
Immunotherapy for Platinum-Resistant Ovarian Cancer as a Glimmer of Hope

 , and
, and
Abstract
1. Introduction
2. Platinum-Resistant Ovarian Cancer
3. Molecular Mechanisms of Platinum Resistance
3.1. Alterations in Drug Influx and Efflux Pathways
3.2. DNA Damage Repair Enhancement
3.3. Reactive Oxygen Species Regulations and Oxidative Stress
3.4. Cell Cycle Disruptions
3.5. Mutations in Apoptotic Pathways and Autophagy
3.6. Epigenetic Changes
3.7. Metabolic Alterations
4. Current Standard of Care for Recurrent Ovarian Cancer
5. The Rise of Cancer Immunotherapy
6. Diverse Immunotherapy Strategies
6.1. Cellular Immunotherapies
6.1.1. Chimeric Antigen Receptor T Cell (CAR-T)
6.1.2. Tumor-Infiltrating Lymphocytes (TILs)
6.1.3. Natural Killer (NK)-Cell Therapies
Memory-like NK Cells
CAR-NK Cells
Allogeneic and Non-CAR NK-Cell Therapies
6.1.4. Macrophage-Based Therapies
CAR-Macrophages (CAR-M)
HER2-Targeted CAR-M Therapy
Macrophage Checkpoint Inhibitors
6.2. Immune Checkpoint Inhibitors
6.2.1. Dual Checkpoint Blockade
6.2.2. Combination Therapy: Checkpoint Inhibitors and Anti-Angiogenics
6.2.3. Combination Therapy: Checkpoint Inhibitors and PARP Inhibitors
6.2.4. Other Novel Checkpoint Targets
6.3. Cancer Vaccines
6.3.1. mRNA-Based Vaccines
6.3.2. Dendritic Cell Vaccines
6.3.3. Peptide and Protein Vaccines
6.3.4. Neoantigen Vaccines
6.3.5. Other Vaccine Modalities
6.4. Bispecific T-Cell Engagers (TCEs) and Dual-Target Antibodies
6.5. Oncolytic Viruses
6.6. Cytokine and Immunomodulatory Therapies
6.7. Engineered T-Cell Receptor (TCR) Therapies
6.8. Miscellaneous Approaches
7. Overcoming Challenges and Future Directions
7.1. Challenge 1: The Immunosuppressive TME
7.2. Challenge 2: Tumor Heterogeneity and Antigen Escape
7.3. Challenge 3: Immune Checkpoint and Exhaustion Pathways
7.4. Challenge 4: Lack of Predictive Biomarkers for Immunotherapy Response
7.5. Challenge 5: Practical Deployment and Persistence of Cellular Therapies
7.6. Challenge 6: Toxicity Management in Combinatorial Regimens
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACT | adoptive cell transfer |
| ADCC | antibody-dependent cellular cytotoxicity |
| AIs | aromatase inhibitors |
| AQPs | aquaporins |
| BER | base excision repair |
| CA-125 | cancer antigen 125 |
| CAR | chimeric antigen receptor |
| CAR-M | chimeric antigen receptor macrophages |
| CAR-T | chimeric antigen receptor T-cell/s |
| CCNE1 | cell cycle regulator cyclin E1 |
| CRS | cytokine release syndrome |
| CSF1R | CSF-1 receptor |
| ctDNA | circulating tumor DNA |
| CTLA-4 | cytotoxic T-cell antigen 4 |
| CTRs | copper transport proteins |
| DC(s) | dendritic cell(s) |
| DDR | DNA damage response |
| EMA | European Medicines Agency |
| EOC(s) | epithelial ovarian cancer(s) |
| ER | estrogen receptor |
| ESMO | European Society For Medical Oncology |
| FDA | Food and Drug Administration |
| FMT | fecal microbiota transplantation |
| FRα | folate receptor alpha |
| FRβ | folate receptor beta |
| FSHR | follicle-stimulating hormone receptor |
| GVHD | graft-versus-host disease |
| HER2 | human epidermal growth factor receptor 2 |
| HGSC(s) | high-grade serous carcinoma(s) |
| HRD | homologous recombination deficiency; homologous recombination-deficient |
| HR | homologous repair |
| ICIs | immune checkpoint inhibitors |
| IFN-α | interferon-alpha |
| IL-2 | interleukin-2 |
| IP | intraperitoneal |
| irAEs | immune-related adverse events |
| IV | intravenous |
| LGSC(s) | low-grade serous carcinoma(s) |
| MDRs | multidrug resistance proteins |
| MDSCs | myeloid-derived suppressor cells |
| MHC | major histocompatibility complex |
| MIRV | Mirvetuximab soravtansine-gynx |
| MMR | mismatch repair |
| MSI-H | microsatellite instability-high |
| NCCN | National Comprehensive Cancer Network |
| NER | nucleotide excision repair |
| NHEJ | non-homologous end joining |
| NK | natural killer |
| OC(s) | ovarian cancer(s) |
| OCDC | oxidized autologous whole-tumor lysates |
| Olvi-Vec | Olvimulogene nanivacirepvec |
| ORR(s) | objective response rate(s) |
| OS | overall survival |
| PARPi | poly (ADP-ribose) polymerase inhibitors |
| PD-1 | programmed death-1 |
| PD-L1 | programmed death-ligand 1 |
| PFI | platinum-free interval |
| PFS | progression-free survival |
| PLD | pegylated liposomal doxorubicin |
| PPROC | primary platinum-resistant ovarian cancer |
| PROC | platinum-resistant ovarian cancer |
| PR | progesterone receptor |
| PSROC | platinum-sensitive recurrent ovarian cancer |
| RB1 | retinoblastoma protein 1 |
| ROC | recurrent ovarian cancer |
| ROS | reactive oxygen species |
| scFvs | single-chain variable fragments |
| SLCs | solute carrier transporters |
| SPROC | secondary platinum-resistant ovarian cancer |
| TAA(s) | tumor-associated antigen(s) |
| TAMs | tumor-associated macrophages |
| TCEs | T-cell engagers |
| TCR | T-cell receptor |
| TIL(s) | tumor-infiltrating lymphocyte(s) |
| TLR | Toll-like receptor |
| TME(s) | tumor microenvironment(s) |
| Tregs | regulatory T cells |
| TRICOM | Triad of Co-stimulatory Molecules |
| T-VEC | talimogene laherparepvec |
| VEGF | vascular endothelial growth factor |
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Trial Name | Phase | Therapy Type | Agents Involved | Patient Population | Key Findings |
|---|---|---|---|---|---|
| IMagyn050 | III | Checkpoint Inhibitor + Chemo | Atezolizumab + bevacizumab + chemo | Frontline OC | No significant PFS benefit in unselected patients |
| JAVELIN Ovarian 100 | III | Checkpoint Inhibitor + Chemo | Avelumab + chemo | Frontline OC | No PFS or OS improvement |
| JAVELIN Ovarian 200 | III | Checkpoint Inhibitor vs. Chemo | Avelumab vs. PLD | PROC | No significant survival benefit |
| KEYNOTE-B96 | III | Checkpoint Inhibitor + Chemo | Pembrolizumab + chemo | PROC (PD-L1+ and all-comers) | Significantly improved PFS vs. chemo alone |
| KEYNOTE-100 | II | ICI monotherapy | Pembrolizumab | Recurrent OC | Low ORR (~8%), better in PD-L1+ tumors |
| DUO-O | III | ICI + PARPi + Chemo | Durvalumab + olaparib + bevacizumab + chemo | Frontline OC | PFS improved in HRD+, no OS benefit yet |
| ATHENA | III | PARPi ± ICI | Rucaparib ± nivolumab | Frontline OC | Rucaparib improved PFS |
| ROSELLA | II | GR modulator | Relacorilant | PROC | Improved PFS when combined with nab-paclitaxel |
| DESKTOP III | III | Surgery | Secondary cytoreduction | PSROC | Improved OS in selected patients |
| NRG-GY003 | II | Dual ICI | Nivolumab + ipilimumab | ROC | Better PFS and ORR vs. nivolumab alone |
| MEDIOLA | II | PARPi + ICI | Durvalumab + olaparib | BRCA-mutant OC | Encouraging ORR and PFS |
| TOPACIO/ KEYNOTE-162 | I/II | PARPi + ICI | Niraparib + pembrolizumab | PROC | Clinical activity in BRCAwt/HRD+ tumors |
| MOONSTONE | III | PARPi + ICI | Niraparib + dostarlimab | PROC | Ongoing |
| ARTISTRY-7 | III | IL-2 agonist ± ICI | Nemvaleukin ± pembrolizumab | PROC | Phase II data showed 28.6% ORR |
| OVERVEIL | III | Oncolytic virus + chemo | Olvi-Vec + chemo | PROC | Improved immune activation |
| City of Hope CAR-T | I | CAR-T | Anti-TAG72 CAR-T | OC | Ongoing |
| BNT211 | I/II | CAR-T | CLDN6-targeted CAR-T | Solid tumors incl. OC | Early efficacy signals |
| LN-145 | II | TIL therapy | LN-145 | PROC | Under evaluation |
| CT-0508 | I | CAR-M | HER2-directed CAR-M | Solid tumors incl. OC | Ongoing |
| SY001 | I | CAR-M | Mesothelin-targeting CAR-M | Recurrent OC | First-in-human report in 2024 |
| FLORA-5 | II | ICI combo | Nivolumab + relatlimab | OC | Testing PD-1 + LAG-3 blockade |
| Tebotelimab trial | I | Bispecific Ab | PD-1 + LAG-3 bsAb | Solid tumors incl. OC | Enhanced T-cell activation |
| NCT04137900 | I | ICI combo | TIM-3/TIGIT + PD-1 blockade | Advanced gynecologic cancers | Ongoing |
| XB002 | I | ADC | B7-H4 targeting ADC | PROC | Early clinical trial |
| SL-172154 | I | Bispecific | CD47 blockade + CD40 activation | PROC | Ongoing |
| GEN-1 | I/II | Gene therapy | IL-12 nanoparticles | PROC | Local immune activation |
References
- Webb, P.M.; Jordan, S.J. Global Epidemiology of Epithelial Ovarian Cancer. Nat. Rev. Clin. Oncol. 2024, 21, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Pejovic, T.; Fitch, K.; Mills, G. Ovarian Cancer Recurrence: “Is the Definition of Platinum Resistance Modified by PARP Inhibitors and Other Intervening Treatments?”. Cancer Drug Resist. 2022, 5, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Tung, K.-H.; Goodman, M.T.; Wu, A.H.; McDuffie, K.; Wilkens, L.R.; Nomura, A.M.Y.; Kolonel, L.N. Aggregation of Ovarian Cancer with Breast, Ovarian, Colorectal, and Prostate Cancer in First-Degree Relatives. Am. J. Epidemiol. 2004, 159, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, K.; Sundquist, J.; Brandt, A. Incidence and Mortality in Epithelial Ovarian Cancer by Family History of Any Cancer. Cancer 2011, 117, 3972–3980. [Google Scholar] [CrossRef]
- Kempf, E.; Desamericq, G.; Vieites, B.; Diaz-Padilla, I.; Calvo, E.; Estevez, P.; Garcia-Arreza, A.; Martinez-Maestre, M.A.; Duran, I. Clinical and Pathologic Features of Patients with Non-Epithelial Ovarian Cancer: Retrospective Analysis of a Single Institution 15-Year Experience. Clin. Transl. Oncol. 2017, 19, 173–179. [Google Scholar] [CrossRef]
- Crum, C.P.; Drapkin, R.; Miron, A.; Ince, T.A.; Muto, M.; Kindelberger, D.W.; Lee, Y. The Distal Fallopian Tube: A New Model for Pelvic Serous Carcinogenesis. Curr. Opin. Obstet. Gynecol. 2007, 19, 3–9. [Google Scholar] [CrossRef]
- Elsherif, S.B.; Bhosale, P.R.; Lall, C.; Menias, C.O.; Itani, M.; Butler, K.A.; Ganeshan, D. Current Update on Malignant Epithelial Ovarian Tumors. Abdom. Radiol. N. Y. 2021, 46, 2264–2280. [Google Scholar] [CrossRef]
- Santoro, A.; Angelico, G.; Travaglino, A.; Inzani, F.; Spadola, S.; Pettinato, A.; Mazzucchelli, M.; Bragantini, E.; Maccio, L.; Zannoni, G.F. The Multiple Facets of Ovarian High Grade Serous Carcinoma: A Review on Morphological, Immunohistochemical and Molecular Features. Crit. Rev. Oncol. Hematol. 2025, 208, 104603. [Google Scholar] [CrossRef]
- Folsom, S.M.; Berger, J.; Soong, T.R.; Rangaswamy, B. Comprehensive Review of Serous Tumors of Tubo-Ovarian Origin: Clinical Behavior, Pathological Correlation, Current Molecular Updates, and Imaging Manifestations. Curr. Probl. Diagn. Radiol. 2023, 52, 425–438. [Google Scholar] [CrossRef]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial Ovarian Cancer. Lancet Lond. Engl. 2019, 393, 1240–1253. [Google Scholar] [CrossRef]
- Oronsky, B.; Ray, C.M.; Spira, A.I.; Trepel, J.B.; Carter, C.A.; Cottrill, H.M. A Brief Review of the Management of Platinum-Resistant-Platinum-Refractory Ovarian Cancer. Med. Oncol. 2017, 34, 103. [Google Scholar] [CrossRef] [PubMed]
- Kajiyama, H.; Mizuno, M.; Shibata, K.; Umezu, T.; Suzuki, S.; Yamamoto, E.; Mitsui, H.; Sekiya, R.; Niimi, K.; Kawai, M.; et al. Oncologic Outcome after Recurrence in Patients with Stage I Epithelial Ovarian Cancer: Are Clear-Cell and Mucinous Histological Types a Different Entities? Eur. J. Obstet. Gynecol. Reprod. Biol. 2014, 181, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Webber, K.; Friedlander, M. Chemotherapy for Epithelial Ovarian, Fallopian Tube and Primary Peritoneal Cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 2017, 41, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Havasi, A.; Cainap, S.S.; Havasi, A.T.; Cainap, C. Ovarian Cancer-Insights into Platinum Resistance and Overcoming It. Med. Kaunas Lith. 2023, 59, 544. [Google Scholar] [CrossRef]
- Hamdan, F.; Cerullo, V. Cancer Immunotherapies: A Hope for the Uncurable? Front. Mol. Med. 2023, 3, 1140977. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Hilpert, F.; Weber, B.; Reuss, A.; Poveda, A.; Kristensen, G.; Sorio, R.; Vergote, I.; Witteveen, P.; Bamias, A.; et al. Bevacizumab Combined with Chemotherapy for Platinum-Resistant Recurrent Ovarian Cancer: The AURELIA Open-Label Randomized Phase III Trial. J. Clin. Oncol. 2014, 32, 1302–1308. [Google Scholar] [CrossRef]
- Trillsch, F.; Mahner, S.; Czogalla, B.; Rottmann, M.; Chekerov, R.; Braicu, E.I.; Oskay-Öczelik, G.; Wimberger, P.; Richter, R.; Sehouli, J. Primary Platinum Resistance and Its Prognostic Impact in Patients with Recurrent Ovarian Cancer: An Analysis of Three Prospective Trials from the NOGGO Study Group. J. Gynecol. Oncol. 2021, 32, e37. [Google Scholar] [CrossRef]
- Elyashiv, O.; Aleohin, N.; Migdan, Z.; Leytes, S.; Peled, O.; Tal, O.; Levy, T. The Poor Prognosis of Acquired Secondary Platinum Resistance in Ovarian Cancer Patients. Cancers 2024, 16, 641. [Google Scholar] [CrossRef] [PubMed]
- Gately, D.P.; Howell, S.B. Cellular Accumulation of the Anticancer Agent Cisplatin: A Review. Br. J. Cancer 1993, 67, 1171–1176. [Google Scholar] [CrossRef]
- Holzer, A.K.; Samimi, G.; Katano, K.; Naerdemann, W.; Lin, X.; Safaei, R.; Howell, S.B. The Copper Influx Transporter Human Copper Transport Protein 1 Regulates the Uptake of Cisplatin in Human Ovarian Carcinoma Cells. Mol. Pharmacol. 2004, 66, 817–823. [Google Scholar] [CrossRef]
- Shen, D.-W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin Resistance: A Cellular Self-Defense Mechanism Resulting from Multiple Epigenetic and Genetic Changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef] [PubMed]
- Harrach, S.; Ciarimboli, G. Role of Transporters in the Distribution of Platinum-Based Drugs. Front. Pharmacol. 2015, 6, 85. [Google Scholar] [CrossRef]
- Johnson, S.W.; Shen, D.; Pastan, I.; Gottesman, M.M.; Hamilton, T.C. Cross-Resistance, Cisplatin Accumulation, and Platinum-DNA Adduct Formation and Removal in Cisplatin-Sensitive and -Resistant Human Hepatoma Cell Lines. Exp. Cell Res. 1996, 226, 133–139. [Google Scholar] [CrossRef]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the Anticancer Drug Cisplatin Mediated by the Copper Transporter Ctr1 in Yeast and Mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef]
- Lin, X.; Okuda, T.; Holzer, A.; Howell, S.B. The Copper Transporter CTR1 Regulates Cisplatin Uptake in Saccharomyces Cerevisiae. Mol. Pharmacol. 2002, 62, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Holzer, A.K.; Manorek, G.H.; Howell, S.B. Contribution of the Major Copper Influx Transporter CTR1 to the Cellular Accumulation of Cisplatin, Carboplatin, and Oxaliplatin. Mol. Pharmacol. 2006, 70, 1390–1394. [Google Scholar] [CrossRef] [PubMed]
- Song, I.-S.; Savaraj, N.; Siddik, Z.H.; Liu, P.; Wei, Y.; Wu, C.J.; Kuo, M.T. Role of Human Copper Transporter Ctr1 in the Transport of Platinum-Based Antitumor Agents in Cisplatin-Sensitive and Cisplatin-Resistant Cells. Mol. Cancer Ther. 2004, 3, 1543–1549. [Google Scholar] [CrossRef]
- Zalewski, M.; Kulbacka, J.; Saczko, J.; Drag-Zalesinska, M.; Choromanska, A. Valspodar-Modulated Chemotherapy in Human Ovarian Cancer Cells SK-OV-3 and MDAH-2774. Biomol. Biomed. 2019, 19, 234–241. [Google Scholar] [CrossRef]
- Baekelandt, M.; Lehne, G.; Tropé, C.G.; Szántó, I.; Pfeiffer, P.; Gustavssson, B.; Kristensen, G.B. Phase I/II Trial of the Multidrug-Resistance Modulator Valspodar Combined with Cisplatin and Doxorubicin in Refractory Ovarian Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2001, 19, 2983–2993. [Google Scholar] [CrossRef]
- Colombo, P.-E.; Fabbro, M.; Theillet, C.; Bibeau, F.; Rouanet, P.; Ray-Coquard, I. Sensitivity and Resistance to Treatment in the Primary Management of Epithelial Ovarian Cancer. Crit. Rev. Oncol. Hematol. 2014, 89, 207–216. [Google Scholar] [CrossRef]
- Damia, G.; Broggini, M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers 2019, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA Repair Dysregulation from Cancer Driver to Therapeutic Target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Ferry, K.V.; Hamilton, T.C.; Johnson, S.W. Increased Nucleotide Excision Repair in Cisplatin-Resistant Ovarian Cancer Cells: Role of ERCC1-XPF. Biochem. Pharmacol. 2000, 60, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; McCorvie, T.J.; Yates, L.A.; Zhang, X. Structural Basis of Homologous Recombination. Cell. Mol. Life Sci. CMLS 2020, 77, 3–18. [Google Scholar] [CrossRef]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA Mutation Frequency and Patterns of Treatment Response in BRCA Mutation–Positive Women with Ovarian Cancer: A Report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef]
- Weigelt, B.; Comino-Méndez, I.; de Bruijn, I.; Tian, L.; Meisel, J.L.; García-Murillas, I.; Fribbens, C.; Cutts, R.; Martelotto, L.G.; Ng, C.K.Y.; et al. Diverse BRCA1 and BRCA2 Reversion Mutations in Circulating Cell-Free DNA of Therapy-Resistant Breast or Ovarian Cancer. Clin. Cancer Res. 2017, 23, 6708–6720. [Google Scholar] [CrossRef]
- Mylavarapu, S.; Das, A.; Roy, M. Role of BRCA Mutations in the Modulation of Response to Platinum Therapy. Front. Oncol. 2018, 8, 16. [Google Scholar] [CrossRef]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary Somatic Mutations Restoring BRCA1/2 Predict Chemotherapy Resistance in Hereditary Ovarian Carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef]
- Wu, X.; Han, L.Y.; Zhang, X.X.; Wang, L. The Study of Nrf2 Signaling Pathway in Ovarian Cancer. Crit. Rev. Eukaryot. Gene Expr. 2018, 28, 329–336. [Google Scholar] [CrossRef]
- Gentric, G.; Kieffer, Y.; Mieulet, V.; Goundiam, O.; Bonneau, C.; Nemati, F.; Hurbain, I.; Raposo, G.; Popova, T.; Stern, M.-H.; et al. PML-Regulated Mitochondrial Metabolism Enhances Chemosensitivity in Human Ovarian Cancers. Cell Metab. 2019, 29, 156–173.e10. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, P.; Chen, X.; Gong, D.; Wu, Y.; Huang, L.; Chen, Y. ROS-Induced DCTPP1 Upregulation Contributes to Cisplatin Resistance in Ovarian Cancer. Front. Mol. Biosci. 2022, 9, 838006. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, X.; Chen, Q.; Liu, T.; Wu, Y.; Huang, L.; Chen, Y. Expression of Human dCTP Pyrophosphatase 1 (DCTPP1) and Its Association with Cisplatin Resistance Characteristics in Ovarian Cancer. J. Cell. Mol. Med. 2024, 28, e18371. [Google Scholar] [CrossRef]
- Kleih, M.; Böpple, K.; Dong, M.; Gaißler, A.; Heine, S.; Olayioye, M.A.; Aulitzky, W.E.; Essmann, F. Direct Impact of Cisplatin on Mitochondria Induces ROS Production That Dictates Cell Fate of Ovarian Cancer Cells. Cell Death Dis. 2019, 10, 851. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Xu, J.; Jia, X. Targeting Mitochondria: A Novel Approach for Treating Platinum-Resistant Ovarian Cancer. J. Transl. Med. 2024, 22, 968. [Google Scholar] [CrossRef]
- Nakayama, N.; Nakayama, K.; Shamima, Y.; Ishikawa, M.; Katagiri, A.; Iida, K.; Miyazaki, K. Gene Amplification CCNE1 Is Related to Poor Survival and Potential Therapeutic Target in Ovarian Cancer. Cancer 2010, 116, 2621–2634. [Google Scholar] [CrossRef]
- Siu, K.T.; Rosner, M.R.; Minella, A.C. An Integrated View of Cyclin E Function and Regulation. Cell Cycle 2012, 11, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Matson, J.P.; Dumitru, R.; Coryell, P.; Baxley, R.M.; Chen, W.; Twaroski, K.; Webber, B.R.; Tolar, J.; Bielinsky, A.-K.; Purvis, J.E.; et al. Rapid DNA Replication Origin Licensing Protects Stem Cell Pluripotency. Elife 2017, 6, e30473. [Google Scholar] [CrossRef]
- da Costa, A.A.B.A.; Chowdhury, D.; Shapiro, G.I.; D’Andrea, A.D.; Konstantinopoulos, P.A. Targeting Replication Stress in Cancer Therapy. Nat. Rev. Drug Discov. 2023, 22, 38–58. [Google Scholar] [CrossRef]
- Zeng, J.; Hills, S.A.; Ozono, E.; Diffley, J.F.X. Cyclin E-Induced Replicative Stress Drives P53-Dependent Whole-Genome Duplication. Cell 2023, 186, 528–542.e14. [Google Scholar] [CrossRef]
- Xu, H.; George, E.; Gallo, D.; Medvedev, S.; Wang, X.; Datta, A.; Kryczka, R.; Hyer, M.L.; Fourtounis, J.; Stocco, R.; et al. Targeting CCNE1 Amplified Ovarian and Endometrial Cancers by Combined Inhibition of PKMYT1 and ATR. Nat. Commun. 2025, 16, 3112. [Google Scholar] [CrossRef]
- Gorski, J.W.; Ueland, F.R.; Kolesar, J.M. CCNE1 Amplification as a Predictive Biomarker of Chemotherapy Resistance in Epithelial Ovarian Cancer. Diagnostics 2020, 10, 279. [Google Scholar] [CrossRef]
- Kim, D.; Chung, H.; Liu, W.; Jeong, K.; Ozmen, T.Y.; Ozmen, F.; Rames, M.J.; Kim, S.; Guo, X.; Jameson, N.; et al. Cyclin E1/CDK2 Activation Defines a Key Vulnerability to WEE1 Kinase Inhibition in Gynecological Cancers. Npj Precis. Oncol. 2025, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Garsed, D.W.; Alsop, K.; Fereday, S.; Emmanuel, C.; Kennedy, C.J.; Etemadmoghadam, D.; Gao, B.; Gebski, V.; Garès, V.; Christie, E.L.; et al. Homologous Recombination DNA Repair Pathway Disruption and Retinoblastoma Protein Loss Are Associated with Exceptional Survival in High-Grade Serous Ovarian Cancer. Clin. Cancer Res. 2018, 24, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Reles, A.; Wen, W.H.; Schmider, A.; Gee, C.; Runnebaum, I.B.; Kilian, U.; Jones, L.A.; El-Naggar, A.; Minguillon, C.; Schönborn, I.; et al. Correlation of P53 Mutations with Resistance to Platinum-Based Chemotherapy and Shortened Survival in Ovarian Cancer. Clin. Cancer Res. 2001, 7, 2984–2997. [Google Scholar]
- Cai, Y.; Tan, X.; Liu, J.; Shen, Y.; Wu, D.; Ren, M.; Huang, P.; Yu, D. Inhibition of PI3K/Akt/mTOR Signaling Pathway Enhances the Sensitivity of the SKOV3/DDP Ovarian Cancer Cell Line to Cisplatin in Vitro. Chin. J. Cancer Res. Chung-Kuo Yen Cheng Yen Chiu 2014, 26, 564–572. [Google Scholar] [CrossRef]
- Deng, J.; Bai, X.; Feng, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Inhibition of PI3K/Akt/mTOR Signaling Pathway Alleviates Ovarian Cancer Chemoresistance through Reversing Epithelial-Mesenchymal Transition and Decreasing Cancer Stem Cell Marker Expression. BMC Cancer 2019, 19, 618. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, J.-Y.; Wu, G.S. ERK-Dependent MKP-1-Mediated Cisplatin Resistance in Human Ovarian Cancer Cells. Cancer Res. 2007, 67, 11933–11941. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, G.S. Role of Autophagy in Cisplatin Resistance in Ovarian Cancer Cells. J. Biol. Chem. 2014, 289, 17163–17173. [Google Scholar] [CrossRef]
- Mohell, N.; Alfredsson, J.; Fransson, Å.; Uustalu, M.; Byström, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.N.; Björklund, U.; Wiman, K.G. APR-246 Overcomes Resistance to Cisplatin and Doxorubicin in Ovarian Cancer Cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef]
- Rada, M.; Nallanthighal, S.; Cha, J.; Ryan, K.; Sage, J.; Eldred, C.; Ullo, M.; Orsulic, S.; Cheon, D.-J. Inhibitor of Apoptosis Proteins (IAPs) Mediate Collagen Type XI Alpha 1-Driven Cisplatin Resistance in Ovarian Cancer. Oncogene 2018, 37, 4809–4820. [Google Scholar] [CrossRef]
- Chan, D.W.; Lam, W.-Y.; Chen, F.; Yung, M.M.H.; Chan, Y.-S.; Chan, W.-S.; He, F.; Liu, S.S.; Chan, K.K.L.; Li, B.; et al. Genome-Wide DNA Methylome Analysis Identifies Methylation Signatures Associated with Survival and Drug Resistance of Ovarian Cancers. Clin. Epigenetics 2021, 13, 142. [Google Scholar] [CrossRef]
- Lund, R.J.; Huhtinen, K.; Salmi, J.; Rantala, J.; Nguyen, E.V.; Moulder, R.; Goodlett, D.R.; Lahesmaa, R.; Carpén, O. DNA Methylation and Transcriptome Changes Associated with Cisplatin Resistance in Ovarian Cancer. Sci. Rep. 2017, 7, 1469. [Google Scholar] [CrossRef]
- Bonito, N.A.; Borley, J.; Wilhelm-Benartzi, C.S.; Ghaem-Maghami, S.; Brown, R. Epigenetic Regulation of the Homeobox Gene MSX1 Associates with Platinum-Resistant Disease in High-Grade Serous Epithelial Ovarian Cancer. Clin. Cancer Res. 2016, 22, 3097–3104. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, S.; Gai, J.; Guan, J.; Li, J.; Li, Y.; Zhao, J.; Zhao, C.; Fu, L.; Li, Q. SIRT5 Promotes Cisplatin Resistance in Ovarian Cancer by Suppressing DNA Damage in a ROS-Dependent Manner via Regulation of the Nrf2/HO-1 Pathway. Front. Oncol. 2019, 9, 754. [Google Scholar] [CrossRef] [PubMed]
- Cacan, E.; Ali, M.W.; Boyd, N.H.; Hooks, S.B.; Greer, S.F. Inhibition of HDAC1 and DNMT1 Modulate RGS10 Expression and Decrease Ovarian Cancer Chemoresistance. PLoS ONE 2014, 9, e87455. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Li, Y.; Wan, Y.; Xue, M. MircroRNA-139 Sensitizes Ovarian Cancer Cell to Cisplatin-Based Chemotherapy through Regulation of ATP7A/B. Cancer Chemother. Pharmacol. 2018, 81, 935–947. [Google Scholar] [CrossRef]
- Wang, T.; Hao, D.; Yang, S.; Ma, J.; Yang, W.; Zhu, Y.; Weng, M.; An, X.; Wang, X.; Li, Y.; et al. miR-211 Facilitates Platinum Chemosensitivity by Blocking the DNA Damage Response (DDR) in Ovarian Cancer. Cell Death Dis. 2019, 10, 495. [Google Scholar] [CrossRef]
- Giacomini, I.; Ragazzi, E.; Pasut, G.; Montopoli, M. The Pentose Phosphate Pathway and Its Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2020, 21, 937. [Google Scholar] [CrossRef]
- Xu, Y.; Gao, W.; Zhang, Y.; Wu, S.; Liu, Y.; Deng, X.; Xie, L.; Yang, J.; Yu, H.; Su, J.; et al. ABT737 Reverses Cisplatin Resistance by Targeting Glucose Metabolism of Human Ovarian Cancer Cells. Int. J. Oncol. 2018, 53, 1055–1068. [Google Scholar] [CrossRef]
- Masamha, C.P.; LaFontaine, P. Molecular Targeting of Glutaminase Sensitizes Ovarian Cancer Cells to Chemotherapy. J. Cell. Biochem. 2018, 119, 6136–6145. [Google Scholar] [CrossRef]
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S.; et al. The Cancer Metabolic Reprogramming and Immune Response. Mol. Cancer 2021, 20, 28. [Google Scholar] [CrossRef]
- Tan, Y.; Li, J.; Zhao, G.; Huang, K.-C.; Cardenas, H.; Wang, Y.; Matei, D.; Cheng, J.-X. Metabolic Reprogramming from Glycolysis to Fatty Acid Uptake and Beta-Oxidation in Platinum-Resistant Cancer Cells. Nat. Commun. 2022, 13, 4554. [Google Scholar] [CrossRef] [PubMed]
- Pervushin, N.V.; Yapryntseva, M.A.; Panteleev, M.A.; Zhivotovsky, B.; Kopeina, G.S. Cisplatin Resistance and Metabolism: Simplification of Complexity. Cancers 2024, 16, 3082. [Google Scholar] [CrossRef]
- Ai, Z.; Lu, Y.; Qiu, S.; Fan, Z. Overcoming Cisplatin Resistance of Ovarian Cancer Cells by Targeting HIF-1-Regulated Cancer Metabolism. Cancer Lett. 2016, 373, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Baert, T.; Ferrero, A.; Sehouli, J.; O’Donnell, D.M.; González-Martín, A.; Joly, F.; van der Velden, J.; Blecharz, P.; Tan, D.S.P.; Querleu, D.; et al. The Systemic Treatment of Recurrent Ovarian Cancer Revisited. Ann. Oncol. 2021, 32, 710–725. [Google Scholar] [CrossRef]
- Pfisterer, J.; Plante, M.; Vergote, I.; du Bois, A.; Hirte, H.; Lacave, A.J.; Wagner, U.; Stähle, A.; Stuart, G.; Kimmig, R.; et al. Gemcitabine Plus Carboplatin Compared With Carboplatin in Patients With Platinum-Sensitive Recurrent Ovarian Cancer: An Intergroup Trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J. Clin. Oncol. 2006, 24, 4699–4707. [Google Scholar] [CrossRef]
- Parmar, M.K.B.; Ledermann, J.A.; Colombo, N.; du Bois, A.; Delaloye, J.-F.; Kristensen, G.B.; Wheeler, S.; Swart, A.M.; Qian, W.; Torri, V.; et al. Paclitaxel plus Platinum-Based Chemotherapy versus Conventional Platinum-Based Chemotherapy in Women with Relapsed Ovarian Cancer: The ICON4/AGO-OVAR-2.2 Trial. Lancet Lond. Engl. 2003, 361, 2099–2106. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Wagner, U.; Aavall-Lundqvist, E.; Gebski, V.; Heywood, M.; Vasey, P.A.; Volgger, B.; Vergote, I.; Pignata, S.; Ferrero, A.; et al. Pegylated Liposomal Doxorubicin and Carboplatin Compared with Paclitaxel and Carboplatin for Patients with Platinum-Sensitive Ovarian Cancer in Late Relapse. J. Clin. Oncol. 2010, 28, 3323–3329. [Google Scholar] [CrossRef]
- Markman, M.; Blessing, J.; Rubin, S.C.; Connor, J.; Hanjani, P.; Waggoner, S. Phase II Trial of Weekly Paclitaxel (80 Mg/M2) in Platinum and Paclitaxel-Resistant Ovarian and Primary Peritoneal Cancers: A Gynecologic Oncology Group Study. Gynecol. Oncol. 2006, 101, 436–440. [Google Scholar] [CrossRef]
- Gordon, A.N.; Fleagle, J.T.; Guthrie, D.; Parkin, D.E.; Gore, M.E.; Lacave, A.J. Recurrent Epithelial Ovarian Carcinoma: A Randomized Phase III Study of Pegylated Liposomal Doxorubicin versus Topotecan. J. Clin. Oncol. 2001, 19, 3312–3322. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Banerjee, S.; Pignata, S. Management of Platinum-Resistant, Relapsed Epithelial Ovarian Cancer and New Drug Perspectives. J. Clin. Oncol. 2019, 37, 2437–2448. [Google Scholar] [CrossRef]
- Aghajanian, C.; Blank, S.V.; Goff, B.A.; Judson, P.L.; Teneriello, M.G.; Husain, A.; Sovak, M.A.; Yi, J.; Nycum, L.R. OCEANS: A Randomized, Double-Blind, Placebo-Controlled Phase III Trial of Chemotherapy with or without Bevacizumab in Patients with Platinum-Sensitive Recurrent Epithelial Ovarian, Primary Peritoneal, or Fallopian Tube Cancer. J. Clin. Oncol. 2012, 30, 2039–2045. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, C.; Goff, B.; Nycum, L.R.; Wang, Y.V.; Husain, A.; Blank, S.V. Final Overall Survival and Safety Analysis of OCEANS, a Phase 3 Trial of Chemotherapy with or without Bevacizumab in Patients with Platinum-Sensitive Recurrent Ovarian Cancer. Gynecol. Oncol. 2015, 139, 10–16. [Google Scholar] [CrossRef]
- Coleman, R.L.; Brady, M.F.; Herzog, T.J.; Sabbatini, P.; Armstrong, D.K.; Walker, J.L.; Kim, B.-G.; Fujiwara, K.; Tewari, K.S.; O’Malley, D.M.; et al. Bevacizumab and Paclitaxel-Carboplatin Chemotherapy and Secondary Cytoreduction in Recurrent, Platinum-Sensitive Ovarian Cancer (NRG Oncology/Gynecologic Oncology Group Study GOG-0213): A Multicentre, Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2017, 18, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib Maintenance Treatment for Recurrent Ovarian Carcinoma after Response to Platinum Therapy (ARIEL3): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib Maintenance Therapy in Platinum-Sensitive Relapsed Ovarian Cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib Tablets as Maintenance Therapy in Patients with Platinum-Sensitive, Relapsed Ovarian Cancer and a BRCA1/2 Mutation (SOLO2/ENGOT-Ov21): A Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Wu, X.H.; Zhu, J.Q.; Yin, R.T.; Yang, J.X.; Liu, J.H.; Wang, J.; Wu, L.Y.; Liu, Z.L.; Gao, Y.N.; Wang, D.B.; et al. Niraparib Maintenance Therapy in Patients with Platinum-Sensitive Recurrent Ovarian Cancer Using an Individualized Starting Dose (NORA): A Randomized, Double-Blind, Placebo-Controlled Phase III Trial☆. Ann. Oncol. 2021, 32, 512–521. [Google Scholar] [CrossRef]
- Moore, K.N.; Angelergues, A.; Konecny, G.E.; García, Y.; Banerjee, S.; Lorusso, D.; Lee, J.-Y.; Moroney, J.W.; Colombo, N.; Roszak, A.; et al. Mirvetuximab Soravtansine in FRα-Positive, Platinum-Resistant Ovarian Cancer. N. Engl. J. Med. 2023, 389, 2162–2174. [Google Scholar] [CrossRef]
- Olawaiye, A.B.; Kim, J.-W.; Bagameri, A.; Bishop, E.; Chudecka-Głaz, A.; Devaux, A.; Gladieff, L.; Gordinier, M.E.; Korach, J.; McCollum, M.E.; et al. Clinical Trial Protocol for ROSELLA: A Phase 3 Study of Relacorilant in Combination with Nab-Paclitaxel versus Nab-Paclitaxel Monotherapy in Advanced Platinum-Resistant Ovarian Cancer. J. Gynecol. Oncol. 2024, 35, e111. [Google Scholar] [CrossRef] [PubMed]
- Climent, M.T.; Serra, A.; Llueca, M.; Llueca, A. Surgery in Recurrent Ovarian Cancer: A Meta-Analysis. Cancers 2023, 15, 3470. [Google Scholar] [CrossRef] [PubMed]
- Chi, D.S.; McCaughty, K.; Diaz, J.P.; Huh, J.; Schwabenbauer, S.; Hummer, A.J.; Venkatraman, E.S.; Aghajanian, C.; Sonoda, Y.; Abu-Rustum, N.R.; et al. Guidelines and Selection Criteria for Secondary Cytoreductive Surgery in Patients with Recurrent, Platinum-Sensitive Epithelial Ovarian Carcinoma. Cancer 2006, 106, 1933–1939. [Google Scholar] [CrossRef]
- Gadducci, A.; Iacconi, P.; Cosio, S.; Fanucchi, A.; Cristofani, R.; Riccardo Genazzani, A. Complete Salvage Surgical Cytoreduction Improves Further Survival of Patients with Late Recurrent Ovarian Cancer. Gynecol. Oncol. 2000, 79, 344–349. [Google Scholar] [CrossRef]
- Benedetti Panici, P.; De Vivo, A.; Bellati, F.; Manci, N.; Perniola, G.; Basile, S.; Muzii, L.; Angioli, R. Secondary Cytoreductive Surgery in Patients with Platinum-Sensitive Recurrent Ovarian Cancer. Ann. Surg. Oncol. 2007, 14, 1136–1142. [Google Scholar] [CrossRef]
- Knipprath-Mészáros, A.; Heinzelmann, V.; Vetter, M. Endocrine Therapy in Epithelial Ovarian Cancer (EOC) New Insights in an Old Target: A Mini Review. J. Cancer Clin. Trials 2018, 3, 1000144. [Google Scholar] [CrossRef]
- Borella, F.; Fucina, S.; Mangherini, L.; Cosma, S.; Carosso, A.R.; Cusato, J.; Cassoni, P.; Bertero, L.; Katsaros, D.; Benedetto, C. Hormone Receptors and Epithelial Ovarian Cancer: Recent Advances in Biology and Treatment Options. Biomedicines 2023, 11, 2157. [Google Scholar] [CrossRef]
- Chan, K.K.L.; Ngu, S.F.; Chu, M.M.Y.; Tse, K.Y.; Ngan, H.Y.S. Tamoxifen Use in Recurrent Ovarian Cancer in a Chinese Population: A 15 -Year Clinical Experience in a Tertiary Referral Center. Asia Pac. J. Clin. Oncol. 2021, 17, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Heinzelmann-Schwarz, V.; Knipprath Mészaros, A.; Stadlmann, S.; Jacob, F.; Schoetzau, A.; Russell, K.; Friedlander, M.; Singer, G.; Vetter, M. Letrozole May Be a Valuable Maintenance Treatment in High-Grade Serous Ovarian Cancer Patients. Gynecol. Oncol. 2018, 148, 79–85. [Google Scholar] [CrossRef]
- Vetter, M.; Stadlmann, S.; Bischof, E.; Georgescu Margarint, E.L.; Schötzau, A.; Singer, G.; Heinzelmann-Schwarz, V.; Montavon, C. Hormone Receptor Expression in Primary and Recurrent High-Grade Serous Ovarian Cancer and Its Implications in Early Maintenance Treatment. Int. J. Mol. Sci. 2022, 23, 14242. [Google Scholar] [CrossRef]
- Ribatti, D. The Concept of Immune Surveillance against Tumors: The First Theories. Oncotarget 2016, 8, 7175–7180. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T. [Interleukin-2 (IL-2)]. Gan Kagaku Ryoho 1994, 21, 719–724. [Google Scholar]
- Rosenberg, S.A. IL-2: The First Effective Immunotherapy for Human Cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef]
- Lembersky, B.C.; Golomb, H.M. Hairy Cell Leukemia: Clinical Features and Therapeutic Advances. Cancer Metastasis Rev. 1987, 6, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive Cell Transfer: A Clinical Path to Effective Cancer Immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef]
- Charkhchi, P.; Cybulski, C.; Gronwald, J.; Wong, F.O.; Narod, S.A.; Akbari, M.R. CA125 and Ovarian Cancer: A Comprehensive Review. Cancers 2020, 12, 3730. [Google Scholar] [CrossRef]
- Xiao, J.; Chen, H.-S. Biological Functions of Melanoma-Associated Antigens. World J. Gastroenterol. 2004, 10, 1849–1853. [Google Scholar] [CrossRef]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Z.; Li, Y.; Zhao, W.; Wu, J.; Zhang, Z. PD-1/PD-L1 Checkpoint Inhibitors in Tumor Immunotherapy. Front. Pharmacol. 2021, 12, 731798. [Google Scholar] [CrossRef]
- Reinartz, S.; Köhler, S.; Schlebusch, H.; Krista, K.; Giffels, P.; Renke, K.; Huober, J.; Möbus, V.; Kreienberg, R.; duBois, A.; et al. Vaccination of Patients with Advanced Ovarian Carcinoma with the Anti-Idiotype ACA125: Immunological Response and Survival (Phase Ib/II). Clin. Cancer Res. 2004, 10, 1580–1587. [Google Scholar] [CrossRef]
- Kandalaft, L.E.; Powell, D.J.; Singh, N.; Coukos, G. Immunotherapy for Ovarian Cancer: What’s Next? J. Clin. Oncol. 2011, 29, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Drake, C.G. Ipilimumab: An Anti-CTLA-4 Antibody for Metastatic Melanoma. Clin. Cancer Res. 2011, 17, 6958–6962. [Google Scholar] [CrossRef] [PubMed]
- Uhara, H.; Tsuchida, T.; Kiyohara, Y.; Akamatsu, A.; Sakamoto, T.; Yamazaki, N. Safety and Effectiveness of Nivolumab in Japanese Patients with Malignant Melanoma: Final Analysis of a Post-marketing Surveillance. J. Dermatol. 2022, 49, 862–871. [Google Scholar] [CrossRef]
- ESMO FDA Approves Pembrolizumab for Advanced Melanoma. Available online: https://www.esmo.org/oncology-news/archive/fda-approves-pembrolizumab-for-advanced-melanoma (accessed on 14 April 2025).
- Matulonis, U.A.; Shapira-Frommer, R.; Santin, A.D.; Lisyanskaya, A.S.; Pignata, S.; Vergote, I.; Raspagliesi, F.; Sonke, G.S.; Birrer, M.; Provencher, D.M.; et al. Antitumor Activity and Safety of Pembrolizumab in Patients with Advanced Recurrent Ovarian Cancer: Results from the Phase II KEYNOTE-100 Study. Ann. Oncol. 2019, 30, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Connor, A.E.; Lyons, P.M.; Kilgallon, A.M.; Simpson, J.C.; Perry, A.S.; Lysaght, J. Examining the Evidence for Immune Checkpoint Therapy in High-Grade Serous Ovarian Cancer. Heliyon 2024, 10, e38888. [Google Scholar] [CrossRef]
- Zamarin, D.; Burger, R.A.; Sill, M.W.; Powell, D.J.; Lankes, H.A.; Feldman, M.D.; Zivanovic, O.; Gunderson, C.; Ko, E.; Mathews, C.; et al. Randomized Phase II Trial of Nivolumab Versus Nivolumab and Ipilimumab for Recurrent or Persistent Ovarian Cancer: An NRG Oncology Study. J. Clin. Oncol. 2020, 38, 1814–1823. [Google Scholar] [CrossRef]
- Zhao, L.; Zhai, Y.; Niu, G. Research Progress of Immune Checkpoint Inhibitors in Ovarian Cancer. Explor. Immunol. 2024, 4, 853–870. [Google Scholar] [CrossRef]
- Drew, Y.; Kim, J.-W.; Penson, R.T.; O’Malley, D.M.; Parkinson, C.; Roxburgh, P.; Plummer, R.; Im, S.-A.; Imbimbo, M.; Ferguson, M.; et al. Olaparib plus Durvalumab, with or without Bevacizumab, as Treatment in PARP Inhibitor-Naïve Platinum-Sensitive Relapsed Ovarian Cancer: A Phase II Multi-Cohort Study. Clin. Cancer Res. 2024, 30, 50–62. [Google Scholar] [CrossRef]
- Schoutrop, E.; El-Serafi, I.; Poiret, T.; Zhao, Y.; Gultekin, O.; He, R.; Moyano-Galceran, L.; Carlson, J.W.; Lehti, K.; Hassan, M.; et al. Mesothelin-Specific CAR T Cells Target Ovarian Cancer. Cancer Res. 2021, 81, 3022–3035. [Google Scholar] [CrossRef]
- Reiss, K.; Yuan, Y.; Ueno, N.; Johnson, M.; Dees, E.C.; Angelos, M.; Chao, J.; Gill, S.; Shestova, O.; Serody, J.; et al. 634 A Phase 1, First-in-Human (FIH) Clinical Trial of the Anti-HER2 CAR Macrophage CT-0508 in Participants with HER2 Overexpressing Solid Tumors. J. Immunother. Cancer 2022, 10, 634. [Google Scholar] [CrossRef]
- Hong, H.; He, Y.; Li, Y.; Shen, Y.; Qu, Y. Clinical Trial Landscape for TIL Therapy: Emerging Insights and Future Directions in Oncology. J. Transl. Med. 2024, 22, 1008. [Google Scholar] [CrossRef]
- Park, J.; Cho, H.W.; Lim, M.C.; Choi, C.H.; Lee, J.-Y. Oregovomab in Combination with Non-Platinum Chemotherapy for the Treatment of PARP Inhibitor– and Platinum-Resistant Ovarian Cancer: A Two-Cohort, Single-Arm Phase 2 Study (OPERA/KGOG3065/APGOT-OV6). J. Clin. Oncol. 2025, 43, 5587. [Google Scholar] [CrossRef]
- Lu, L.; Ma, W.; Johnson, C.H.; Khan, S.A.; Irwin, M.L.; Pusztai, L. In Silico Designed mRNA Vaccines Targeting CA-125 Neoantigen in Breast and Ovarian Cancer. Vaccine 2023, 41, 2073–2083. [Google Scholar] [CrossRef] [PubMed]
- Cutri-French, C.; Nasioudis, D.; George, E.; Tanyi, J.L. CAR-T Cell Therapy in Ovarian Cancer: Where Are We Now? Diagnostics 2024, 14, 819. [Google Scholar] [CrossRef]
- Anixa Biosciences Inc Anixa Biosciences and Moffitt Cancer Center Complete Dosing in Third Cohort in Ovarian Cancer CAR-T Clinical Trial. Available online: https://www.prnewswire.com/news-releases/anixa-biosciences-and-moffitt-cancer-center-complete-dosing-in-third-cohort-in-ovarian-cancer-car-t-clinical-trial-302385536.html (accessed on 14 April 2025).
- Anixa Biosciences Inc Anixa Biosciences Commences Treatment of Fifth Patient in Ovarian Cancer CAR-T Clinical Trial. Available online: https://www.prnewswire.com/news-releases/anixa-biosciences-commences-treatment-of-fifth-patient-in-ovarian-cancer-car-t-clinical-trial-302150758.html (accessed on 14 April 2025).
- Guo, J.; Zeng, X.; Zhu, Y.; Yang, D.; Zhao, X. Mesothelin-Based CAR-T Cells Exhibit Potent Antitumor Activity against Ovarian Cancer. J. Transl. Med. 2024, 22, 367. [Google Scholar] [CrossRef] [PubMed]
- CAR T Cell Therapy Trial Targets Advanced Ovarian Cancer|City of Hope. Available online: https://www.cityofhope.org/car-t-cell-therapy-trial-targets-advanced-ovarian-cancer (accessed on 14 April 2025).
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T Cells Enhance Antitumor Efficacy and Overcome the Tumor Microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef]
- BioNTech to Present Clinical Data Updates Across mRNA and Immunomodulatory Oncology Portfolio at ESMO Congress 2024|BioNTech. Available online: https://investors.biontech.de/news-releases/news-release-details/biontech-present-clinical-data-updates-across-mrna-and/ (accessed on 14 April 2025).
- Gitto, S.B.; Ihewulezi, C.J.N.; Powell, D.J. Adoptive T Cell Therapy for Ovarian Cancer. Gynecol. Oncol. 2024, 186, 77–84. [Google Scholar] [CrossRef]
- FDA Grants Novel TIL Therapy Breakthrough Therapy Designation for Advanced Cervical Cancer. Available online: https://www.targetedonc.com/view/fda-grants-novel-til-therapy-breakthrough-therapy-designation-for-advanced-cervical-cancer (accessed on 14 April 2025).
- Iovance Biotherapeutics, Inc. A Phase 2, Multicenter Study to Evaluate the Efficacy and Safety Using Autologous Tumor Infiltrating Lymphocytes (LN-145) in Patients with Recurrent, Metastatic or Persistent Cervical Carcinoma; Iovance Biotherapeutics, Inc.: San Carlos, CA, USA, 2024. [Google Scholar]
- Gang, M.; Wong, P.; Berrien-Elliott, M.M.; Fehniger, T.A. Memory-like Natural Killer Cells for Cancer Immunotherapy. Semin. Hematol. 2020, 57, 185–193. [Google Scholar] [CrossRef]
- Terrén, I.; Orrantia, A.; Astarloa-Pando, G.; Amarilla-Irusta, A.; Zenarruzabeitia, O.; Borrego, F. Cytokine-Induced Memory-Like NK Cells: From the Basics to Clinical Applications. Front. Immunol. 2022, 13, 884648. [Google Scholar] [CrossRef]
- A Phase 1b Study of Cytokine-Induced Memory like (CIML) Natural Killer (NK) Cell Therapy in Recurrent Ovarian Cancer|Dana-Farber Cancer Institute. Available online: https://www.dana-farber.org/clinical-trials/23-493 (accessed on 14 April 2025).
- Pan, S.; Wang, F.; Jiang, J.; Lin, Z.; Chen, Z.; Cao, T.; Yang, L. Chimeric Antigen Receptor-Natural Killer Cells: A New Breakthrough in the Treatment of Solid Tumours. Clin. Oncol. 2023, 35, 153–162. [Google Scholar] [CrossRef]
- Phase I/II Study of TROP2 CAR Engineered IL15-Transduced Cord Blood-Derived NK Cells Delivered Intraperitoneally for the Management of Platinum Resistant Ovarian Cancer, Mesonephric-like Adenocarcinoma, and Pancreatic Cancer. ASCO. Available online: https://www.asco.org/abstracts-presentations/ABSTRACT442670 (accessed on 14 April 2025).
- Hoogstad-van Evert, J.S.; Bekkers, R.; Ottevanger, N.; Jansen, J.H.; Massuger, L.; Dolstra, H. Harnessing Natural Killer Cells for the Treatment of Ovarian Cancer. Gynecol. Oncol. 2020, 157, 810–816. [Google Scholar] [CrossRef]
- Shi, Y.; Li, X.; Dong, Y.; Yuan, H.; Wang, Y.; Yang, R. Exploring the Potential of CAR-Macrophage Therapy. Life Sci. 2025, 361, 123300. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Wang, H.; Zuo, D.; Xu, J.; Feng, Y.; Xue, D.; Zhang, L.; Lin, L.; Zhang, J. A Clinical Study of Autologous Chimeric Antigen Receptor Macrophage Targeting Mesothelin Shows Safety in Ovarian Cancer Therapy. J. Hematol. Oncol. 2024, 17, 116. [Google Scholar] [CrossRef] [PubMed]
- Maute, R.; Xu, J.; Weissman, I.L. CD47–SIRPα-Targeted Therapeutics: Status and Prospects. Immuno-Oncol. Technol. 2022, 13, 100070. [Google Scholar] [CrossRef] [PubMed]
- Osborn, G.; Stavraka, C.; Adams, R.; Sayasneh, A.; Ghosh, S.; Montes, A.; Lacy, K.E.; Kristeleit, R.; Spicer, J.; Josephs, D.H.; et al. Macrophages in Ovarian Cancer and Their Interactions with Monoclonal Antibody Therapies. Clin. Exp. Immunol. 2021, 209, 4–21. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yan, L.; Wang, Q. Efficacy of PD-1/PD-L1 Inhibitors in Ovarian Cancer: A Single-Arm Meta-Analysis. J. Ovarian Res. 2021, 14, 112. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Fu, Y.; Cui, Z.; Abidin, Z.; Yuan, J.; Zhang, X.; Li, R.; Zhao, C. Relatlimab: A Novel Drug Targeting Immune Checkpoint LAG-3 in Melanoma Therapy. Front. Pharmacol. 2024, 14, 1349081. [Google Scholar] [CrossRef]
- Luke, J.J.; Patel, M.R.; Blumenschein, G.R.; Hamilton, E.; Chmielowski, B.; Ulahannan, S.V.; Connolly, R.M.; Santa-Maria, C.A.; Wang, J.; Bahadur, S.W.; et al. The PD-1- and LAG-3-Targeting Bispecific Molecule Tebotelimab in Solid Tumors and Hematologic Cancers: A Phase 1 Trial. Nat. Med. 2023, 29, 2814–2824. [Google Scholar] [CrossRef]
- Weimer, P.; Wellbrock, J.; Sturmheit, T.; Oliveira-Ferrer, L.; Ding, Y.; Menzel, S.; Witt, M.; Hell, L.; Schmalfeldt, B.; Bokemeyer, C.; et al. Tissue-Specific Expression of TIGIT, PD-1, TIM-3, and CD39 by Γδ T Cells in Ovarian Cancer. Cells 2022, 11, 964. [Google Scholar] [CrossRef]
- Dai, T.; Sun, H.; Liban, T.; Vicente-Suarez, I.; Zhang, B.; Song, Y.; Jiang, Z.; Yu, J.; Sheng, J.; Lv, B. A Novel Anti-LAG-3/TIGIT Bispecific Antibody Exhibits Potent Anti-Tumor Efficacy in Mouse Models as Monotherapy or in Combination with PD-1 Antibody. Sci. Rep. 2024, 14, 10661. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in Cancer Treatment: A Review of 15 Years of Clinical Experience and Future Outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.-L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A Modulates Expression of Inhibitory Checkpoints on CD8+ T Cells in Tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Benmebarek, M.-R.; Oguz, C.; Ruf, B.; Myojin, Y.; Huang, P.; Ma, C.; Bauer, K.; Paya, M.V.; Seifert, M.; Lack, J.; et al. 472 Anti-VEGF Treatment Amplifies Immune Checkpoint Inhibitor Induced Immune Responses by Targeting B and Regulatory T Cells. J. Immunother. Cancer 2023, 11, 472. [Google Scholar] [CrossRef]
- Kurtz, J.-E.; Pujade-Lauraine, E.; Oaknin, A.; Belin, L.; Leitner, K.; Cibula, D.; Denys, H.; Rosengarten, O.; Rodrigues, M.; de Gregorio, N.; et al. Atezolizumab Combined with Bevacizumab and Platinum-Based Therapy for Platinum-Sensitive Ovarian Cancer: Placebo-Controlled Randomized Phase III ATALANTE/ENGOT-Ov29 Trial. J. Clin. Oncol. 2023, 41, 4768–4778. [Google Scholar] [CrossRef] [PubMed]
- Gotlieb, W.H.; Tzur, Y. Atezolizumab Does Not Improve Progression-Free Survival in Patients with Recurrent Platinum-Sensitive Ovarian Cancer. Gynecol. Pelvic Med. 2024, 7, 8. [Google Scholar] [CrossRef]
- Zsiros, E.; Lynam, S.; Attwood, K.M.; Wang, C.; Chilakapati, S.; Gomez, E.C.; Liu, S.; Akers, S.; Lele, S.; Frederick, P.J.; et al. Efficacy and Safety of Pembrolizumab in Combination with Bevacizumab and Oral Metronomic Cyclophosphamide in the Treatment of Recurrent Ovarian Cancer. JAMA Oncol. 2021, 7, 78–85. [Google Scholar] [CrossRef]
- Merck Sharp & Dohme LLC. A Randomized Phase 3, Double-Blind Study of Chemotherapy with or Without Pembrolizumab Followed by Maintenance with Olaparib or Placebo for the First-Line Treatment of BRCA Non-Mutated Advanced Epithelial Ovarian Cancer (EOC) (KEYLYNK-001; ENGOT-Ov43; GOG-3036); 2024. Available online: https://clinicaltrials.gov/study/NCT03740165 (accessed on 26 June 2025).
- Merck Announces Phase 3 KEYNOTE-B96 Trial Met Primary Endpoint of Progression-Free Survival (PFS) in Patients with Platinum-Resistant Recurrent Ovarian Cancer Whose Tumors Expressed PD-L1 and in All Comers. Available online: https://www.merck.com/news/merck-announces-phase-3-keynote-b96-trial-met-primary-endpoint-of-progression-free-survival-pfs-in-patients-with-platinum-resistant-recurrent-ovarian-cancer-whose-tumors-expressed-pd-l1-and-in-all-c/ (accessed on 26 June 2025).
- Merck Sharp & Dohme LLC A Phase 3, Randomized, Double-Blind Study of Pembrolizumab Versus Placebo in Combination with Paclitaxel with or Without Bevacizumab for the Treatment of Platinum-Resistant Recurrent Ovarian Cancer (KEYNOTE-B96; ENGOT-Ov65). 2025. Available online: https://clinicaltrials.gov/study/NCT05116189 (accessed on 26 June 2025).
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef]
- Randall, L.M.; O’Malley, D.M.; Monk, B.J.; Coleman, R.L.; Gaillard, S.; Adams, S.; Duska, L.R.; Dalton, H.; Holloway, R.W.; Huang, M.; et al. Niraparib and Dostarlimab for the Treatment of Recurrent Platinum-Resistant Ovarian Cancer: Results of a Phase II Study (MOONSTONE/GOG-3032). Gynecol. Oncol. 2023, 178, 161–169. [Google Scholar] [CrossRef]
- Harter, P.; Trillsch, F.; Okamoto, A.; Reuss, A.; Kim, J.-W.; Rubio-Pérez, M.J.; Vardar, M.A.; Scambia, G.; Tredan, O.; Nyvang, G.-B.; et al. Durvalumab with Paclitaxel/Carboplatin (PC) and Bevacizumab (Bev), Followed by Maintenance Durvalumab, Bev, and Olaparib in Patients (Pts) with Newly Diagnosed Advanced Ovarian Cancer (AOC) without a Tumor BRCA1/2 Mutation (Non-tBRCAm): Results from the Randomized, Placebo (Pbo)-Controlled Phase III DUO-O Trial. J. Clin. Oncol. 2023, 41, LBA5506. [Google Scholar] [CrossRef]
- Westin, S.N.; Moore, K.; Chon, H.S.; Lee, J.-Y.; Thomes Pepin, J.; Sundborg, M.; Shai, A.; de la Garza, J.; Nishio, S.; Gold, M.A.; et al. Durvalumab Plus Carboplatin/Paclitaxel Followed by Maintenance Durvalumab With or Without Olaparib as First-Line Treatment for Advanced Endometrial Cancer: The Phase III DUO-E Trial. J. Clin. Oncol. 2024, 42, 283–299. [Google Scholar] [CrossRef]
- Monk, B.J.; Coleman, R.L.; Fujiwara, K.; Wilson, M.K.; Oza, A.M.; Oaknin, A.; O’Malley, D.M.; Lorusso, D.; Westin, S.N.; Safra, T.; et al. ATHENA (GOG-3020/ENGOT-Ov45): A Randomized, Phase III Trial to Evaluate Rucaparib as Monotherapy (ATHENA-MONO) and Rucaparib in Combination with Nivolumab (ATHENA-COMBO) as Maintenance Treatment Following Frontline Platinum-Based Chemotherapy in Ovarian Cancer. Int. J. Gynecol. Cancer 2021, 31, 1589–1594. [Google Scholar] [CrossRef] [PubMed]
- Phipps, M.; Falchook, G.S. B7 Homolog 4 (B7-H4)-Directed Agents in Oncology Clinical Trials: A Review. J. Immunother. Precis. Oncol. 2025, 8, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Fakih, M.; Schneider, C.; Chiorean, E.G.; Bendell, J.; Kristeleit, R.; Kurzrock, R.; Blagden, S.P.; Brana, I.; Goff, L.W.; et al. Phase I/II Sequencing Study of Azacitidine, Epacadostat, and Pembrolizumab in Advanced Solid Tumors. Br. J. Cancer 2023, 128, 2227–2235. [Google Scholar] [CrossRef]
- Clinical Results with Combination of Anti-CD27 Agonist Antibody, Varlilumab, with Anti-PD1 Antibody Nivolumab in Advanced Cancer Patients. ASCO. Available online: https://www.asco.org/abstracts-presentations/ABSTRACT185799?utm_source=chatgpt.com (accessed on 14 April 2025).
- Sallman, D.A.; Al Malki, M.M.; Asch, A.S.; Wang, E.S.; Jurcic, J.G.; Bradley, T.J.; Flinn, I.W.; Pollyea, D.A.; Kambhampati, S.; Tanaka, T.N.; et al. Magrolimab in Combination with Azacitidine in Patients with Higher-Risk Myelodysplastic Syndromes: Final Results of a Phase Ib Study. J. Clin. Oncol. 2023, 41, 2815–2826. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.; Parisi, C.; Barlesi, F. Anti-TIGIT Therapies for Solid Tumors: A Systematic Review. ESMO Open 2023, 8, 101184. [Google Scholar] [CrossRef]
- Clinical Trial: NCT04590326—My Cancer Genome. Available online: https://www.mycancergenome.org/content/clinical_trials/NCT04590326/ (accessed on 14 April 2025).
- BioNTech Presents Positive Phase 1/2 Data Update for CAR-T Cell Therapy Candidate BNT211 in Advanced Solid Tumors at ESMO Congress 2023|BioNTech. Available online: https://investors.biontech.de/news-releases/news-release-details/biontech-presents-positive-phase-12-data-update-car-t-cell/ (accessed on 15 April 2025).
- BioNTech|Technologies for Customized Treatment Approaches. Available online: https://www.biontech.com/int/en/home/pipeline-and-products/pipeline.html (accessed on 15 April 2025).
- The ModiFY Study. Available online: https://modi-1-study.digitrial.com/ (accessed on 15 April 2025).
- Oxford Researchers Secure Funding for World’s First Ovarian Cancer Prevention Vaccine|University of Oxford. Available online: https://www.ox.ac.uk/news/2024-10-04-oxford-researchers-secure-funding-worlds-first-ovarian-cancer-prevention-vaccine (accessed on 15 April 2025).
- Three-Year Phase 1 Follow-Up Data for mRNA-Based Individualized Immunotherapy Candidate Show Persistence of Immune Response and Delayed Tumor Recurrence in Some Patients with Resected Pancreatic Cancer|BioNTech. Available online: https://investors.biontech.de/news-releases/news-release-details/three-year-phase-1-follow-data-mrna-based-individualized/ (accessed on 15 April 2025).
- Martin, S.D.; Brown, S.D.; Wick, D.A.; Nielsen, J.S.; Kroeger, D.R.; Twumasi-Boateng, K.; Holt, R.A.; Nelson, B.H. Low Mutation Burden in Ovarian Cancer May Limit the Utility of Neoantigen-Targeted Vaccines. PLoS ONE 2016, 11, e0155189. [Google Scholar] [CrossRef]
- Cibula, D.; Rob, L.; Mallmann, P.; Knapp, P.; Klat, J.; Chovanec, J.; Minar, L.; Melichar, B.; Hein, A.; Kieszko, D.; et al. Dendritic Cell-Based Immunotherapy (DCVAC/OvCa) Combined with Second-Line Chemotherapy in Platinum-Sensitive Ovarian Cancer (SOV02): A Randomized, Open-Label, Phase 2 Trial. Gynecol. Oncol. 2021, 162, 652–660. [Google Scholar] [CrossRef]
- Tanyi, J.L.; Chiang, C.L.-L.; Chiffelle, J.; Thierry, A.-C.; Baumgartener, P.; Huber, F.; Goepfert, C.; Tarussio, D.; Tissot, S.; Torigian, D.A.; et al. Personalized Cancer Vaccine Strategy Elicits Polyfunctional T Cells and Demonstrates Clinical Benefits in Ovarian Cancer. NPJ Vaccines 2021, 6, 68. [Google Scholar] [CrossRef]
- Chu, C.S.; Boyer, J.; Schullery, D.S.; Gimotty, P.A.; Gamerman, V.; Bender, J.; Levine, B.L.; Coukos, G.; Rubin, S.C.; Morgan, M.A.; et al. Phase I/II Randomized Trial of Dendritic Cell Vaccination with or without Cyclophosphamide for Consolidation Therapy of Advanced Ovarian Cancer in First or Second Remission. Cancer Immunol. Immunother. CII 2011, 61, 629–641. [Google Scholar] [CrossRef]
- Manning-Geist, B.L.; Gnjatic, S.; Aghajanian, C.; Konner, J.; Kim, S.H.; Sarasohn, D.; Soldan, K.; Tew, W.P.; Sarlis, N.J.; Zamarin, D.; et al. Phase I Study of a Multivalent WT1 Peptide Vaccine (Galinpepimut-S) in Combination with Nivolumab in Patients with WT1-Expressing Ovarian Cancer in Second or Third Remission. Cancers 2023, 15, 1458. [Google Scholar] [CrossRef]
- Smithy, J.W.; Pianko, M.J.; Maher, C.; Postow, M.A.; Shoushtari, A.N.; Momtaz, P.; Chapman, P.B.; Wolchok, J.D.; Park, J.H.; Callahan, M.K. Checkpoint Blockade in Melanoma Patients with Underlying Chronic Lymphocytic Leukemia. J. Immunother. 2021, 44, 9. [Google Scholar] [CrossRef]
- Abagovomab—An Overview|ScienceDirect Topics. Available online: https://www.sciencedirect.com/topics/medicine-and-dentistry/abagovomab?utm_source=chatgpt.com (accessed on 15 April 2025).
- Pavlick, A.; Blazquez, A.B.; Meseck, M.; Lattanzi, M.; Ott, P.A.; Marron, T.U.; Holman, R.M.; Mandeli, J.; Salazar, A.; McClain, C.; et al. Combined Vaccination with NY-ESO-1 Protein, Poly-ICLC, and Montanide Improves Humoral and Cellular Immune Responses in High-Risk Melanoma Patients. Cancer Immunol. Res. 2020, 8, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Vreeland, T.J.; Litton, J.K.; Qiao, N.; Philips, A.V.; Alatrash, G.; Hale, D.F.; Jackson, D.O.; Peace, K.M.; Greene, J.M.; Berry, J.S.; et al. Phase Ib Trial of Folate Binding Protein (FBP)-Derived Peptide Vaccines, E39 and an Attenuated Version, E39’: An Analysis of Safety and Immune Response. Clin. Immunol. 2018, 192, 6–13. [Google Scholar] [CrossRef]
- 14MB098 Cancer Vaccine: Full-Length Variant Survivin Vaccine Potentiates Autologous Hematopoietic Stem Cell Transplantation in Multiple Myeloma. Available online: https://www.moffitt.org/research-science/academic-and-industry-partnerships/office-of-innovation/available-technologies/immunotherapies/14mb098-cancer-vaccine-full-length-variant-survivin-vaccine-potentiates-autologous-hematopoietic-stem-cell-transplantation-in-multiple-myeloma/ (accessed on 15 April 2025).
- Melssen, M.M.; Petroni, G.R.; Chianese-Bullock, K.A.; Wages, N.A.; Grosh, W.W.; Varhegyi, N.; Smolkin, M.E.; Smith, K.T.; Galeassi, N.V.; Deacon, D.H.; et al. A Multipeptide Vaccine plus Toll-like Receptor Agonists LPS or polyICLC in Combination with Incomplete Freund’s Adjuvant in Melanoma Patients. J. Immunother. Cancer 2019, 7, 163. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Jaffee, E.M. Regulatory T Cell Modulation Using Cyclophosphamide in Vaccine Approaches: A Current Perspective. Cancer Res. 2012, 72, 3439–3444. [Google Scholar] [CrossRef] [PubMed]
- Peter, R.M. Ovarian Cancer: BioNTech, Curelab, OvarianVax and More Bring Vaccines to the Clinic. Available online: https://www.labiotech.eu/in-depth/ovarian-cancer-biontech-curelab-ovarianvax-vaccines-clinic/ (accessed on 26 June 2025).
- Krasny, S.; Baranau, Y.; Polyakov, S.; Zharkova, E.; Streltsova, O.; Filimonava, A.; Siarheyeva, V.; Kazlouskaya, S.; Khorau, A.; Gabai, V.; et al. Clinical Efficacy of Plasmid Encoding P62/SQSTM1 (Elenagen) in Combination with Gemcitabine in Patients with Platinum-Resistant Ovarian Cancer: A Randomized Controlled Trial. Front. Oncol. 2024, 14, 1343023. [Google Scholar] [CrossRef]
- Elenagen, a Novel DNA Immunotherapy for the Deadliest Form of Ovarian Cancer, Delays Disease Progression—Ecancer. Available online: http://ecancer.org/en/news/24279-elenagen-a-novel-dna-immunotherapy-for-the-deadliest-form-of-ovarian-cancer-delays-disease-progression (accessed on 15 April 2025).
- Hung, S.-I.; Chu, M.-T.; Hou, M.-M.; Lee, Y.-S.; Yang, C.-K.; Chu, S.-Y.; Liu, F.-Y.; Hsu, H.-C.; Pao, S.-C.; Teng, Y.-C.; et al. Personalized Neoantigen-Based T Cell Therapy Triggers Cytotoxic Lymphocytes Expressing Polyclonal TCR against Metastatic Ovarian Cancer. Biomed. Pharmacother. 2023, 169, 115928. [Google Scholar] [CrossRef]
- Mangogna, A.; Munari, G.; Pepe, F.; Maffii, E.; Giampaolino, P.; Ricci, G.; Fassan, M.; Malapelle, U.; Biffi, S. Homologous Recombination Deficiency in Ovarian Cancer: From the Biological Rationale to Current Diagnostic Approaches. J. Pers. Med. 2023, 13, 284. [Google Scholar] [CrossRef]
- Sobhani, N.; Scaggiante, B.; Morris, R.; Chai, D.; Catalano, M.; Tardiel-Cyril, D.R.; Neeli, P.; Roviello, G.; Mondani, G.; Li, Y. Therapeutic Cancer Vaccines: From Biological Mechanisms and Engineering to Ongoing Clinical Trials. Cancer Treat. Rev. 2022, 109, 102429. [Google Scholar] [CrossRef]
- Biswas, N.; Chakrabarti, S.; Padul, V.; Jones, L.D.; Ashili, S. Designing Neoantigen Cancer Vaccines, Trials, and Outcomes. Front. Immunol. 2023, 14, 1105420. [Google Scholar] [CrossRef] [PubMed]
- Mat, A. ISA Pharmaceuticals Announces the Start of Clinical Trial Using Its Novel AMPLIVANT® Technology; ISA Ther. BV: Oegstgeest, The Netherlands, 2022. [Google Scholar]
- Oosting, L.T.; Franke, K.; Martin, M.V.; Kloosterman, W.P.; Jamieson, J.A.; Glenn, L.A.; de Jager, M.W.; van Zanten, J.; Allersma, D.P.; Gareb, B. Development of a Personalized Tumor Neoantigen Based Vaccine Formulation (FRAME-001) for Use in a Phase II Trial for the Treatment of Advanced Non-Small Cell Lung Cancer. Pharmaceutics 2022, 14, 1515. [Google Scholar] [CrossRef] [PubMed]
- Deniger, D.C.; Pasetto, A.; Robbins, P.F.; Gartner, J.J.; Prickett, T.D.; Paria, B.C.; Malekzadeh, P.; Jia, L.; Yossef, R.; Langhan, M.M.; et al. T-Cell Responses to TP53 “Hotspot” Mutations and Unique Neoantigens Expressed by Human Ovarian Cancers. Clin. Cancer Res. 2018, 24, 5562–5573. [Google Scholar] [CrossRef]
- Hao, Q.; Long, Y.; Yang, Y.; Deng, Y.; Ding, Z.; Yang, L.; Shu, Y.; Xu, H. Development and Clinical Applications of Therapeutic Cancer Vaccines with Individualized and Shared Neoantigens. Vaccines 2024, 12, 717. [Google Scholar] [CrossRef]
- Mohebtash, M.; Tsang, K.-Y.; Madan, R.A.; Huen, N.-Y.; Poole, D.J.; Jochems, C.; Jones, J.; Ferrara, T.; Heery, C.R.; Arlen, P.M.; et al. A Pilot Study of MUC-1/CEA/TRICOM Poxviral-Based Vaccine in Patients with Metastatic Breast and Ovarian Cancer. Clin. Cancer Res. 2011, 17, 7164–7173. [Google Scholar] [CrossRef] [PubMed]
- FDA Clears Clinical Study of Vaccine Therapy for Ovarian Cancer. Available online: https://www.targetedonc.com/view/fda-clears-clinical-study-of-vaccine-therapy-for-ovarian-cancer (accessed on 15 April 2025).
- Liang, S.C.; Moskalenko, M.; Van Roey, M.; Jooss, K. Depletion of Regulatory T Cells by Targeting Folate Receptor 4 Enhances the Potency of a GM-CSF-Secreting Tumor Cell Immunotherapy. Clin. Immunol. 2013, 148, 287–298. [Google Scholar] [CrossRef]
- Moore, K.N.; Bouberhan, S.; Hamilton, E.P.; Liu, J.F.; O’Cearbhaill, R.E.; O’Malley, D.M.; Papadamitriou, K.; Schröder, D.; Van Nieuwenhuysen, E.; Yoo, S.-Y.; et al. First-in-Human Phase 1/2 Study of Ubamatamab, a MUC16xCD3 Bispecific Antibody, Administered Alone or in Combination with Cemiplimab in Patients with Recurrent Ovarian Cancer. J. Clin. Oncol. 2023, 41, TPS5624. [Google Scholar] [CrossRef]
- MacroGenics Announces Partial Clinical Hold on MGD009 Phase 1 Studies|MacroGenics, Inc. Available online: https://ir.macrogenics.com/news-releases/news-release-details/macrogenics-announces-partial-clinical-hold-mgd009-phase-1 (accessed on 15 April 2025).
- Trispecifics Improve Immune-Cell Engagement. Cancer Discov. 2022, 12, 1404. [CrossRef]
- Holloway, R.W.; Mendivil, A.A.; Kendrick, J.E.; Abaid, L.N.; Brown, J.V.; LeBlanc, J.; McKenzie, N.D.; Mori, K.M.; Ahmad, S. Clinical Activity of Olvimulogene Nanivacirepvec–Primed Immunochemotherapy in Heavily Pretreated Patients With Platinum-Resistant or Platinum-Refractory Ovarian Cancer. JAMA Oncol. 2023, 9, 903–908. [Google Scholar] [CrossRef]
- Holloway, R.W.; Thaker, P.; Mendivil, A.A.; Ahmad, S.; Al-Niaimi, A.N.; Barter, J.; Beck, T.; Chambers, S.K.; Coleman, R.L.; Crafton, S.M.; et al. A Phase III, Multicenter, Randomized Study of Olvimulogene Nanivacirepvec Followed by Platinum-Doublet Chemotherapy and Bevacizumab Compared with Platinum-Doublet Chemotherapy and Bevacizumab in Women with Platinum-Resistant/Refractory Ovarian Cancer. Int. J. Gynecol. Cancer 2023, 33, 1458–1463. [Google Scholar] [CrossRef]
- Cohn, D.; Sill, M.; Walker, J.; O’Malley, D.; Nagel, C.; Rutledge, T.; Bradley, W.; Richardson, D.; Moxley, K.; Aghajanian, C. Randomized Phase IIB Evaluation of Weekly Paclitaxel versus Weekly Paclitaxel with Oncolytic Reovirus (Reolysin®) in Recurrent Ovarian, Tubal, or Peritoneal Cancer: An NRG Oncology/Gynecologic Oncology Group Study. Gynecol. Oncol. 2017, 146, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Odunsi, K.; Slomovitz, B.; Hubbard-Lucey, V.M.; McCabe, D.; Shohara, L.; Schwarzenberger, P.; Ricciardi, T.; Macri, M.; Ryan, A.; et al. Abstract A022: Phase 1/2 Study to Evaluate Systemic Durvalumab (Durva) + Intraperitoneal ONCOS-102 in Patients with Peritoneal Disease Who Have Epithelial Ovarian (OC) or Metastatic Colorectal Cancer (CRC). Cancer Immunol. Res. 2019, 7, A022. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Dummer, R.; Gyorki, D.E.; Berger, A.C.; Conry, R.M.; Demidov, L.V.; Chan, E.; Treichel, S.; Faries, M.B.; Ross, M.I. Interim Analysis of a Randomized, Open-Label Phase 2 Study of Talimogene Laherparepvec (T-VEC) Neoadjuvant Treatment (Neotx) plus Surgery (Surgx) vs Surgx for Resectable Stage IIIB-IVM1a Melanoma (MEL). J. Clin. Oncol. 2018, 36, 9508. [Google Scholar] [CrossRef]
- Study of ONCR-177 Alone and in Combination with PD-1 Blockade in Adult Subjects with Advanced and/or Refractory Cutaneous, Subcutaneous or Metastatic Nodal Solid Tumors or with Liver Metastases of Solid Tumors—NCI. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/search/v?id=NCI-2020-02978 (accessed on 15 April 2025).
- Vaishampayan, U.N.; Muzaffar, J.; Winer, I.; Rosen, S.D.; Hoimes, C.J.; Chauhan, A.; Spreafico, A.; Lewis, K.D.; Bruno, D.S.; Dumas, O.; et al. Nemvaleukin Alfa, a Modified Interleukin-2 Cytokine, as Monotherapy and with Pembrolizumab in Patients with Advanced Solid Tumors (ARTISTRY-1). J. Immunother. Cancer 2024, 12, e010143. [Google Scholar] [CrossRef]
- Felices, M.; Chu, S.; Kodal, B.; Bendzick, L.; Ryan, C.; Lenvik, A.J.; Boylan, K.L.M.; Wong, H.C.; Skubitz, A.P.N.; Miller, J.S.; et al. IL-15 Super-Agonist (ALT-803) Enhances Natural Killer (NK) Cell Function against Ovarian Cancer. Gynecol. Oncol. 2017, 145, 453–461. [Google Scholar] [CrossRef]
- Indraccolo, S.; Tisato, V.; Tosello, V.; Habeler, W.; Esposito, G.; Moserle, L.; Stievano, L.; Persano, L.; Chieco-Bianchi, L.; Amadori, A. Interferon-Alpha Gene Therapy by Lentiviral Vectors Contrasts Ovarian Cancer Growth through Angiogenesis Inhibition. Hum. Gene Ther. 2005, 16, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Kment, J.; Newsted, D.; Young, S.; Vermeulen, M.C.; Laight, B.J.; Greer, P.A.; Lan, Y.; Craig, A.W. Blockade of TGF-β and PD-L1 by Bintrafusp Alfa Promotes Survival in Preclinical Ovarian Cancer Models by Promoting T Effector and NK Cell Responses. Br. J. Cancer 2024, 130, 2003–2015. [Google Scholar] [CrossRef]
- Flood, B.A.; Higgs, E.F.; Li, S.; Luke, J.J.; Gajewski, T.F. STING Pathway Agonism as a Cancer Therapeutic. Immunol. Rev. 2019, 290, 24–38. [Google Scholar] [CrossRef]
- Monk, B.J.; Brady, M.F.; Aghajanian, C.; Lankes, H.A.; Rizack, T.; Leach, J.; Fowler, J.M.; Higgins, R.; Hanjani, P.; Morgan, M.; et al. A Phase 2, Randomized, Double-Blind, Placebo- Controlled Study of Chemo-Immunotherapy Combination Using Motolimod with Pegylated Liposomal Doxorubicin in Recurrent or Persistent Ovarian Cancer: A Gynecologic Oncology Group Partners Study. Ann. Oncol. 2017, 28, 996–1004. [Google Scholar] [CrossRef]
- Willingham, S.B.; Ho, P.Y.; Hotson, A.; Hill, C.; Piccione, E.C.; Hsieh, J.; Liu, L.; Buggy, J.J.; McCaffery, I.; Miller, R.A. A2AR Antagonism with CPI-444 Induces Antitumor Responses and Augments Efficacy to Anti-PD-(L)1 and Anti-CTLA-4 in Preclinical Models. Cancer Immunol. Res. 2018, 6, 1136–1149. [Google Scholar] [CrossRef]
- Ishihara, M.; Kitano, S.; Kageyama, S.; Miyahara, Y.; Yamamoto, N.; Kato, H.; Mishima, H.; Hattori, H.; Funakoshi, T.; Kojima, T.; et al. NY-ESO-1-Specific Redirected T Cells with Endogenous TCR Knockdown Mediate Tumor Response and Cytokine Release Syndrome. J. Immunother. Cancer 2022, 10, e003811. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.P.; Vale, N.R.; Zacharakis, N.; Krishna, S.; Yu, Z.; Gasmi, B.; Gartner, J.J.; Sindiri, S.; Malekzadeh, P.; Deniger, D.C.; et al. Adoptive Cellular Therapy with Autologous Tumor-Infiltrating Lymphocytes and T-Cell Receptor–Engineered T Cells Targeting Common P53 Neoantigens in Human Solid Tumors. Cancer Immunol. Res. 2022, 10, 932–946. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.; Khan, B.; Payumo, F.; Chiorean, E.G.; Gahvari, Z.; Hecht, J.R.; Hurwitz, M.E.; Leidner, R.; Lenz, H.-J.; Pelster, M.; et al. AFNT-211: A Phase 1 Study of Autologous CD4+ and CD8+ T Cells Engineered to Express a High Avidity HLA-A*11:01-Restricted, KRAS G12V-Specific, Transgenic TCR, a CD8α/β Coreceptor, and a FAS41BB Switch Receptor in Patients with Advanced/Metastatic Solid Tumors. J. Clin. Oncol. 2024, 42, TPS8650. [Google Scholar] [CrossRef]
- Rodriguez, G.M.; Yakubovich, E.; Vanderhyden, B.C. Unveiling the Immunogenicity of Ovarian Tumors as the Crucial Catalyst for Therapeutic Success. Cancers 2023, 15, 5694. [Google Scholar] [CrossRef]
- Wang, C.; Xiong, C.; Hsu, Y.-C.; Wang, X.; Chen, L. Human Leukocyte Antigen (HLA) and Cancer Immunotherapy: HLA-Dependent and -Independent Adoptive Immunotherapies. Ann. Blood 2020, 5, 14. [Google Scholar] [CrossRef]
- Roy, A.G.; Robinson, J.M.; Sharma, P.; Rodriguez-Garcia, A.; Poussin, M.A.; Nickerson-Nutter, C.; Powell, D.J. Folate Receptor Beta as a Direct and Indirect Target for Antibody-Based Cancer Immunotherapy. Int. J. Mol. Sci. 2021, 22, 5572. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Sweis, R.F.; Kasper, S.; Hamid, O.; Bhatia, S.; Dummer, R.; Stradella, A.; Long, G.V.; Spreafico, A.; Shimizu, T.; et al. Combination of the STING Agonist MIW815 (ADU-S100) and PD-1 Inhibitor Spartalizumab in Advanced/Metastatic Solid Tumors or Lymphomas: An Open-Label, Multicenter, Phase Ib Study. Clin. Cancer Res. 2023, 29, 110–121. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Schuetz, T.J.; Blumenstein, B.A.; Glode, L.M.; Bilhartz, D.L.; Wyand, M.; Manson, K.; Panicali, D.L.; Laus, R.; Schlom, J.; et al. Overall Survival Analysis of a Phase II Randomized Controlled Trial of a Poxviral-Based PSA-Targeted Immunotherapy in Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2010, 28, 1099–1105. [Google Scholar] [CrossRef]
- Lakhani, N.J.; Stewart, D.; Richardson, D.L.; Dockery, L.E.; Van Le, L.; Call, J.; Rangwala, F.; Wang, G.; Ma, B.; Metenou, S.; et al. First-in-Human Phase I Trial of the Bispecific CD47 Inhibitor and CD40 Agonist Fc-Fusion Protein, SL-172154 in Patients with Platinum-Resistant Ovarian Cancer. J. Immunother. Cancer 2025, 13, e010565. [Google Scholar] [CrossRef]
- Jamal, R.; Messaoudene, M.; de Figuieredo, M.; Routy, B. Future Indications and Clinical Management for Fecal Microbiota Transplantation (FMT) in Immuno-Oncology. Semin. Immunol. 2023, 67, 101754. [Google Scholar] [CrossRef]
- Pawłowska, A.; Rekowska, A.; Kuryło, W.; Pańczyszyn, A.; Kotarski, J.; Wertel, I. Current Understanding on Why Ovarian Cancer Is Resistant to Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2023, 24, 10859. [Google Scholar] [CrossRef] [PubMed]
- Bullock, K.; Richmond, A. Suppressing MDSC Recruitment to the Tumor Microenvironment by Antagonizing CXCR2 to Enhance the Efficacy of Immunotherapy. Cancers 2021, 13, 6293. [Google Scholar] [CrossRef] [PubMed]
- Thaker, P.H.; Brady, W.E.; Lankes, H.A.; Odunsi, K.; Bradley, W.H.; Moore, K.N.; Muller, C.Y.; Anwer, K.; Schilder, R.J.; Alvarez, R.D.; et al. A Phase I Trial of Intraperitoneal GEN-1, an IL-12 Plasmid Formulated with PEG-PEI-Cholesterol Lipopolymer, Administered with Pegylated Liposomal Doxorubicin in Patients with Recurrent or Persistent Epithelial Ovarian, Fallopian Tube or Primary Peritoneal Cancers: An NRG Oncology/Gynecologic Oncology Group Study. Gynecol. Oncol. 2017, 147, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.M.; Coleman, R.L.; Sood, A.K. Targeting the Tumor Microenvironment in Ovarian Cancer. Eur. J. Cancer 1990 2016, 56, 131–143. [Google Scholar] [CrossRef]
- Fields, E.C.; McGuire, W.P.; Lin, L.; Temkin, S.M. Radiation Treatment in Women with Ovarian Cancer: Past, Present, and Future. Front. Oncol. 2017, 7, 177. [Google Scholar] [CrossRef]
- Domínguez-Prieto, V.; Qian, S.; Villarejo-Campos, P.; Meliga, C.; González-Soares, S.; Guijo Castellano, I.; Jiménez-Galanes, S.; García-Arranz, M.; Guadalajara, H.; García-Olmo, D. Understanding CAR T Cell Therapy and Its Role in Ovarian Cancer and Peritoneal Carcinomatosis from Ovarian Cancer. Front. Oncol. 2023, 13, 1104547. [Google Scholar] [CrossRef]
- Gómez-Melero, S.; Hassouneh, F.; Vallejo-Bermúdez, I.M.; Agüera-Morales, E.; Solana, R.; Caballero-Villarraso, J. Tandem CAR-T Cell Therapy: Recent Advances and Current Challenges. Front. Immunol. 2025, 16, 1546172. [Google Scholar] [CrossRef]
- Hou, J.Y.; Chapman, J.S.; Kalashnikova, E.; Pierson, W.; Smith-McCune, K.; Pineda, G.; Vattakalam, R.M.; Ross, A.; Mills, M.; Suarez, C.J.; et al. Circulating Tumor DNA Monitoring for Early Recurrence Detection in Epithelial Ovarian Cancer. Gynecol. Oncol. 2022, 167, 334–341. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The Future of Immune Checkpoint Therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Chamberlain, C.A.; Bennett, E.P.; Kverneland, A.H.; Svane, I.M.; Donia, M.; Met, Ö. Highly Efficient PD-1-Targeted CRISPR-Cas9 for Tumor-Infiltrating Lymphocyte-Based Adoptive T Cell Therapy. Mol. Ther. Oncolytics 2022, 24, 417–428. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted Delivery of a PD-1-Blocking scFv by CAR-T Cells Enhances Anti-Tumor Efficacy in Vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Yuan, Y.; Huang, L.; Pu, C.; Ding, T.; Xiao, X.; Cao, Z.; Wu, T.; Ding, L.; Sun, H.; et al. Dominant-Negative PD1-Armored CART Cells Induce Remission in Refractory Diffuse Large B-Cell Lymphoma (DLBCL) Patients. J. Clin. Oncol. 2019, 37, e19028. [Google Scholar] [CrossRef]
- Sosnowska, A.; Czystowska-Kuzmicz, M.; Golab, J. Extracellular Vesicles Released by Ovarian Carcinoma Contain Arginase 1 That Mitigates Antitumor Immune Response. Oncoimmunology 2019, 8, e1655370. [Google Scholar] [CrossRef]
- FDA. Center for Drug Evaluation and Research FDA Grants Accelerated Approval to Pembrolizumab for First Tissue/Site Agnostic Indication; FDA: Silver Spring, MD, USA, 2024. [Google Scholar]
- Park, J.; Kim, J.C.; Lee, Y.J.; Kim, S.; Kim, S.W.; Shin, E.-C.; Lee, J.Y.; Park, S.-H. Unique Immune Characteristics and Differential Anti-PD-1-Mediated Reinvigoration Potential of CD8+ TILs Based on BRCA1/2 Mutation Status in Epithelial Ovarian Cancers. J. Immunother. Cancer 2024, 12, e009058. [Google Scholar] [CrossRef]
- O’Malley, D.M.; Krivak, T.C.; Kabil, N.; Munley, J.; Moore, K.N. PARP Inhibitors in Ovarian Cancer: A Review. Target. Oncol. 2023, 18, 471–503. [Google Scholar] [CrossRef]
- Xue, S.; Su, X.; Ke, L.; Huang, Y. CXCL9 Correlates with Antitumor Immunity and Is Predictive of a Favorable Prognosis in Uterine Corpus Endometrial Carcinoma. Front. Oncol. 2023, 13, 1077780. [Google Scholar] [CrossRef] [PubMed]
- Kasikova, L.; Rakova, J.; Hensler, M.; Lanickova, T.; Tomankova, J.; Pasulka, J.; Drozenova, J.; Mojzisova, K.; Fialova, A.; Vosahlikova, S.; et al. Tertiary Lymphoid Structures and B Cells Determine Clinically Relevant T Cell Phenotypes in Ovarian Cancer. Nat. Commun. 2024, 15, 2528. [Google Scholar] [CrossRef]
- Liu, S.; Matsuzaki, J.; Wei, L.; Tsuji, T.; Battaglia, S.; Hu, Q.; Cortes, E.; Wong, L.; Yan, L.; Long, M.; et al. Efficient Identification of Neoantigen-Specific T-Cell Responses in Advanced Human Ovarian Cancer. J. Immunother. Cancer 2019, 7, 156. [Google Scholar] [CrossRef]
- Ali, L.; Garrido-Castro, A.; Lenehan, P.; Bollenrucher, N.; Stump, C.; Dougan, M.; Goel, S.; Shapiro, G.; Tolaney, S.; Dougan, S. PD-1 Blockade and CDK4/6 Inhibition Augment Nonoverlapping Features of T Cell Activation in Cancer. J. Exp. Med. 2023, 220, e20220729. [Google Scholar] [CrossRef]
- Xie, T.; Huang, A.; Yan, H.; Ju, X.; Xiang, L.; Yuan, J. Artificial Intelligence: Illuminating the Depths of the Tumor Microenvironment. J. Transl. Med. 2024, 22, 799. [Google Scholar] [CrossRef]
- Cai, Y.; Chen, R.; Gao, S.; Li, W.; Liu, Y.; Su, G.; Song, M.; Jiang, M.; Jiang, C.; Zhang, X. Artificial Intelligence Applied in Neoantigen Identification Facilitates Personalized Cancer Immunotherapy. Front. Oncol. 2023, 12, 1054231. [Google Scholar] [CrossRef]
- Amaria, R.; Knisely, A.; Vining, D.; Kopetz, S.; Overman, M.J.; Javle, M.; Antonoff, M.B.; Tzeng, C.-W.D.; Wolff, R.A.; Pant, S.; et al. Efficacy and Safety of Autologous Tumor-Infiltrating Lymphocytes in Recurrent or Refractory Ovarian Cancer, Colorectal Cancer, and Pancreatic Ductal Adenocarcinoma. J. Immunother. Cancer 2024, 12, e006822. [Google Scholar] [CrossRef]
- Zhu, X.; Cai, H.; Zhao, L.; Ning, L.; Lang, J. CAR-T Cell Therapy in Ovarian Cancer: From the Bench to the Bedside. Oncotarget 2017, 8, 64607–64621. [Google Scholar] [CrossRef]
- Sánchez-Moreno, I.; Lasarte-Cia, A.; Martín-Otal, C.; Casares, N.; Navarro, F.; Gorraiz, M.; Sarrión, P.; Hervas-Stubbs, S.; Jordana, L.; Rodriguez-Madoz, J.R.; et al. Tethered IL15-IL15Rα Augments Antitumor Activity of CD19 CAR-T Cells but Displays Long-Term Toxicity in an Immunocompetent Lymphoma Mouse Model. J. Immunother. Cancer 2024, 12, e008572. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.-Y.; Kim, Y.-Y.; Yu, H.-S.; Lee, M.; Kim, S.; Lee, J. CRISPR/Cas9-Mediated Knockout of DGK Improves Antitumor Activities of Human T Cells. Cancer Res. 2018, 78, 4692–4703. [Google Scholar] [CrossRef] [PubMed]
- Implanted “Drug Factories” Deliver Immunotherapy to Tumors—NCI. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2022/implanted-drug-factories-il2-ovarian-cancer (accessed on 15 April 2025).
- Astašauskaitė, S.; Kupčinskaitė-Noreikienė, R.; Zaborienė, I.; Vaičiūnienė, R.; Vanagas, T.; Pranys, D.; Poškienė, L.; Juozaitytė, E. Multiorgan Toxicity from Dual Checkpoint Inhibitor Therapy, Resulting in a Complete Response—A Case Report. Medicina 2024, 60, 1129. [Google Scholar] [CrossRef] [PubMed]
- Kotch, C.; Barrett, D.; Teachey, D.T. Tocilizumab for the Treatment of Chimeric Antigen Receptor T Cell-Induced Cytokine Release Syndrome. Expert Rev. Clin. Immunol. 2019, 15, 813–822. [Google Scholar] [CrossRef]
- Yabuno, A.; Matsushita, H.; Hamano, T.; Tan, T.Z.; Shintani, D.; Fujieda, N.; Tan, D.S.P.; Huang, R.Y.-J.; Fujiwara, K.; Kakimi, K.; et al. Identification of Serum Cytokine Clusters Associated with Outcomes in Ovarian Clear Cell Carcinoma. Sci. Rep. 2020, 10, 18503. [Google Scholar] [CrossRef]
- Ball, K.; Dovedi, S.J.; Vajjah, P.; Phipps, A. Strategies for Clinical Dose Optimization of T Cell-Engaging Therapies in Oncology. MAbs 2023, 15, 2181016. [Google Scholar] [CrossRef]
- Hoare, J.; Campbell, N.; Carapuça, E. Oncolytic Virus Immunotherapies in Ovarian Cancer: Moving beyond Adenoviruses. Porto Biomed. J. 2018, 3, e7. [Google Scholar] [CrossRef]
- Gargett, T.; Brown, M.P. The Inducible Caspase-9 Suicide Gene System as a “Safety Switch” to Limit on-Target, off-Tumor Toxicities of Chimeric Antigen Receptor T Cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef] [PubMed]
- AbbVie and Turnstone Biologics Announce Global Collaboration on Viral Immunotherapies in Oncology. Available online: https://news.abbvie.com/2017-10-10-AbbVie-and-Turnstone-Biologics-Announce-Global-Collaboration-on-Viral-Immunotherapies-in-Oncology (accessed on 15 April 2025).
- Iovance Biotherapeutics Announces Clinical Data in Frontline Advanced Melanoma at ASCO 2024 Annual Meeting. Available online: https://www.biospace.com/iovance-biotherapeutics-announces-clinical-data-in-frontline-advanced-melanoma-at-asco-2024-annual-meeting (accessed on 15 April 2025).
- Bedard, M.; Salio, M.; Cerundolo, V. Harnessing the Power of Invariant Natural Killer T Cells in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1829. [Google Scholar] [CrossRef] [PubMed]


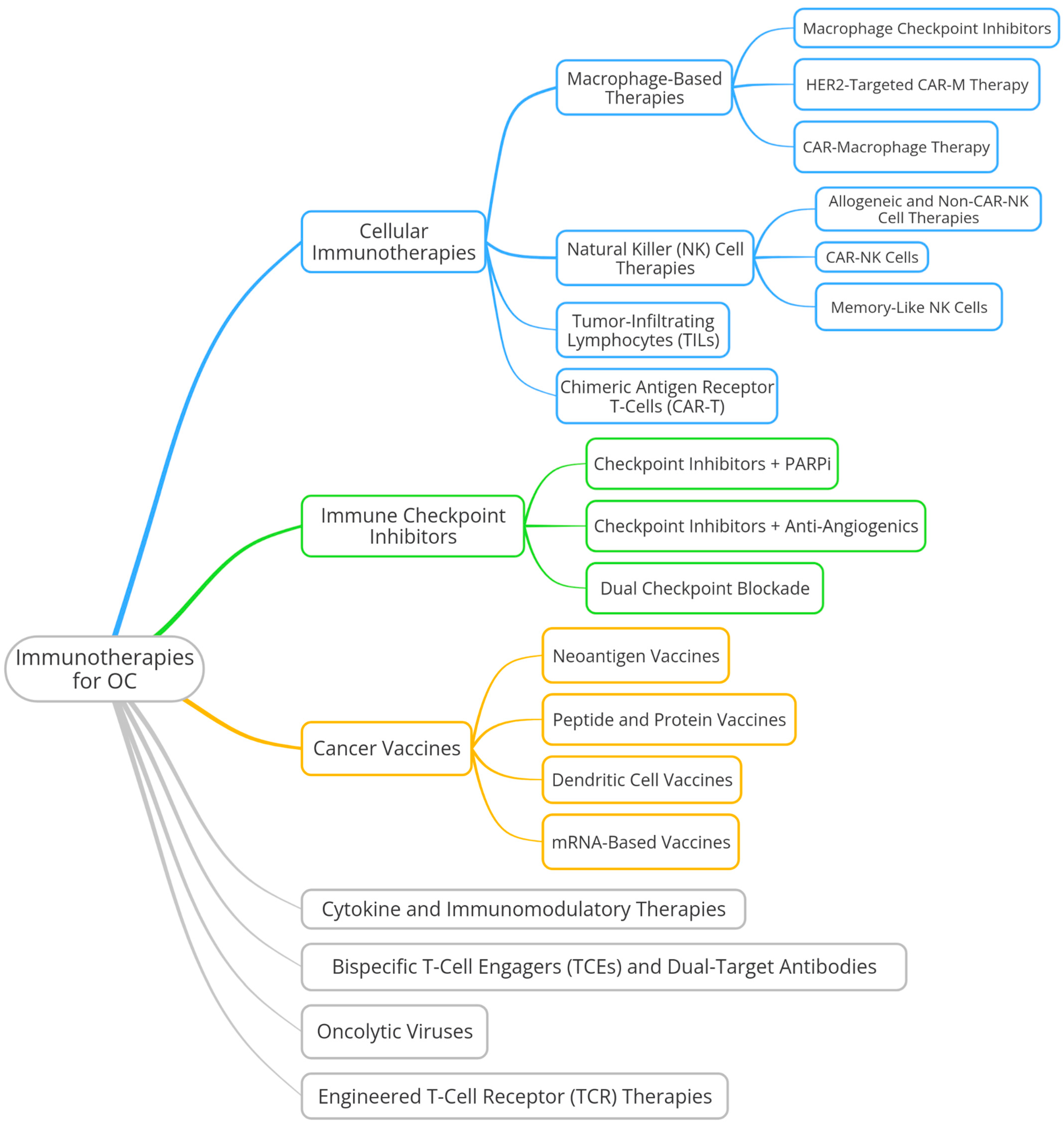
| Treatment Phase | Modality | Patient Subgroup | Examples |
|---|---|---|---|
| Surgical Options | Cytoreduction | PSROC (platinum-sensitive, resectable) 1 | Secondary cytoreduction; palliative surgery for symptom relief |
| Chemotherapy | Platinum-based | PSROC | Carboplatin + paclitaxel, gemcitabine, PLD |
| Non-platinum | PROC or platinum-refractory ROC | Weekly paclitaxel, PLD, gemcitabine, topotecan | |
| Targeted Therapy | Anti-VEGF 2 | General population (regardless of HRD) | Bevacizumab + chemotherapy for PSROC/PROC |
| FRα overexpression | PROC with folate receptor alpha (FRα+) | Mirvetuximab soravtansine (MIRV) | |
| Hormonal Therapy | Hormone receptor-positive | ER+/PR+ ROC 3 | Tamoxifen, letrozole, anastrozole |
| Maintenance Therapy | PARP inhibitors (PARPi) | BRCA1/2 mutated (germline or somatic) | Olaparib, niraparib, rucaparib |
| HRD-positive, BRCA wild-type | - | Niraparib, rucaparib (selected indications) | |
| HR-proficient (HRp) | No biomarker approval, limited benefit | Used with caution or in trial settings | |
| Treatment Strategy | Based on PFI | PSROC (PFI ≥ 6 months) | Platinum rechallenge |
| PROC (PFI < 6 months) | Non-platinum regimens; considered targeted/hormonal therapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brancewicz, J.; Padzińska-Pruszyńska, I.B.; Kubiak, M.; Kucharzewska, P. Immunotherapy for Platinum-Resistant Ovarian Cancer as a Glimmer of Hope. Cells 2025, 14, 995. https://doi.org/10.3390/cells14130995
Brancewicz J, Padzińska-Pruszyńska IB, Kubiak M, Kucharzewska P. Immunotherapy for Platinum-Resistant Ovarian Cancer as a Glimmer of Hope. Cells. 2025; 14(13):995. https://doi.org/10.3390/cells14130995
Chicago/Turabian StyleBrancewicz, Jan, Irena Barbara Padzińska-Pruszyńska, Małgorzata Kubiak, and Paulina Kucharzewska. 2025. "Immunotherapy for Platinum-Resistant Ovarian Cancer as a Glimmer of Hope" Cells 14, no. 13: 995. https://doi.org/10.3390/cells14130995
APA StyleBrancewicz, J., Padzińska-Pruszyńska, I. B., Kubiak, M., & Kucharzewska, P. (2025). Immunotherapy for Platinum-Resistant Ovarian Cancer as a Glimmer of Hope. Cells, 14(13), 995. https://doi.org/10.3390/cells14130995