

Epigenetic Changes Regulating Epithelial–Mesenchymal Plasticity in Human Trophoblast Differentiation

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
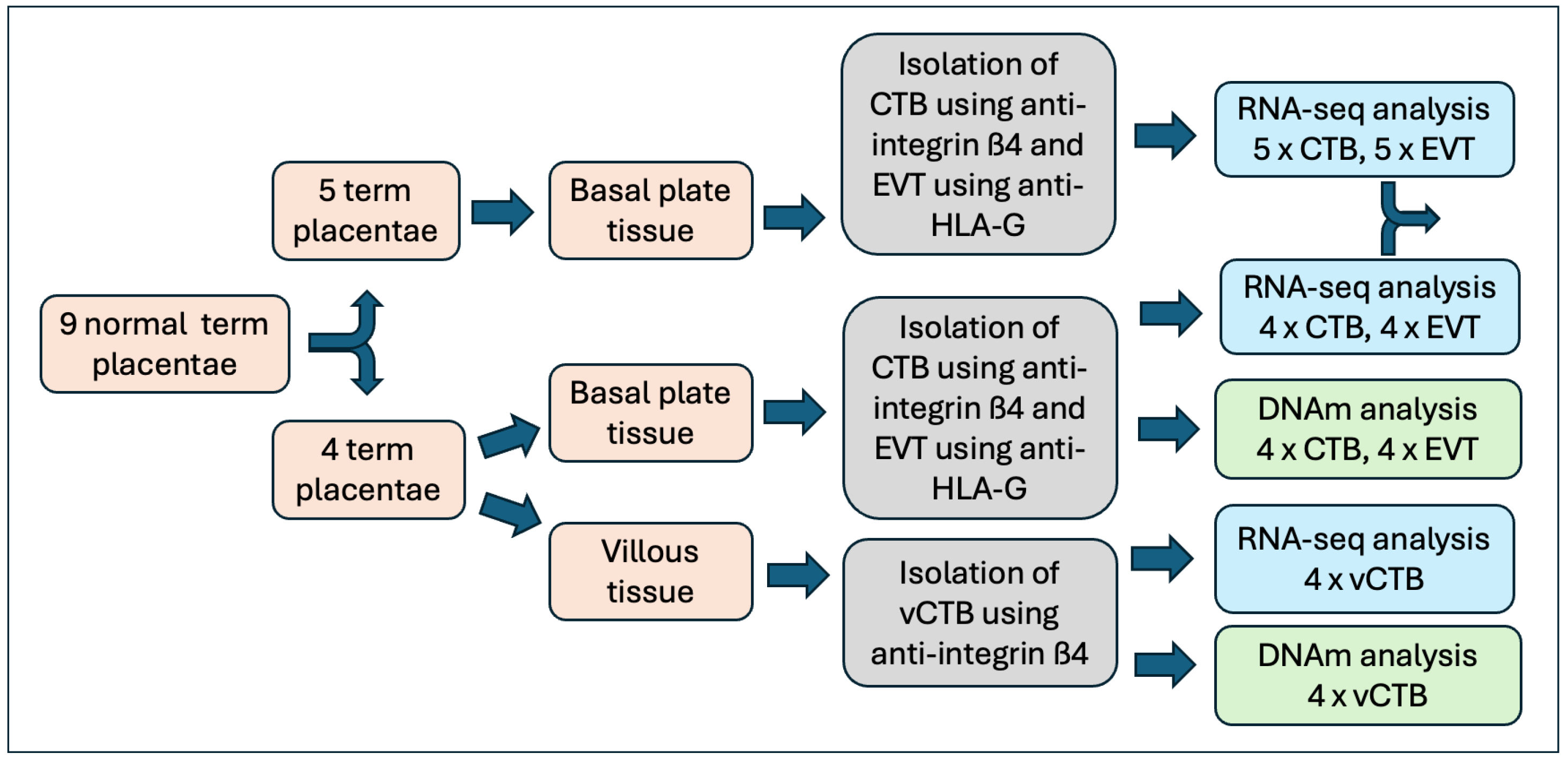
2.1. Placental Tissue and Cell Isolation
2.2. siRNA Treatment
2.3. Cell Migration and Invasion Assays
2.4. RNA Sequencing
2.5. RNA Sequencing Data Analysis
2.6. Differential Abundance Analysis
2.7. Functional Enrichment Analysis and Visualization
2.8. DNA Methylation
2.9. Expression/Methylation Correlation Analysis
2.10. PCR
2.11. Western Blotting
2.12. Statistical Analysis
3. Results
3.1. Sample Characterization
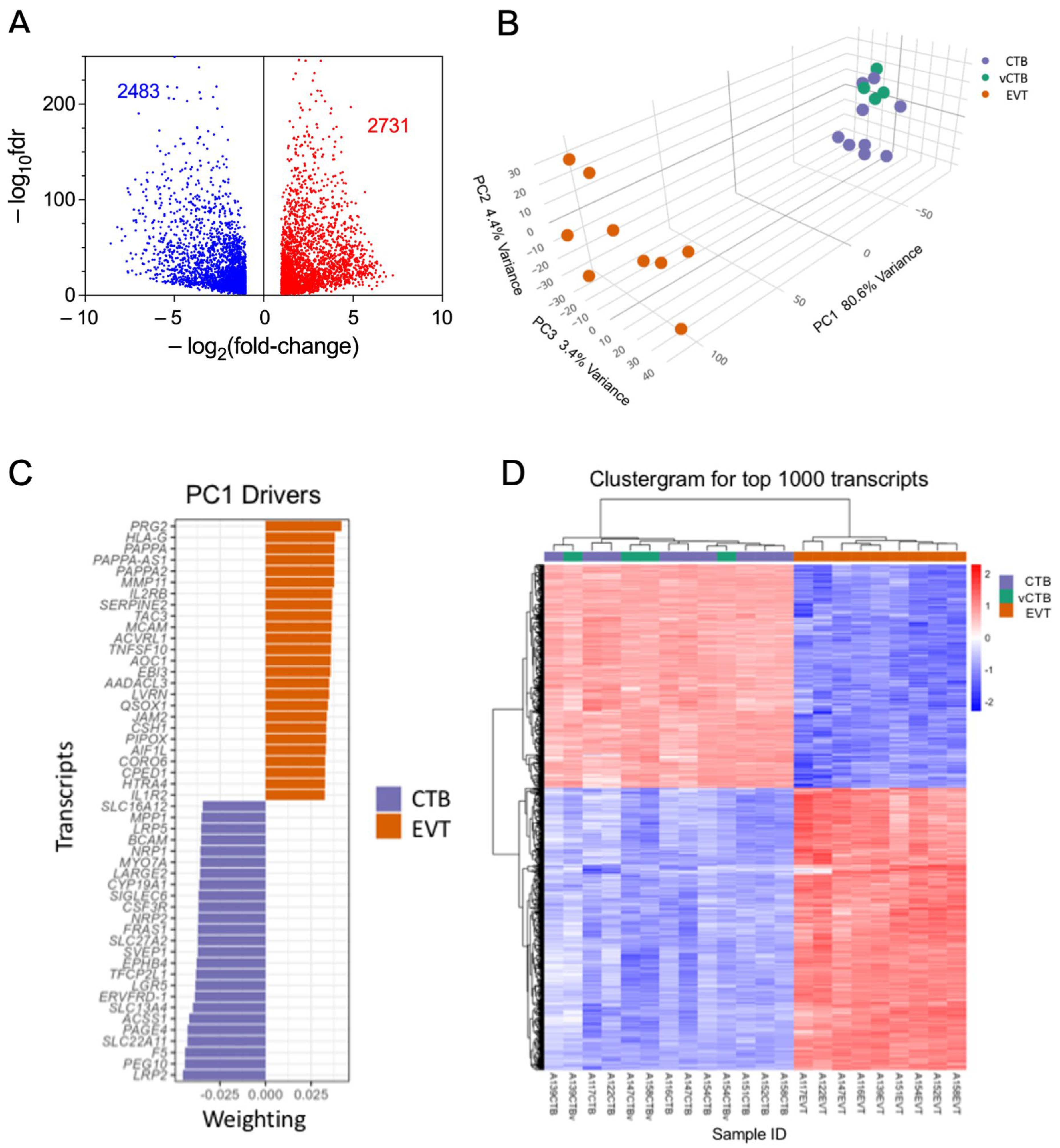
3.2. Comparison of CTB and EVT Transcriptomes Highlights Differential Expression of Genes Involved in Trophoblast Differentiation and EMP
3.3. EMP in Trophoblast Differentiation
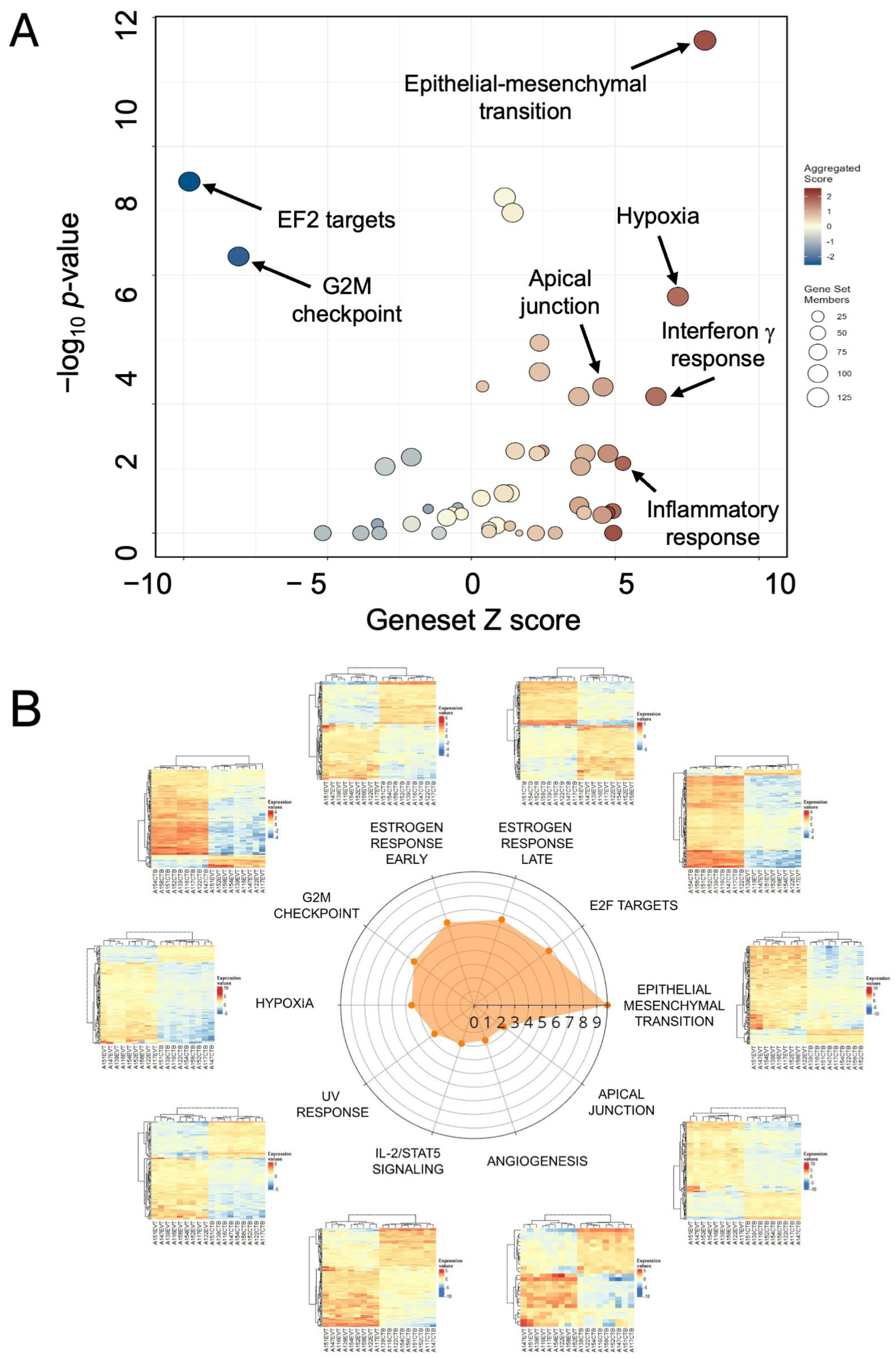
3.4. Functional Enrichment of the EMP Mechanisms in Differentiation
3.5. Differences in EMP Genes Between Basal Plate and Villous Tree CTB
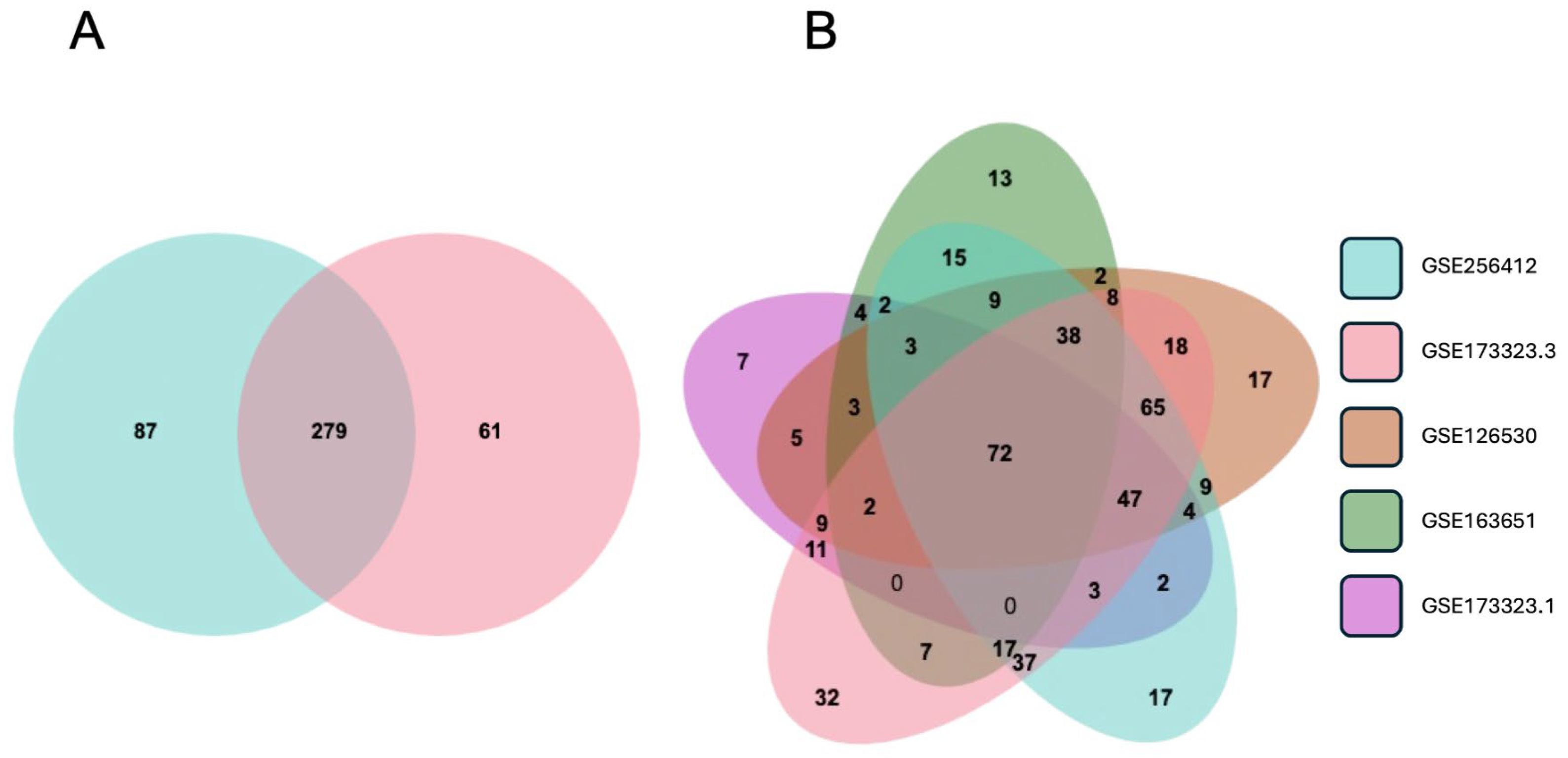
3.6. Validation of the EMP and Designation of a Trophoblast EMP Signature
3.7. Is EVT Gene Expression Reversibly Modified by In Vivo Conditions?
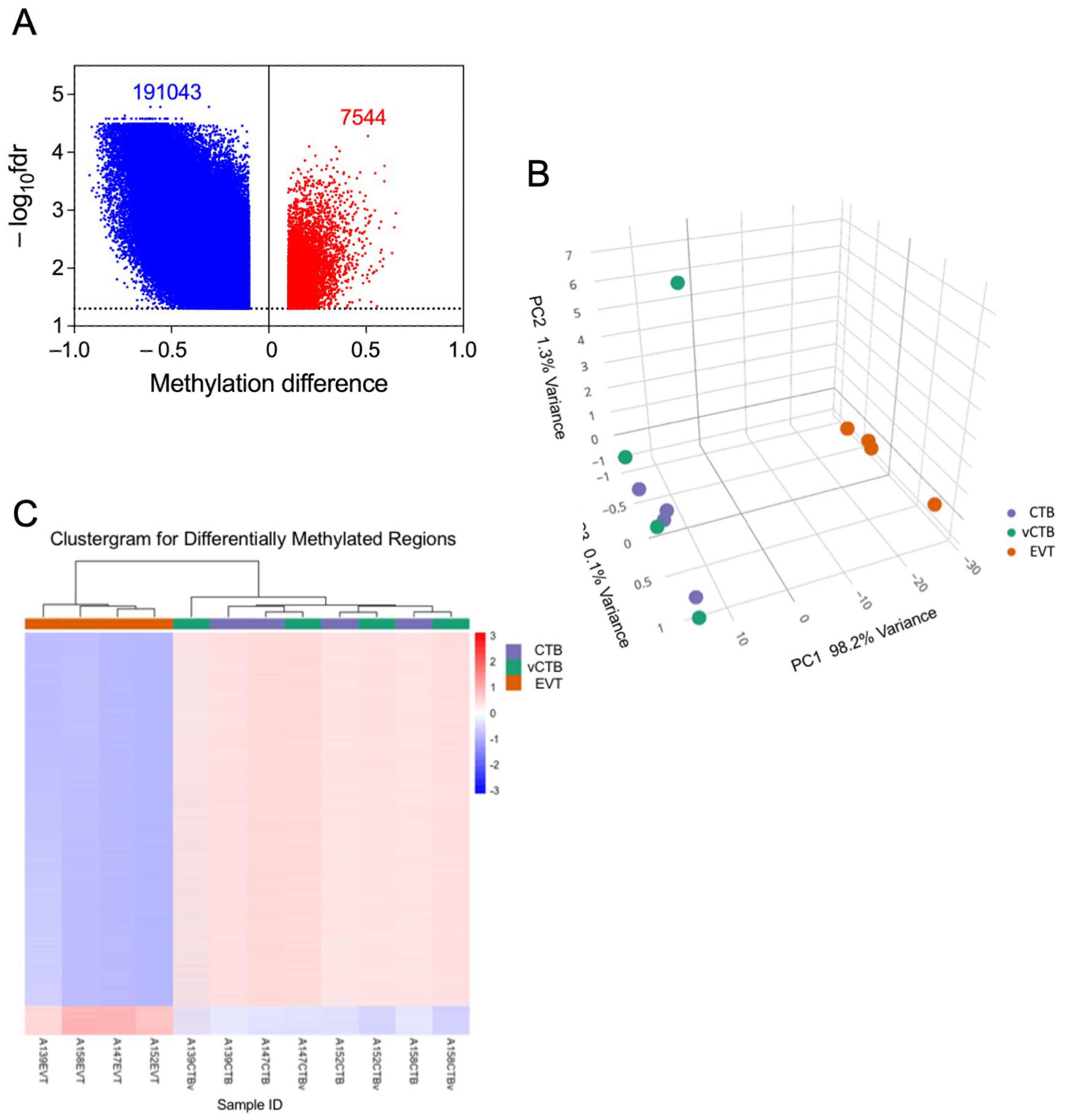
3.8. Comparison of CTB and EVT Methylomes Highlights Widespread Losses and Focal Gains of CpG Methylation in EVT
3.9. Correlation Between Gene Expression and Methylation
3.10. Genes Showing Gain of Methylation, Differential Expression, and Involvement in EMP
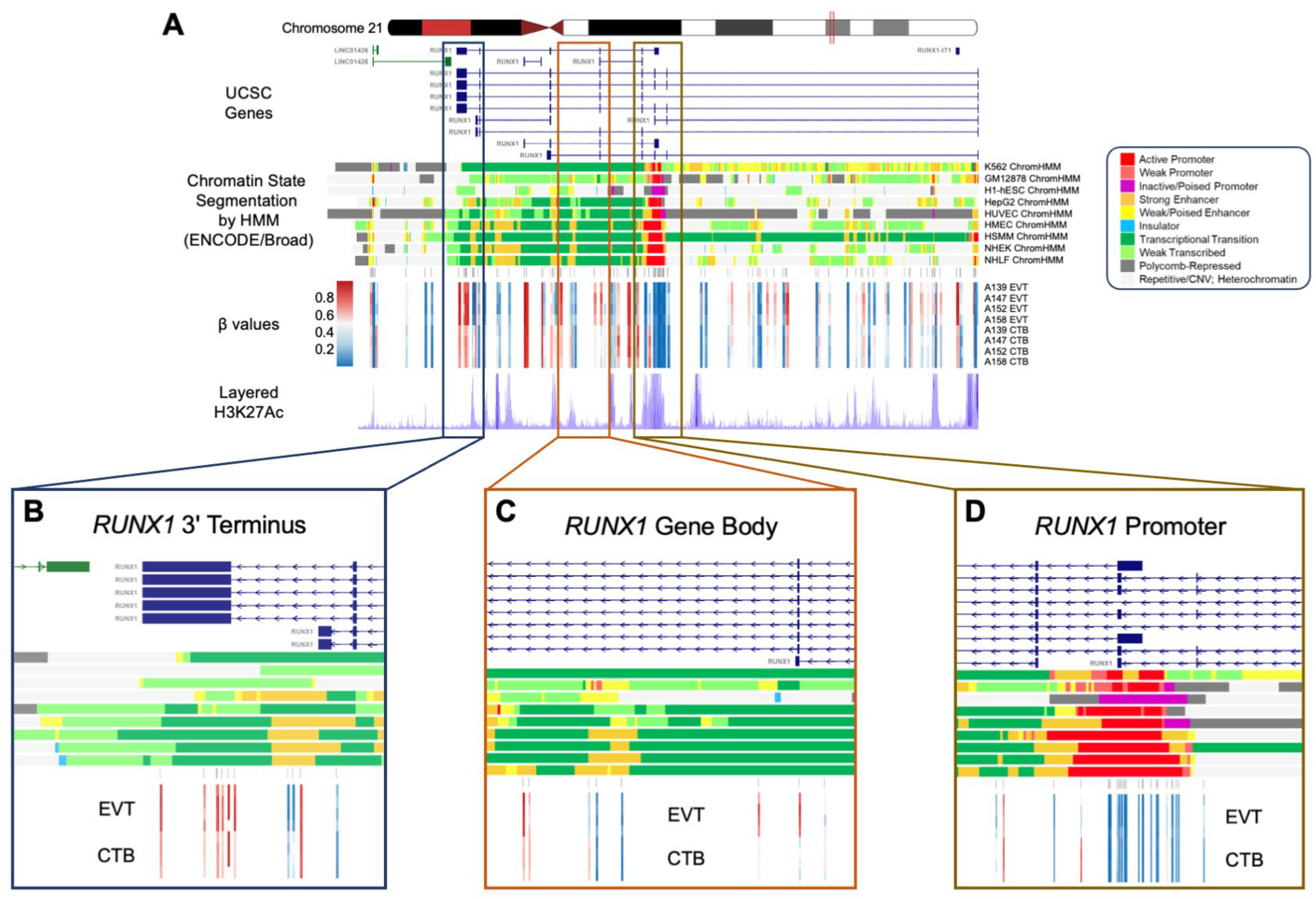
3.11. A 1.3 Mb Domain Gain of Methylation in EVT Spans the EMT-Associated RUNX1 Gene
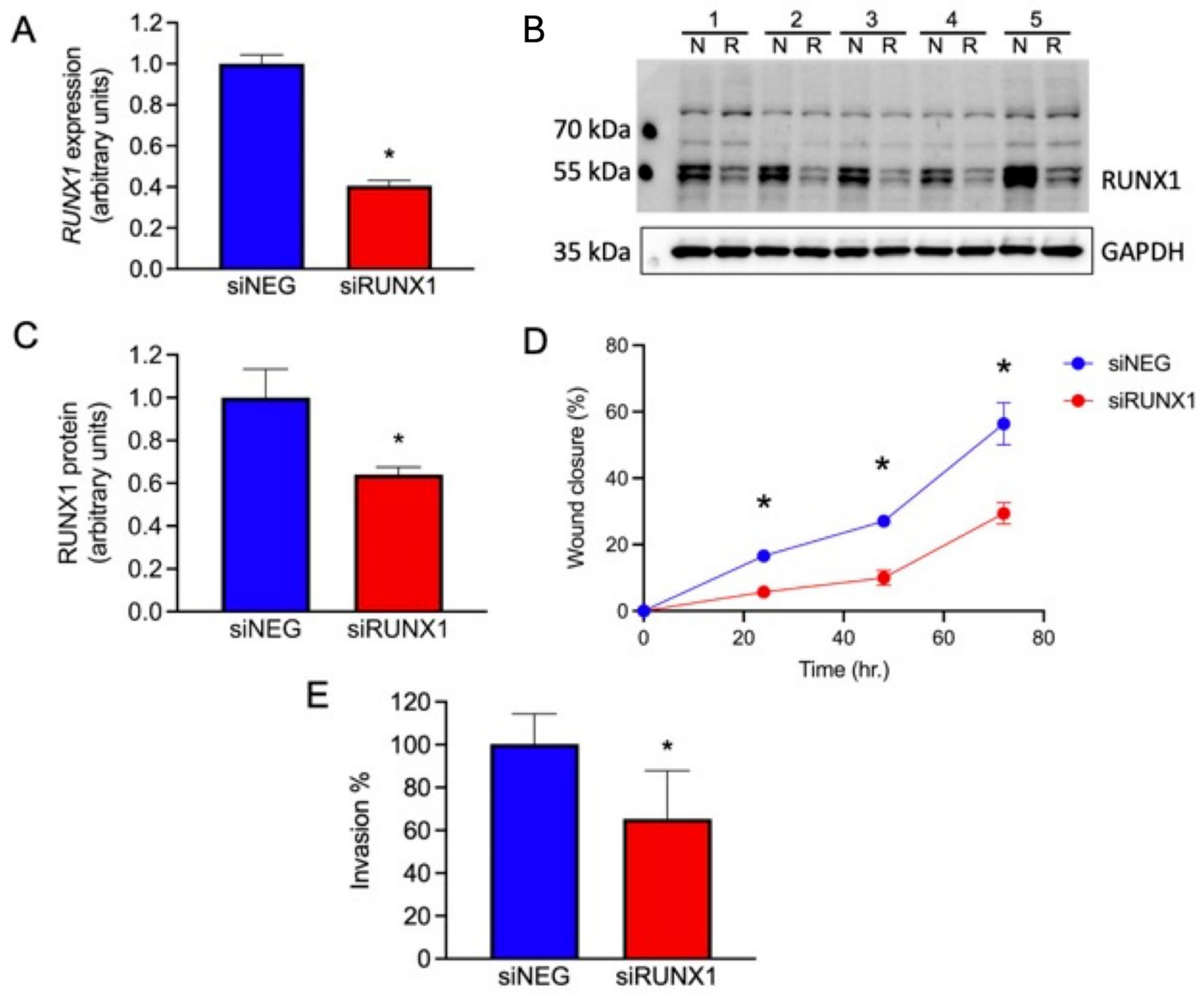
3.12. Knockdown of RUNX1 Leads to Altered Cell Migration and Invasion
4. Discussion
4.1. Confirmation of EMP in Trophoblast Differentiation
4.2. Changes in the DNA Methylation Profile
4.3. Gain of Methylation and Trophoblast EMP
4.4. Functional Effects of Methylation
4.5. Consequences of Methylation and Gene Expression Changes
4.6. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Knofler, M.; Haider, S.; Saleh, L.; Pollheimer, J.; Gamage, T.; James, J. Human placenta and trophoblast development: Key molecular mechanisms and model systems. Cell Mol. Life Sci. 2019, 76, 3479–3496. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Sundl, M.; Glasner, A.; Huppertz, B.; Moser, G. The trophoblast plug during early pregnancy: A deeper insight. Histochem. Cell Biol. 2016, 146, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Phipps, E.A.; Thadhani, R.; Benzing, T.; Karumanchi, S.A. Pre-eclampsia: Pathogenesis, novel diagnostics and therapies. Nat. Rev. Nephrol. 2019, 15, 275–289. [Google Scholar] [CrossRef]
- Roberts, J.M.; Rich-Edwards, J.W.; McElrath, T.F.; Garmire, L.; Myatt, L.; Global Pregnancy, C. Subtypes of Preeclampsia: Recognition and Determining Clinical Usefulness. Hypertension 2021, 77, 1430–1441. [Google Scholar] [CrossRef] [PubMed]
- Tantbirojn, P.; Crum, C.P.; Parast, M.M. Pathophysiology of placenta creta: The role of decidua and extravillous trophoblast. Placenta 2008, 29, 639–645. [Google Scholar] [CrossRef]
- Vicovac, L.; Aplin, J.D. Epithelial-mesenchymal transition during trophoblast differentiation. Acta Anat. 1996, 156, 202–216. [Google Scholar] [CrossRef]
- Kokkinos, M.I.; Murthi, P.; Wafai, R.; Thompson, E.W.; Newgreen, D.F. Cadherins in the human placenta--epithelial-mesenchymal transition (EMT) and placental development. Placenta 2010, 31, 747–755. [Google Scholar] [CrossRef]
- Davies, J.E.; Pollheimer, J.; Yong, H.E.; Kokkinos, M.I.; Kalionis, B.; Knofler, M.; Murthi, P. Epithelial-mesenchymal transition during extravillous trophoblast differentiation. Cell Adhes. Migr. 2016, 10, 310–321. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- DaSilva-Arnold, S.; James, J.L.; Al-Khan, A.; Zamudio, S.; Illsley, N.P. Differentiation of first trimester cytotrophoblast to extravillous trophoblast involves an epithelial-mesenchymal transition. Placenta 2015, 36, 1412–1418. [Google Scholar] [CrossRef]
- DaSilva-Arnold, S.; Zamudio, S.; Al-Khan, A.; Alvarez-Perez, J.; Mannion, C.; Koenig, C.; Luke, D.; Perez, A.; Petroff, M.; Alvarez, M.; et al. Human trophoblast epithelial-mesenchymal transition in abnormally invasive placenta. Biol. Reprod. 2018, 99, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Illsley, N.P.; DaSilva-Arnold, S.C.; Zamudio, S.; Alvarez, M.; Al-Khan, A. Trophoblast invasion: Lessons from abnormally invasive placenta (placenta accreta). Placenta 2020, 102, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Oh, S.; Abdelnabi, J.; Al-Dulaimi, R.; Aggarwal, A.; Ramos, M.; Davis, S.; Riester, M.; Waldron, L. HGNChelper: Identification and correction of invalid gene symbols for human and mouse. F1000Research 2020, 9, 1493. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Marini, F.; Ludt, A.; Linke, J.; Strauch, K. GeneTonic: An R/Bioconductor package for streamlining the interpretation of RNA-seq data. BMC Bioinform. 2021, 22, 610. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef]
- Eden, E.; Lipson, D.; Yogev, S.; Yakhini, Z. Discovering motifs in ranked lists of DNA sequences. PLoS Comput. Biol. 2007, 3, e39. [Google Scholar] [CrossRef]
- Mendioroz, M.; Do, C.; Jiang, X.; Liu, C.; Darbary, H.K.; Lang, C.F.; Lin, J.; Thomas, A.; Abu-Amero, S.; Stanier, P.; et al. Trans effects of chromosome aneuploidies on DNA methylation patterns in human Down syndrome and mouse models. Genome Biol. 2015, 16, 263. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.P.; Triche, T.J., Jr.; Hansen, K.D. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 2017, 33, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Arenas, C.; Gonzalez, J. MEAL: Perform Methylation Analysis; R Package Version 1.37.0. 2024. Available online: https://bioconductor.org/packages/MEAL (accessed on 11 November 2024).
- Fenton, T.R.; Kim, J.H. A systematic review and meta-analysis to revise the Fenton growth chart for preterm infants. BMC Pediatr. 2013, 13, 59. [Google Scholar] [CrossRef]
- Natenzon, A.; McFadden, P.; DaSilva-Arnold, S.C.; Zamudio, S.; Illsley, N.P. Diminished trophoblast differentiation in early onset preeclampsia. Placenta 2022, 120, 25–31. [Google Scholar] [CrossRef]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef] [PubMed]
- Groger, C.J.; Grubinger, M.; Waldhor, T.; Vierlinger, K.; Mikulits, W. Meta-analysis of gene expression signatures defining the epithelial to mesenchymal transition during cancer progression. PLoS ONE 2012, 7, e51136. [Google Scholar] [CrossRef]
- Zhao, M.; Kong, L.; Liu, Y.; Qu, H. dbEMT: An epithelial-mesenchymal transition associated gene resource. Sci. Rep. 2015, 5, 11459. [Google Scholar] [CrossRef]
- Cheng, W.Y.; Kandel, J.J.; Yamashiro, D.J.; Canoll, P.; Anastassiou, D. A multi-cancer mesenchymal transition gene expression signature is associated with prolonged time to recurrence in glioblastoma. PLoS ONE 2012, 7, e34705. [Google Scholar] [CrossRef]
- Gamage, T.; Schierding, W.; Tsai, P.; Ludgate, J.L.; Chamley, L.W.; Weeks, R.J.; Macaulay, E.C.; James, J.L. Human trophoblasts are primarily distinguished from somatic cells by differences in the pattern rather than the degree of global CpG methylation. Biol. Open 2018, 7, bio034884. [Google Scholar] [CrossRef]
- Lu, X.; He, Y.; Zhu, C.; Wang, H.; Chen, S.; Lin, H.Y. Twist1 is involved in trophoblast syncytialization by regulating GCM1. Placenta 2016, 39, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef]
- Debnath, P.; Huirem, R.S.; Dutta, P.; Palchaudhuri, S. Epithelial-mesenchymal transition and its transcription factors. Biosci. Rep. 2022, 42, BSR20211754. [Google Scholar] [CrossRef]
- Rawn, S.M.; Cross, J.C. The evolution, regulation, and function of placenta-specific genes. Annu. Rev. Cell Dev. Biol. 2008, 24, 159–181. [Google Scholar] [CrossRef] [PubMed]
- Morey, R.; Farah, O.; Kallol, S.; Requena, D.F.; Meads, M.; Moretto-Zita, M.; Soncin, F.; Laurent, L.C.; Parast, M.M. Transcriptomic Drivers of Differentiation, Maturation, and Polyploidy in Human Extravillous Trophoblast. Front. Cell Dev. Biol. 2021, 9, 702046. [Google Scholar] [CrossRef] [PubMed]
- Vondra, S.; Kunihs, V.; Eberhart, T.; Eigner, K.; Bauer, R.; Haslinger, P.; Haider, S.; Windsperger, K.; Klambauer, G.; Schutz, B.; et al. Metabolism of cholesterol and progesterone is differentially regulated in primary trophoblastic subtypes and might be disturbed in recurrent miscarriages. J. Lipid Res. 2019, 60, 1922–1934. [Google Scholar] [CrossRef]
- Ogoyama, M.; Ohkuchi, A.; Takahashi, H.; Zhao, D.; Matsubara, S.; Takizawa, T. LncRNA H19-Derived miR-675-5p Accelerates the Invasion of Extravillous Trophoblast Cells by Inhibiting GATA2 and Subsequently Activating Matrix Metalloproteinases. Int. J. Mol. Sci. 2021, 22, 1237. [Google Scholar] [CrossRef]
- Zhou, Y.; Gormley, M.J.; Hunkapiller, N.M.; Kapidzic, M.; Stolyarov, Y.; Feng, V.; Nishida, M.; Drake, P.M.; Bianco, K.; Wang, F.; et al. Reversal of gene dysregulation in cultured cytotrophoblasts reveals possible causes of preeclampsia. J. Clin. Investig. 2013, 123, 2862–2872. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, K.; Qian, C.N.; Leach, R. DNA methylation is associated with transcription of Snail and Slug genes. Biochem. Biophys. Res. Commun. 2013, 430, 1083–1090. [Google Scholar] [CrossRef]
- Anton, L.; Brown, A.G.; Bartolomei, M.S.; Elovitz, M.A. Differential methylation of genes associated with cell adhesion in preeclamptic placentas. PLoS ONE 2014, 9, e100148. [Google Scholar] [CrossRef]
- Nordor, A.V.; Nehar-Belaid, D.; Richon, S.; Klatzmann, D.; Bellet, D.; Dangles-Marie, V.; Fournier, T.; Aryee, M.J. The early pregnancy placenta foreshadows DNA methylation alterations of solid tumors. Epigenetics 2017, 12, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kim, M.Y.; Elliot, G.; Zhou, Y.; Zhao, G.; Li, D.; Lowdon, R.F.; Gormley, M.; Kapidzic, M.; Robinson, J.F.; et al. Human placental cytotrophoblast epigenome dynamics over gestation and alterations in placental disease. Dev. Cell 2021, 56, 1238–1252 e1235. [Google Scholar] [CrossRef]
- Ehrlich, M.; Gama-Sosa, M.A.; Huang, L.H.; Midgett, R.M.; Kuo, K.C.; McCune, R.A.; Gehrke, C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982, 10, 2709–2721. [Google Scholar] [CrossRef]
- Grigoriu, A.; Ferreira, J.C.; Choufani, S.; Baczyk, D.; Kingdom, J.; Weksberg, R. Cell specific patterns of methylation in the human placenta. Epigenetics 2011, 6, 368–379. [Google Scholar] [CrossRef]
- Novakovic, B.; Evain-Brion, D.; Murthi, P.; Fournier, T.; Saffery, R. Variable DAXX gene methylation is a common feature of placental trophoblast differentiation, preeclampsia, and response to hypoxia. FASEB J. 2017, 31, 2380–2392. [Google Scholar] [CrossRef]
- Schroeder, D.I.; Blair, J.D.; Lott, P.; Yu, H.O.; Hong, D.; Crary, F.; Ashwood, P.; Walker, C.; Korf, I.; Robinson, W.P.; et al. The human placenta methylome. Proc. Natl. Acad. Sci. USA 2013, 110, 6037–6042. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Macaulay, E.C.; Rodger, E.J.; Stockwell, P.A.; Parry, M.F.; Roberts, H.E.; Slatter, T.L.; Hung, N.A.; Devenish, C.J.; Morison, I.M. Placental Hypomethylation Is More Pronounced in Genomic Loci Devoid of Retroelements. G3 2016, 6, 1911–1921. [Google Scholar] [CrossRef]
- Delgado-Tirado, S.; Amarnani, D.; Zhao, G.; Rossin, E.J.; Eliott, D.; Miller, J.B.; Greene, W.A.; Ramos, L.; Arevalo-Alquichire, S.; Leyton-Cifuentes, D.; et al. Topical delivery of a small molecule RUNX1 transcription factor inhibitor for the treatment of proliferative vitreoretinopathy. Sci. Rep. 2020, 10, 20554. [Google Scholar] [CrossRef] [PubMed]
- Fritz, A.J.; Hong, D.; Boyd, J.; Kost, J.; Finstaad, K.H.; Fitzgerald, M.P.; Hanna, S.; Abuarqoub, A.H.; Malik, M.; Bushweller, J.; et al. RUNX1 and RUNX2 transcription factors function in opposing roles to regulate breast cancer stem cells. J. Cell. Physiol. 2020, 235, 7261–7272. [Google Scholar] [CrossRef]
- Lu, C.; Yang, Z.; Yu, D.; Lin, J.; Cai, W. RUNX1 regulates TGF-beta induced migration and EMT in colorectal cancer. Pathol. Res. Pract. 2020, 216, 153142. [Google Scholar] [CrossRef]
- VanOudenhove, J.J.; Medina, R.; Ghule, P.N.; Lian, J.B.; Stein, J.L.; Zaidi, S.K.; Stein, G.S. Transient RUNX1 Expression during Early Mesendodermal Differentiation of hESCs Promotes Epithelial to Mesenchymal Transition through TGFB2 Signaling. Stem Cell Rep. 2016, 7, 884–896. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- DaSilva-Arnold, S.C.; Kuo, C.Y.; Davra, V.; Remache, Y.; Kim, P.C.W.; Fisher, J.P.; Zamudio, S.; Al-Khan, A.; Birge, R.B.; Illsley, N.P. ZEB2, a master regulator of the epithelial-mesenchymal transition, mediates trophoblast differentiation. Mol. Hum. Reprod. 2019, 25, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Y.; Fu, Z.; Huang, W.; Yu, Z.; Wang, J.; Zheng, K.; Zhang, S.; Li, S.; Chen, J. MiR-29b-3p Inhibits Migration and Invasion of Papillary Thyroid Carcinoma by Downregulating COL1A1 and COL5A1. Front. Oncol. 2022, 12, 837581. [Google Scholar] [CrossRef]
- Dejardin, T.; Carollo, P.S.; Sipieter, F.; Davidson, P.M.; Seiler, C.; Cuvelier, D.; Cadot, B.; Sykes, C.; Gomes, E.R.; Borghi, N. Nesprins are mechanotransducers that discriminate epithelial-mesenchymal transition programs. J. Cell Biol. 2020, 219, e201908036. [Google Scholar] [CrossRef]
- Bermudez, L.G.; Madariaga, I.; Zuniga, M.I.; Olaya, M.; Canas, A.; Rodriguez, L.S.; Moreno, O.M.; Rojas, A. RUNX1 gene expression changes in the placentas of women smokers. Exp. Ther. Med. 2021, 22, 902. [Google Scholar] [CrossRef]
- Ponder, K.L.; Barcena, A.; Bos, F.L.; Gormley, M.; Zhou, Y.; Ona, K.; Kapidzic, M.; Zovein, A.C.; Fisher, S.J. Preeclampsia and Inflammatory Preterm Labor Alter the Human Placental Hematopoietic Niche. Reprod. Sci. 2016, 23, 1179–1192. [Google Scholar] [CrossRef]
- Varberg, K.M.; Iqbal, K.; Muto, M.; Simon, M.E.; Scott, R.L.; Kozai, K.; Choudhury, R.H.; Aplin, J.D.; Biswell, R.; Gibson, M.; et al. ASCL2 reciprocally controls key trophoblast lineage decisions during hemochorial placenta development. Proc. Natl. Acad. Sci. USA 2021, 118, e2016517118. [Google Scholar] [CrossRef]
- Horii, M.; Morey, R.; Bui, T.; Touma, O.; Nelson, K.K.; Cho, H.Y.; Rishik, H.; Laurent, L.C.; Parast, M.M. Modeling preeclampsia using human induced pluripotent stem cells. Sci. Rep. 2021, 11, 5877. [Google Scholar] [CrossRef] [PubMed]
- Goffin, F.; Munaut, C.; Malassine, A.; Evain-Brion, D.; Frankenne, F.; Fridman, V.; Dubois, M.; Uzan, S.; Merviel, P.; Foidart, J.M. Evidence of a limited contribution of feto-maternal interactions to trophoblast differentiation along the invasive pathway. Tissue Antigens 2003, 62, 104–116. [Google Scholar] [CrossRef] [PubMed]
- von Rango, U.; Krusche, C.A.; Kertschanska, S.; Alfer, J.; Kaufmann, P.; Beier, H.M. Apoptosis of extravillous trophoblast cells limits the trophoblast invasion in uterine but not in tubal pregnancy during first trimester. Placenta 2003, 24, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fan, X.; Wang, R.; Lu, X.; Dang, Y.L.; Wang, H.; Lin, H.Y.; Zhu, C.; Ge, H.; Cross, J.C.; et al. Single-cell RNA-seq reveals the diversity of trophoblast subtypes and patterns of differentiation in the human placenta. Cell Res. 2018, 28, 819–832. [Google Scholar] [CrossRef]
- Marsh, B.; Zhou, Y.; Kapidzic, M.; Fisher, S.; Blelloch, R. Regionally distinct trophoblast regulate barrier function and invasion in the human placenta. eLife 2022, 11, e78829. [Google Scholar] [CrossRef]
- Tsang, J.C.H.; Vong, J.S.L.; Ji, L.; Poon, L.C.Y.; Jiang, P.; Lui, K.O.; Ni, Y.B.; To, K.F.; Cheng, Y.K.Y.; Chiu, R.W.K.; et al. Integrative single-cell and cell-free plasma RNA transcriptomics elucidates placental cellular dynamics. Proc. Natl. Acad. Sci. USA 2017, 114, E7786–E7795. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Process | Z Score | Adj. p Value |
|---|---|---|
| HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION | 8.12 | 1.71 × 10−10 |
| HALLMARK_HYPOXIA | 7.18 | 2.63 × 10−5 |
| HALLMARK_INTERFERON_GAMMA_RESPONSE | 6.42 | 2.77 × 10−3 |
| HALLMARK_INFLAMMATORY_RESPONSE | 4.75 | 4.15 × 10−2 |
| HALLMARK_APICAL_JUNCTION | 4.58 | 1.99 × 10−3 |
| HALLMARK_TNFA_SIGNALING_VIA_NFKB | 3.96 | 4.15 × 10−2 |
| HALLMARK_GLYCOLYSIS | 3.74 | 2.77 × 10−3 |
| HALLMARK_TGF_BETA_SIGNALING | 2.47 | 4.15 × 10−2 |
| HALLMARK_IL2_STAT5_SIGNALING | 2.37 | 1.11 × 10−3 |
| HALLMARK_UV_RESPONSE_DN | 2.37 | 2.68 × 10−4 |
| HALLMARK_ANDROGEN_RESPONSE | 2.29 | 4.15 × 10−2 |
| HALLMARK_APOPTOSIS | 1.53 | 4.15 × 10−2 |
| HALLMARK_ESTROGEN_RESPONSE_EARLY | 1.44 | 4.38 × 10−7 |
| HALLMARK_MITOTIC_SPINDLE | −2.09 | 4.76 × 10−2 |
| HALLMARK_G2M_CHECKPOINT | −8.09 | 3.66 × 10−6 |
| Gene ID | Fold Change | CpG Gain | Gene ID | Fold Change | CpG Gain | Gene ID | Fold Change | CpG Gain |
|---|---|---|---|---|---|---|---|---|
| HSPG2 | 38.4 | 3 | PTPN14 | −2.4 | 3 | FGFR2 | −19.1 | 9 |
| ASCL2 | 37.3 | 3 | NOTCH1 | −3.0 | 2 | BMP7 | −24.0 | 5 |
| LAIR2 | 31.7 | 2 | TPD52L1 | −2.9 | 2 | MSX2 | −46.8 | 2 |
| PMEPA1 | 27.0 | 2 | MAP3K4 | −3.0 | 5 | LRP5 | −91.4 | 6 |
| TGFB1 | 17.5 | 3 | TGM2 | −3.1 | 2 | SVEP1 | −93.6 | 2 |
| FLT4 | 13.9 | 8 | SYK | −3.7 | 2 | ZBTB7C | −104.2 | 6 |
| MICALL2 | 12.5 | 12 | IGF1R | −4.2 | 2 | LARGE2 | −114.9 | 2 |
| LPCAT1 | 10.2 | 2 | FOXK1 | −4.8 | 2 | NRP2 | −123.0 | 2 |
| TWIST2 | 10.2 | 2 | BCL2 | −6.9 | 2 | FRAS1 | −128.1 | 2 |
| COL5A1 | 9.9 | 47 | MET | −7.73 | 2 | EPHB4 | −140.9 | 5 |
| RUNX1 | 7.5 | 21 | FBN2 | −12.3 | 2 | ACSS1 | −189.9 | 7 |
| FSCN1 | 6.2 | 7 | FXYD3 | −13.1 | 3 | PEG10 | −195.8 | 5 |
| COL4A2 | 4.4 | 4 | SPINT1 | −14.3 | 3 | SLC27A2 | −248.2 | 3 |
| BMP1 | 3.8 | 2 | ALDH1A3 | −14.8 | 2 | LRP2 | −293.8 | 2 |
| PLOD1 | 3.5 | 2 | JAG1 | −15.8 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ackerman IV, W.E.; Rigo, M.M.; DaSilva-Arnold, S.C.; Do, C.; Tariq, M.; Salas, M.; Castano, A.; Zamudio, S.; Tycko, B.; Illsley, N.P. Epigenetic Changes Regulating Epithelial–Mesenchymal Plasticity in Human Trophoblast Differentiation. Cells 2025, 14, 970. https://doi.org/10.3390/cells14130970
Ackerman IV WE, Rigo MM, DaSilva-Arnold SC, Do C, Tariq M, Salas M, Castano A, Zamudio S, Tycko B, Illsley NP. Epigenetic Changes Regulating Epithelial–Mesenchymal Plasticity in Human Trophoblast Differentiation. Cells. 2025; 14(13):970. https://doi.org/10.3390/cells14130970
Chicago/Turabian StyleAckerman IV, William E., Mauricio M. Rigo, Sonia C. DaSilva-Arnold, Catherine Do, Mariam Tariq, Martha Salas, Angelica Castano, Stacy Zamudio, Benjamin Tycko, and Nicholas P. Illsley. 2025. "Epigenetic Changes Regulating Epithelial–Mesenchymal Plasticity in Human Trophoblast Differentiation" Cells 14, no. 13: 970. https://doi.org/10.3390/cells14130970
APA StyleAckerman IV, W. E., Rigo, M. M., DaSilva-Arnold, S. C., Do, C., Tariq, M., Salas, M., Castano, A., Zamudio, S., Tycko, B., & Illsley, N. P. (2025). Epigenetic Changes Regulating Epithelial–Mesenchymal Plasticity in Human Trophoblast Differentiation. Cells, 14(13), 970. https://doi.org/10.3390/cells14130970