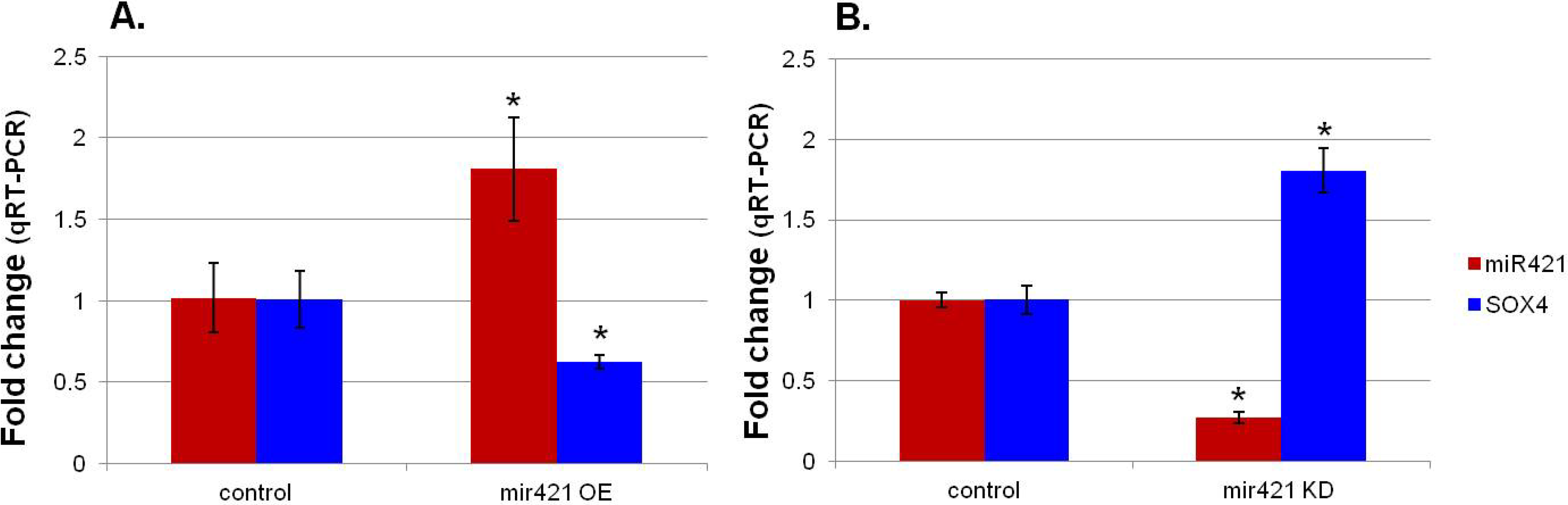
| Sample Name | miR-421 | SOX4 |
|---|---|---|
| CP neo cells | 1.02 ± 0.21 | 1.01 ± 0.18 |
| miR-421 OE | 1.81 ± 0.32 * | 0.63 ± 0.04 * |
| p value | 0.023 | 0.021 |
| TOF cells | 1.00 ± 0.05 | 1.00 ± 0.09 |
| miR-421 KD | 0.27 ± 0.03 * | 1.81 ± 0.14 * |
| p value | 0.004 | 0.011 |
4. Conclusions
Abbreviations
| CHD | congenital heart disease |
| FDR | false discovery rate |
| ncRNA | noncoding RNA |
| qRT-PCR | quantitative reverse transcription-PCR |
| scaRNAs | small cajal associated RNAs |
| snRNA | small nuclear RNA |
| snoRNA | small nucleolar RNA |
| TOF | tetralogy of Fallot |
| VSD | ventricular septal defect |
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Bruneau, B.G. The developmental genetics of congenital heart disease. Nature 2008, 451, 943–948. [Google Scholar]
- Mitchell, S.C.; Korones, S.B.; Berendes, H.W. Congenital heart disease in 56,109 births. Incidence and natural history. Circulation 1971, 43, 323–332. [Google Scholar]
- McElhinney, D.B.; Geiger, E.; Blinder, J.; Benson, D.W.; Goldmuntz, E. Nkx2.5 mutations in patients with congenital heart disease. J. Am. Coll Cardiol. 2003, 42, 1650–1655. [Google Scholar] [CrossRef]
- Hartman, J.L.T.; Garvik, B.; Hartwell, L. Principles for the buffering of genetic variation. Science 2001, 291, 1001–1004. [Google Scholar]
- Rutherford, S.L.; Henikoff, S. Quantitative epigenetics. Nat. Genet. 2003, 33, 6–8. [Google Scholar]
- Thomas, M.; Lieberman, J.; Lal, A. Desperately seeking microrna targets. Nat. Struct. Mol. Biol. 2010, 17, 1169–1174. [Google Scholar] [CrossRef]
- Liu, N.; Olson, E.N. Microrna regulatory networks in cardiovascular development. Dev. Cell. 2010, 18, 510–525. [Google Scholar] [CrossRef]
- Ivey, K.N.; Muth, A.; Arnold, J.; King, F.W.; Yeh, R.F.; Fish, J.E.; Hsiao, E.C.; Schwartz, R.J.; Conklin, B.R.; Bernstein, H.S.; et al. Microrna regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell 2008, 2, 219–229. [Google Scholar] [CrossRef]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microrna that targets hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking mirna-1–2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef]
- Liu, N.; Bezprozvannaya, S.; Williams, A.H.; Qi, X.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Microrna-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008, 22, 3242–3254. [Google Scholar]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted deletion reveals essential and overlapping functions of the mir-17 through 92 family of mirna clusters. Cell 2008, 132, 875–886. [Google Scholar] [CrossRef]
- Kuhn, D.E.; Nuovo, G.J.; Martin, M.M.; Malana, G.E.; Pleister, A.P.; Jiang, J.; Schmittgen, T.D.; Terry, A.V., Jr.; Gardiner, K.; Head, E.; et al. Human chromosome 21-derived mirnas are overexpressed in down syndrome brains and hearts. Biochem. Biophys. Res. Commun. 2008, 370, 473–477. [Google Scholar] [CrossRef]
- Xu, J.; Hu, Z.; Xu, Z.; Gu, H.; Yi, L.; Cao, H.; Chen, J.; Tian, T.; Liang, J.; Lin, Y.; et al. Functional variant in microrna-196a2 contributes to the susceptibility of congenital heart disease in a chinese population. Hum. Mutat. 2009, 30, 1231–1236. [Google Scholar]
- O’Brien, J.E., Jr.; Kibiryeva, N.; Zhou, X.G.; Marshall, J.A.; Lofland, G.K.; Artman, M.; Chen, J.; Bittel, D.C. Noncoding rna expression in myocardium from infants with tetralogy of fallot. Circ. Cardiovasc. Genet. 2012, 5, 279–286. [Google Scholar]
- Bittel, D.C.; Butler, M.G.; Kibiryeva, N.; Marshall, J.A.; Chen, J.; Lofland, G.K.; O'Brien, J.E., Jr. Gene expression in cardiac tissues from infants with idiopathic conotruncal defects. BMC Med. Genomics. 2011, 4, 1. [Google Scholar] [CrossRef]
- Scharer, C.D.; McCabe, C.D.; Ali-Seyed, M.; Berger, M.F.; Bulyk, M.L.; Moreno, C.S. Genome-wide promoter analysis of the sox4 transcriptional network in prostate cancer cells. Cancer Res. 2009, 69, 709–717. [Google Scholar] [CrossRef]
- Paul, M.H.; Harvey, R.P.; Wegner, M.; Sock, E. Cardiac outflow tract development relies on the complex function of sox4 and sox11 in multiple cell types. Cell. Mol. Life Sci. 2013. [Google Scholar] [CrossRef]
- Schilham, M.W.; Oosterwegel, M.A.; Moerer, P.; Ya, J.; de Boer, P.A.; van de Wetering, M.; Verbeek, S.; Lamers, W.H.; Kruisbeek, A.M.; Cumano, A.; et al. Defects in cardiac outflow tract formation and pro-b-lymphocyte expansion in mice lacking sox-4. Nature 1996, 380, 711–714. [Google Scholar]
- Chen, L.; Tang, Y.; Wang, J.; Yan, Z.; Xu, R. Mir-421 induces cell proliferation and apoptosis resistance in human nasopharyngeal carcinoma via downregulation of foxo4. Biochem Biophys Res. Commun. 2013, 435, 745–750. [Google Scholar] [CrossRef]
- Zhang, Y.; Gong, W.; Dai, S.; Huang, G.; Shen, X.; Gao, M.; Xu, Z.; Zeng, Y.; He, F. Downregulation of human farnesoid x receptor by mir-421 promotes proliferation and migration of hepatocellular carcinoma cells. Mol. Cancer Res. 2012, 10, 516–522. [Google Scholar]
- LifeNet Health. Available online: www.Lifenethealth.Org (accessed on 8 July 2014).
- Bittel, D.C.; Zhou, X.G.; Kibiryeva, N.; Fiedler, S.; O’Brien, J.E., Jr.; Marshall, J.; Yu, S.; Liu, H.Y. Ultra high-resolution gene centric genomic structural analysis of a non-syndromic congenital heart defect, tetralogy of fallot. PLoS One 2014, 9, e87472. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bittel, D.C.; Kibiryeva, N.; Marshall, J.A.; O'Brien, J.E., Jr. MicroRNA-421 Dysregulation is Associated with Tetralogy of Fallot. Cells 2014, 3, 713-723. https://doi.org/10.3390/cells3030713
Bittel DC, Kibiryeva N, Marshall JA, O'Brien JE Jr. MicroRNA-421 Dysregulation is Associated with Tetralogy of Fallot. Cells. 2014; 3(3):713-723. https://doi.org/10.3390/cells3030713
Chicago/Turabian StyleBittel, Douglas C., Nataliya Kibiryeva, Jennifer A. Marshall, and James E. O'Brien, Jr. 2014. "MicroRNA-421 Dysregulation is Associated with Tetralogy of Fallot" Cells 3, no. 3: 713-723. https://doi.org/10.3390/cells3030713