
Transmembrane Protein-184A Interacts with Syndecan-4 and Rab GTPases and Is Required to Maintain VE-Cadherin Levels

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Zebrafish Strains, Housing, and Husbandry
2.2. Morpholino Injections
2.3. Fin Imaging Measurements and Statistical Analysis
2.4. Materials—Antibodies
2.5. Culturing and Transfections
2.6. Immunofluorescent Staining
2.7. Co-Immunoprecipitation and Western Blotting
2.8. WCL Harvest and Cell Fractionation
2.9. RNA Purification and RT-qPCR
2.10. VEGF Treatment and Vesicle Staining
2.11. Corrected Total Cell Fluorescence (CTCF) and Image J Fiji Quantification
2.12. RStudio Visualization of Immunofluorescent Cell Experiments
2.13. Scratch Wounding and Leading-Edge Analysis
3. Results
3.1. TMEM184A Interacts with Sdc4 in Vascular ECs
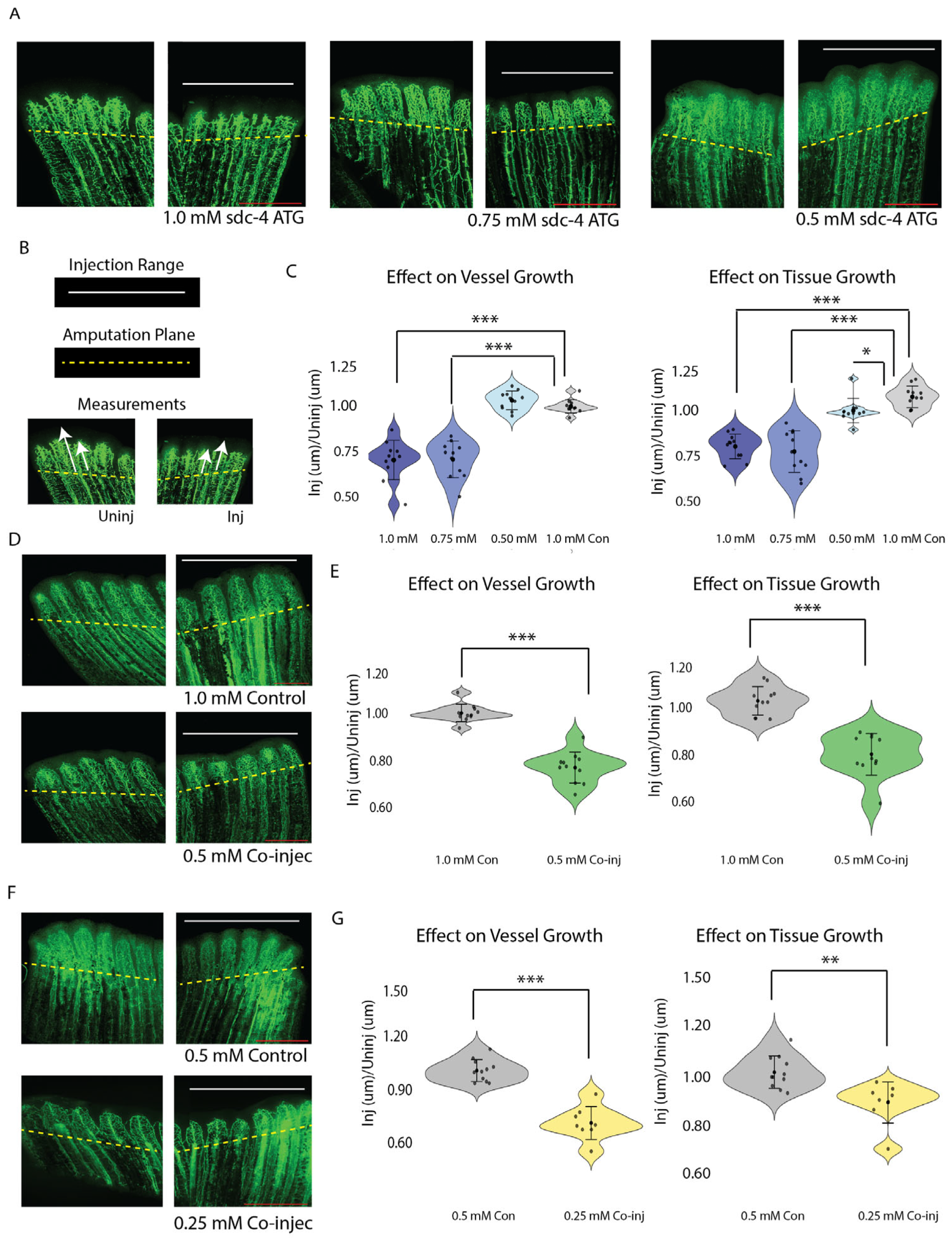
3.2. Sdc4 and Tmem184a Function Cooperatively to Promote Vessel and Tissue Outgrowth
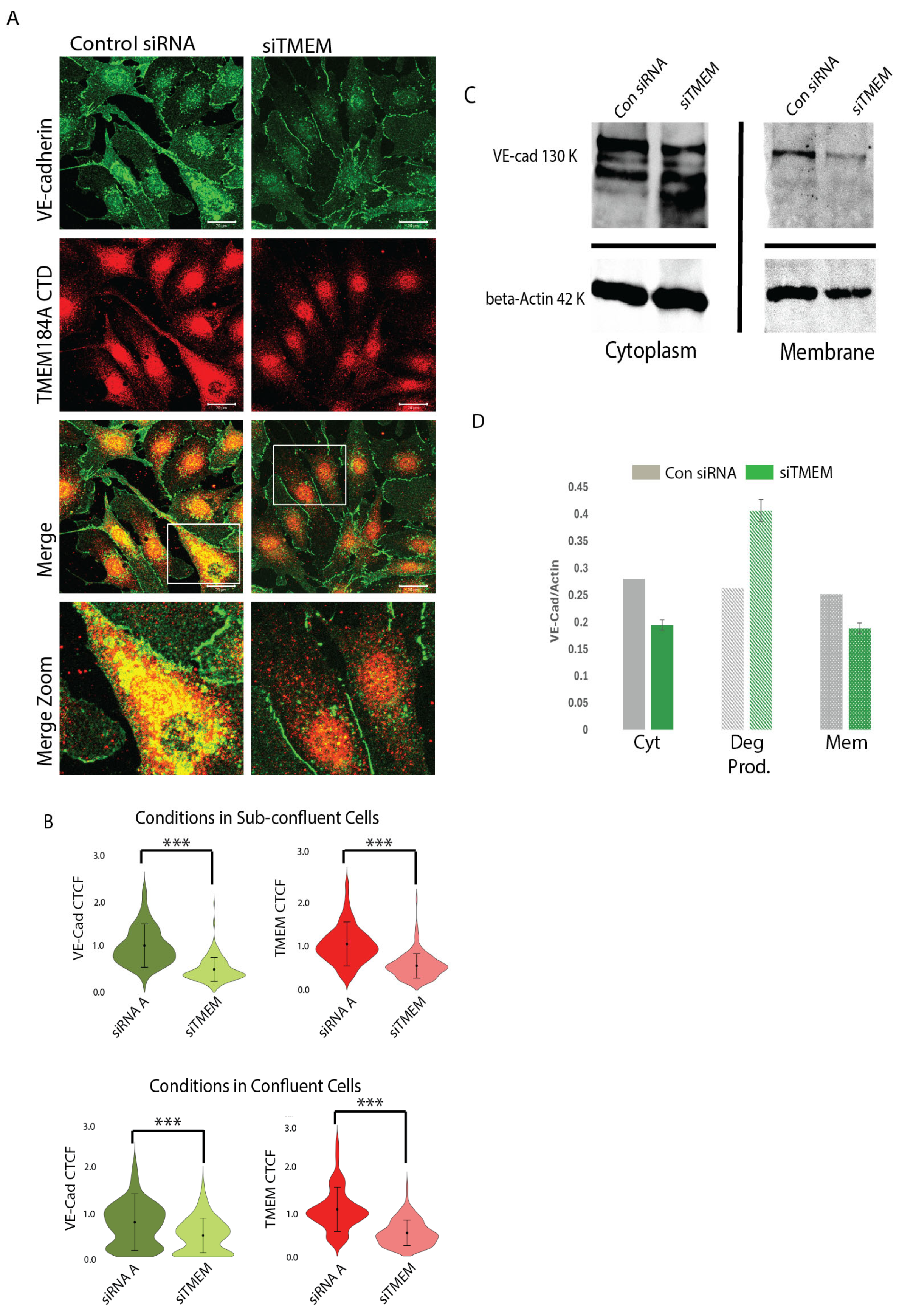
3.3. TMEM184A Is Required to Maintain Post-Translational VE-Cad Levels in Sub-Confluent ECs
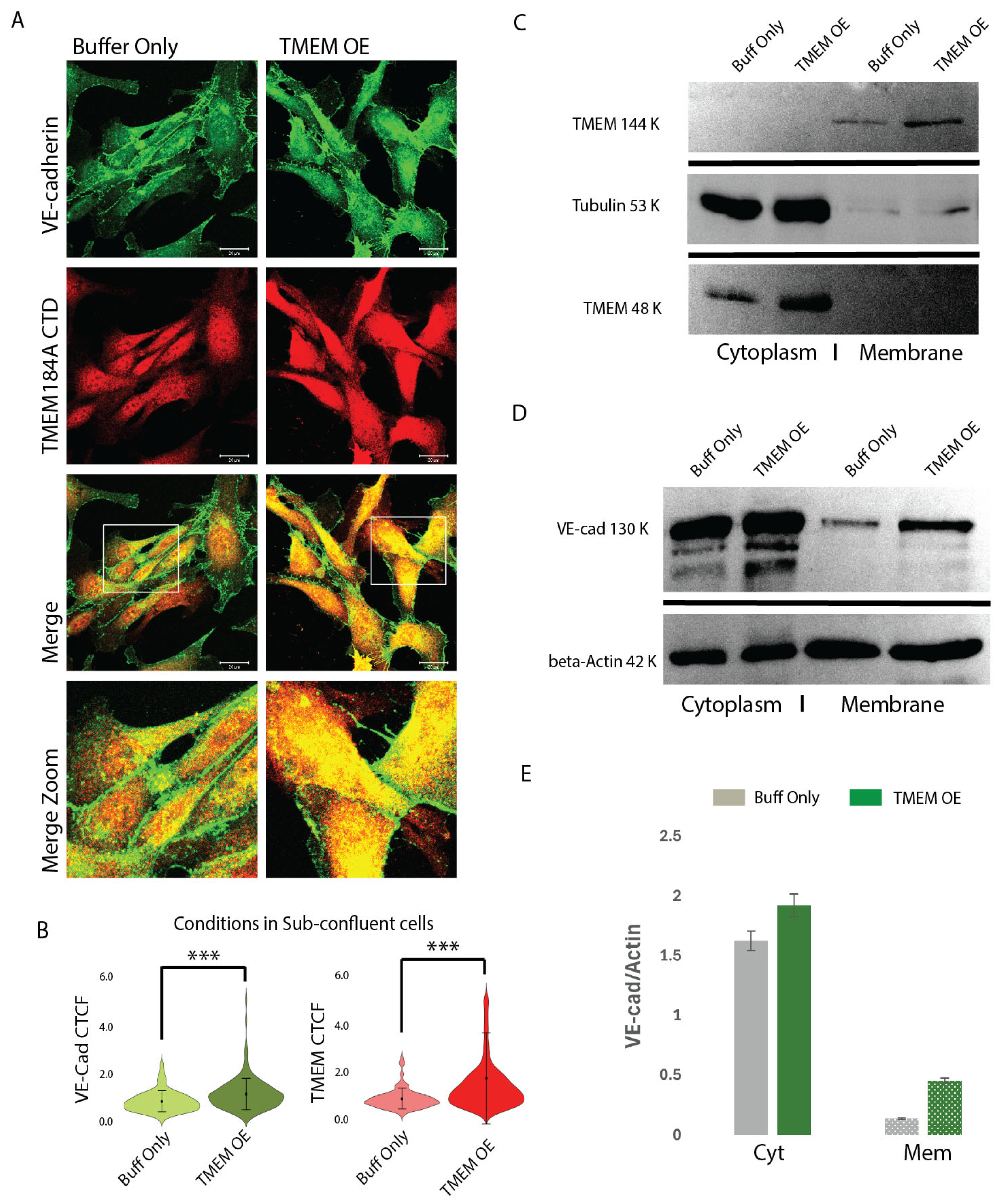
3.4. TMEM184A-tGFP Expression Colocalizes in VE-Cad Puncta and Increases Total VE-Cad in Sub-Confluent ECs
3.5. TMEM184A Colocalizes with Rab-GTPases in Response to VEGF
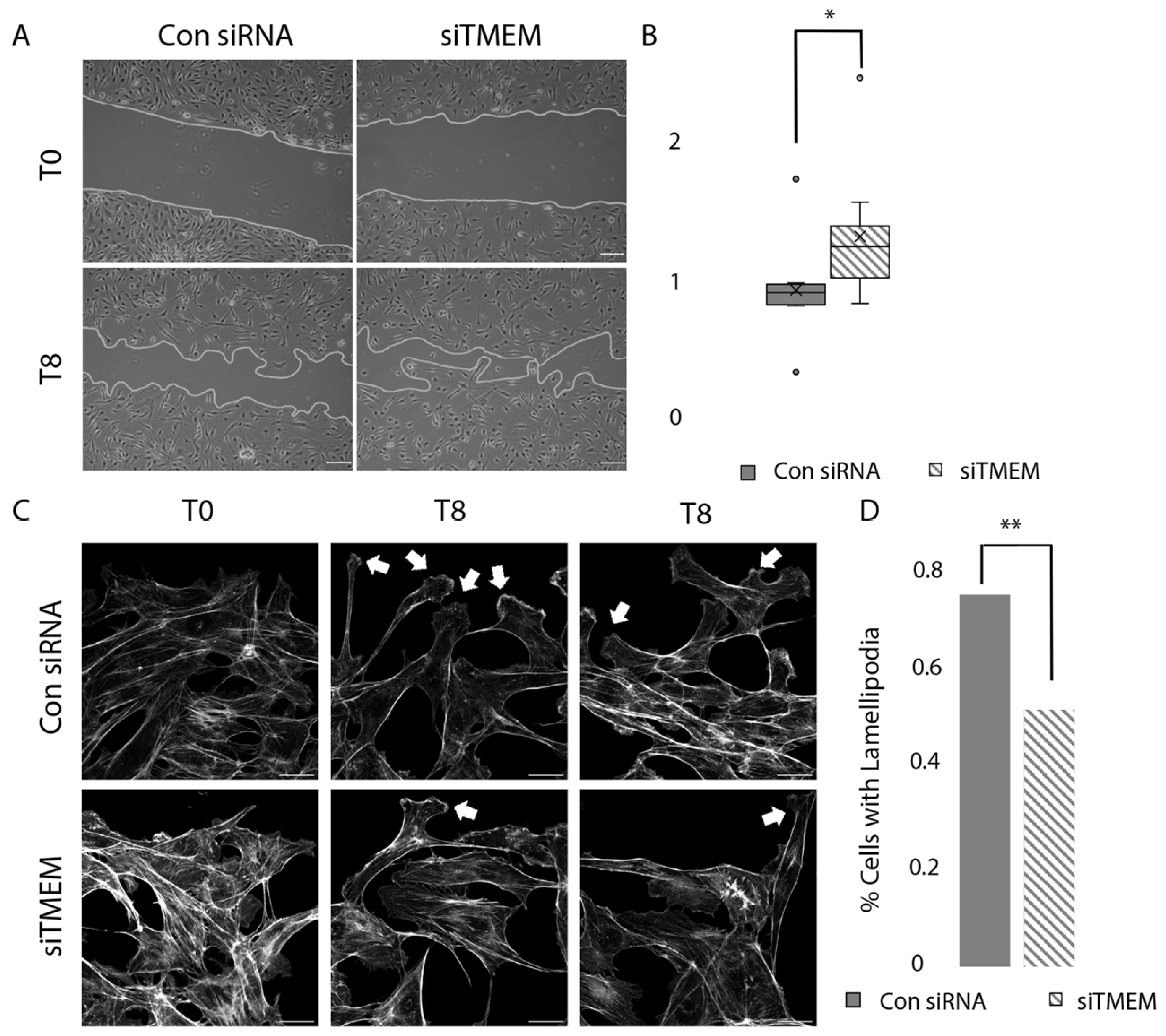
3.6. TMEM184A KD Cells Migrate Faster Compared to Control siRNA Cells in Wounding
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Farwell, S.L.; Kanyi, D.; Hamel, M.; Slee, J.B.; Miller, E.A.; Cipolle, M.D.; Lowe-Krentz, L.J. Heparin Decreases in Tumor Necrosis Factor alpha (TNFalpha)-induced Endothelial Stress Responses Require Transmembrane Protein 184A and Induction of Dual Specificity Phosphatase 1. J. Biol. Chem. 2016, 291, 5342–5354. [Google Scholar] [CrossRef] [PubMed]
- Gilotti, A.C.; Nimlamool, W.; Pugh, R.; Slee, J.B.; Barthol, T.C.; Miller, E.A.; Lowe-Krentz, L.J. Heparin responses in vascular smooth muscle cells involve cGMP-dependent protein kinase (PKG). J. Cell. Physiol. 2014, 229, 2142–2152. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Talotta-Altenburg, L.M.; Silimperi, K.A.; Ciabattoni, G.O.; Lowe-Krentz, L.J. Endothelial nitric oxide synthase activation is required for heparin receptor effects on vascular smooth muscle cells. American journal of physiology. Cell Physiol. 2020, 318, C463–C475. [Google Scholar] [CrossRef] [PubMed]
- Pugh, R.J.; Slee, J.B.; Farwell, S.L.; Li, Y.; Barthol, T.; Patton, W.A.; Lowe-Krentz, L.J. Transmembrane Protein 184A Is a Receptor Required for Vascular Smooth Muscle Cell Responses to Heparin. J. Biol. Chem. 2016, 291, 5326–5341. [Google Scholar] [CrossRef]
- Pukac, L.A.; Carter, J.E.; Ottlinger, M.E.; Karnovsky, M.J. Mechanisms of inhibition by heparin of PDGF stimulated MAP kinase activation in vascular smooth muscle cells. J. Cell. Physiol. 1997, 172, 69–78. [Google Scholar] [CrossRef]
- Savage, J.M.; Gilotti, A.C.; Granzow, C.A.; Molina, F.; Lowe-Krentz, L.J. Antibodies against a putative heparin receptor slow cell proliferation and decrease MAPK activation in vascular smooth muscle cells. J. Cell. Physiol. 2001, 187, 283–293. [Google Scholar] [CrossRef]
- Thourani, V.H.; Brar, S.S.; Kennedy, T.P.; Thornton, L.R.; Watts, J.A.; Ronson, R.S.; Zhao, Z.Q.; Sturrock, A.L.; Hoidal, J.R.; Vinten-Johansen, J. Nonanticoagulant heparin inhibits NF-kappaB activation and attenuates myocardial reperfusion injury. Am. J. Physiology. Heart Circ. Physiol. 2000, 278, H2084–H2093. [Google Scholar] [CrossRef]
- Barry, A.K.; Wang, N.; Leckband, D.E. Local VE-cadherin mechanotransduction triggers long-ranged remodeling of endothelial monolayers. J. Cell Sci. 2015, 128, 1341–1351. [Google Scholar] [CrossRef]
- Dorland, Y.L.; Huveneers, S. Cell-cell junctional mechanotransduction in endothelial remodeling. Cell Mol. Life Sci. 2017, 74, 279–292. [Google Scholar] [CrossRef]
- Libby, P. Inflammation during the life cycle of the atherosclerotic plaque. Cardiovasc. Res. 2021, 117, 2525–2536. [Google Scholar] [CrossRef]
- Gavard, J.; Gutkind, J.S. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 2006, 8, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Tugues, S.; Li, X.; Gualandi, L.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 2011, 437, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Mansouri, M.; Rizk, A.; Berger, P. Regulation of VEGFR2 trafficking and signaling by Rab GTPase-activating proteins. Sci. Rep. 2019, 9, 13342. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Hermanson, S.; Ekker, S.C. Syndecan-2 is essential for angiogenic sprouting during zebrafish development. Blood 2004, 103, 1710–1719. [Google Scholar] [CrossRef]
- Corti, F.; Wang, Y.; Rhodes, J.M.; Atri, D.; Archer-Hartmann, S.; Zhang, J.; Zhuang, Z.W.; Chen, D.; Wang, T.; Wang, Z.; et al. Publisher Correction: N-terminal syndecan-2 domain selectively enhances 6-O heparan sulfate chains sulfation and promotes VEGFA165-dependent neovascularization. Nat. Commun. 2019, 10, 2124. [Google Scholar] [CrossRef]
- Vuong, T.T.; Reine, T.M.; Sudworth, A.; Jenssen, T.G.; Kolset, S.O. Syndecan-4 is a major syndecan in primary human endothelial cells in vitro, modulated by inflammatory stimuli and involved in wound healing. J. Histochem. Cytochem. 2015, 63, 280–292. [Google Scholar] [CrossRef]
- Nunes, S.S.; Outeiro-Bernstein, M.A.; Juliano, L.; Vardiero, F.; Nader, H.B.; Woods, A.; Legrand, C.; Morandi, V. Syndecan-4 contributes to endothelial tubulogenesis through interactions with two motifs inside the pro-angiogenic N-terminal domain of thrombospondin-1. J. Cell. Physiol. 2008, 214, 828–837. [Google Scholar] [CrossRef]
- Morgan, M.R.; Hamidi, H.; Bass, M.D.; Warwood, S.; Ballestrem, C.; Humphries, M.J. Syndecan-4 phosphorylation is a control point for integrin recycling. Dev. Cell 2013, 24, 472–485. [Google Scholar] [CrossRef]
- Matthews, H.K.; Marchant, L.; Carmona-Fontaine, C.; Kuriyama, S.; Larrain, J.; Holt, M.R.; Parsons, M.; Mayor, R. Directional migration of neural crest cells in vivo is regulated by Syndecan-4/Rac1 and non-canonical Wnt signaling/RhoA. Development 2008, 135, 1771–1780. [Google Scholar] [CrossRef]
- Williamson, R.C.; Cowell, C.A.M.; Reville, T.; Roper, J.A.; Rendall, T.C.S.; Bass, M.D. Coronin-1C Protein and Caveolin Protein Provide Constitutive and Inducible Mechanisms of Rac1 Protein Trafficking. J. Biol. Chem. 2015, 290, 15437–15449. [Google Scholar] [CrossRef]
- De Rossi, G.; Vahatupa, M.; Cristante, E.; Arokiasamy, S.; Liyanage, S.E.; May, U.; Pellinen, L.; Uusitalo-Jarvinen, H.; Bainbridge, J.W.; Jarvinen, T.A.H.; et al. Pathological Angiogenesis Requires Syndecan-4 for Efficient VEGFA-Induced VE-Cadherin Internalization. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1374–1389. [Google Scholar] [CrossRef] [PubMed]
- Echtermeyer, F.; Streit, M.; Wilcox-Adelman, S.; Saoncella, S.; Denhez, F.; Detmar, M.; Goetinck, P. Delayed wound repair and impaired angiogenesis in mice lacking syndecan-4. J. Clin. Investig. 2001, 107, R9–R14. [Google Scholar] [CrossRef] [PubMed]
- Sauteur, L.; Krudewig, A.; Herwig, L.; Ehrenfeuchter, N.; Lenard, A.; Affolter, M.; Belting, H.G. Cdh5/VE-cadherin promotes endothelial cell interface elongation via cortical actin polymerization during angiogenic sprouting. Cell Rep. 2014, 9, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Grimsley-Myers, C.M.; Isaacson, R.H.; Cadwell, C.M.; Campos, J.; Hernandes, M.S.; Myers, K.R.; Seo, T.; Giang, W.; Griendling, K.K.; Kowalczyk, A.P. VE-cadherin endocytosis controls vascular integrity and patterning during development. J. Cell Biol. 2020, 219, e201909081. [Google Scholar] [CrossRef]
- Delva, E.; Kowalczyk, A.P. Regulation of cadherin trafficking. Traffic 2009, 10, 259–267. [Google Scholar] [CrossRef]
- Su, W.; Kowalczyk, A.P. The VE-cadherin cytoplasmic domain undergoes proteolytic processing during endocytosis. Mol. Biol. Cell 2017, 28, 76–84. [Google Scholar] [CrossRef]
- Chrifi, I.; Louzao-Martinez, L.; Brandt, M.M.; van Dijk, C.G.M.; Burgisser, P.E.; Zhu, C.; Kros, J.M.; Verhaar, M.C.; Duncker, D.J.; Cheng, C. CMTM4 regulates angiogenesis by promoting cell surface recycling of VE-cadherin to endothelial adherens junctions. Angiogenesis 2019, 22, 75–93. [Google Scholar] [CrossRef]
- Best, D.; Adams, I.R. Sdmg1 is a component of secretory granules in mouse secretory exocrine tissues. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2009, 238, 223–231. [Google Scholar] [CrossRef]
- Field, C.J.; Perez, A.M.; Samet, T.; Ricles, V.; Iovine, M.K.; Lowe-Krentz, L.J. Involvement of transmembrane protein 184a during angiogenesis in zebrafish embryos. Front. Physiol. 2022, 13, 845407. [Google Scholar] [CrossRef]
- Farwell, S.L.N.; Reylander, K.G.; Iovine, M.K.; Lowe-Krentz, L.J. Novel Heparin Receptor Transmembrane Protein 184a Regulates Angiogenesis in the Adult Zebrafish Caudal Fin. Front. Physiol. 2017, 8, 671. [Google Scholar] [CrossRef]
- Rawls, J.F.; Frieda, M.R.; McAdow, A.R.; Gross, J.P.; Clayton, C.M.; Heyen, C.K.; Johnson, S.L. Coupled mutagenesis screens and genetic mapping in zebrafish. Genetics 2003, 163, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Lawson, N.D.; Weinstein, B.M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 2002, 248, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Thummel, R.; Kathryn Iovine, M. Using Morpholinos to Examine Gene Function During Fin Regeneration. Methods Mol. Biol. 2017, 1565, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Divari, S.; Berio, E.; Biolatti, B.; Cannizzo, F.T. Reference Gene Selection and Prednisolone Target Gene Expression in Adipose Tissues of Friesian Cattle. J. Agric. Food Chem. 2017, 65, 11140–11145. [Google Scholar] [CrossRef]
- Kuntz, M.; Mysiorek, C.; Petrault, O.; Boucau, M.C.; Aijjou, R.; Uzbekov, R.; Berezowski, V. Transient oxygen-glucose deprivation sensitizes brain capillary endothelial cells to rtPA at 4h of reoxygenation. Microvasc. Res. 2014, 91, 44–57. [Google Scholar] [CrossRef]
- Suarez-Arnedo, A.; Torres Figueroa, F.; Clavijo, C.; Arbelaez, P.; Cruz, J.C.; Munoz-Camargo, C. An image J plugin for the high throughput image analysis of in vitro scratch wound healing assays. PLoS ONE 2020, 15, e0232565. [Google Scholar] [CrossRef]
- Patton, W.A., 2nd; Granzow, C.A.; Getts, L.A.; Thomas, S.C.; Zotter, L.M.; Gunzel, K.A.; Lowe-Krentz, L.J. Identification of a heparin-binding protein using monoclonal antibodies that block heparin binding to porcine aortic endothelial cells. Biochem. J. 1995, 311 Pt 2, 461–469. [Google Scholar] [CrossRef]
- Givens, C.; Tzima, E. Endothelial Mechanosignaling: Does One Sensor Fit All? Antioxid. Redox Signal. 2016, 25, 373–388. [Google Scholar] [CrossRef]
- Lim, X.R.; Harraz, O.F. Mechanosensing by Vascular Endothelium. Annu. Rev. Physiol. 2024, 86, 71–97. [Google Scholar] [CrossRef]
- Gopal, S.; Multhaupt, H.A.B.; Pocock, R.; Couchman, J.R. Cell-extracellular matrix and cell-cell adhesion are linked by syndecan-4. Matrix Biol. 2017, 60–61, 57–69. [Google Scholar] [CrossRef]
- Lambert, J.; Makin, K.; Akbareian, S.; Johnson, R.; Alghamdi, A.A.A.; Robinson, S.D.; Edwards, D.R. ADAMTS-1 and syndecan-4 intersect in the regulation of cell migration and angiogenesis. J. Cell Sci. 2020, 133, jcs235762. [Google Scholar] [CrossRef] [PubMed]
- Malinova, T.S.; Angulo-Urarte, A.; Nuchel, J.; Tauber, M.; van der Stoel, M.M.; Janssen, V.; de Haan, A.; Groenen, A.G.; Tebbens, M.; Graupera, M.; et al. A junctional PACSIN2/EHD4/MICAL-L1 complex coordinates VE-cadherin trafficking for endothelial migration and angiogenesis. Nat. Commun. 2021, 12, 2610. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, E.; Sneh, T.; Moeendarbary, E.; Javanmardi, Y.; Efimova, N.; Yang, C.; Marino-Bravante, G.E.; Chen, X.; Escribano, J.; Spill, F.; et al. Feedback between mechanosensitive signaling and active forces governs endothelial junction integrity. Nat. Commun. 2022, 13, 7089. [Google Scholar] [CrossRef] [PubMed]
- Gulino-Debrac, D. Mechanotransduction at the basis of endothelial barrier function. Tissue Barriers 2013, 1, e24180. [Google Scholar] [CrossRef]
- Jin, Y.; Ding, Y.; Richards, M.; Kaakinen, M.; Giese, W.; Baumann, E.; Szymborska, A.; Rosa, A.; Nordling, S.; Schimmel, L.; et al. Tyrosine-protein kinase Yes controls endothelial junctional plasticity and barrier integrity by regulating VE-cadherin phosphorylation and endocytosis. Nat. Cardiovasc. Res. 2022, 1, 1156–1173. [Google Scholar] [CrossRef]
- Xiao, K.; Garner, J.; Buckley, K.M.; Vincent, P.A.; Chiasson, C.M.; Dejana, E.; Faundez, V.; Kowalczyk, A.P. p120-Catenin regulates clathrin-dependent endocytosis of VE-cadherin. Mol. Biol. Cell 2005, 16, 5141–5151. [Google Scholar] [CrossRef]
- Vestweber, D. Vascular Endothelial Protein Tyrosine Phosphatase Regulates Endothelial Function. Physiology 2021, 36, 84–93. [Google Scholar] [CrossRef]
- Juettner, V.V.; Kruse, K.; Dan, A.; Vu, V.H.; Khan, Y.; Le, J.; Leckband, D.; Komarova, Y.; Malik, A.B. VE-PTP stabilizes VE-cadherin junctions and the endothelial barrier via a phosphatase-independent mechanism. J. Cell Biol. 2019, 218, 1725–1742. [Google Scholar] [CrossRef]
- Best, D.; Sahlender, D.A.; Walther, N.; Peden, A.A.; Adams, I.R. Sdmg1 is a conserved transmembrane protein associated with germ cell sex determination and germline-soma interactions in mice. Development 2008, 135, 1415–1425. [Google Scholar] [CrossRef]
- Francis, C.R.; Kushner, E.J. Trafficking in blood vessel development. Angiogenesis 2022, 25, 291–305. [Google Scholar] [CrossRef]
- Yan, Z.; Wang, Z.G.; Segev, N.; Hu, S.; Minshall, R.D.; Dull, R.O.; Zhang, M.; Malik, A.B.; Hu, G. Rab11a Mediates Vascular Endothelial-Cadherin Recycling and Controls Endothelial Barrier Function. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Cantini, M.; Reboud, J.; Cooper, J.M.; Roca-Cusachs, P.; Salmeron-Sanchez, M. Molecular clutch drives cell response to surface viscosity. Proc. Natl. Acad. Sci. USA 2018, 115, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Ciobanasu, C.; Wang, H.; Henriot, V.; Mathieu, C.; Fente, A.; Csillag, S.; Vigouroux, C.; Faivre, B.; Le Clainche, C. Integrin-bound talin head inhibits actin filament barbed-end elongation. J. Biol. Chem. 2018, 293, 2586–2596. [Google Scholar] [CrossRef] [PubMed]
- Chronopoulos, A.; Thorpe, S.D.; Cortes, E.; Lachowski, D.; Rice, A.J.; Mykuliak, V.V.; Rog, T.; Lee, D.A.; Hytonen, V.P.; Del Rio Hernandez, A.E. Syndecan-4 tunes cell mechanics by activating the kindlin-integrin-RhoA pathway. Nat. Mater. 2020, 19, 669–678. [Google Scholar] [CrossRef]
- van der Flier, A.; Badu-Nkansah, K.; Whittaker, C.A.; Crowley, D.; Bronson, R.T.; Lacy-Hulbert, A.; Hynes, R.O. Endothelial alpha5 and alphav integrins cooperate in remodeling of the vasculature during development. Development 2010, 137, 2439–2449. [Google Scholar] [CrossRef]
- Zimmermann, P.; Meerschaert, K.; Reekmans, G.; Leenaerts, I.; Small, J.V.; Vandekerckhove, J.; David, G.; Gettemans, J. PIP(2)-PDZ domain binding controls the association of syntenin with the plasma membrane. Mol. Cell 2002, 9, 1215–1225. [Google Scholar] [CrossRef]
- Baratchi, S.; Knoerzer, M.; Khoshmanesh, K.; Mitchell, A.; McIntyre, P. Shear Stress Regulates TRPV4 Channel Clustering and Translocation from Adherens Junctions to the Basal Membrane. Sci. Rep. 2017, 7, 15942. [Google Scholar] [CrossRef]
- Vellino, S.; Oddou, C.; Rivier, P.; Boyault, C.; Hiriart-Bryant, E.; Kraut, A.; Martin, R.; Coute, Y.; Knolker, H.J.; Valverde, M.A.; et al. Cross-talk between the calcium channel TRPV4 and reactive oxygen species interlocks adhesive and degradative functions of invadosomes. J. Cell Biol. 2021, 220, e201910079. [Google Scholar] [CrossRef]
- Cappelli, H.C.; Kanugula, A.K.; Adapala, R.K.; Amin, V.; Sharma, P.; Midha, P.; Paruchuri, S.; Thodeti, C.K. Mechanosensitive TRPV4 channels stabilize VE-cadherin junctions to regulate tumor vascular integrity and metastasis. Cancer Lett. 2019, 442, 15–20. [Google Scholar] [CrossRef]
- Troyanovsky, R.B.; Sergeeva, A.P.; Indra, I.; Chen, C.S.; Kato, R.; Shapiro, L.; Honig, B.; Troyanovsky, S.M. Sorting of cadherin-catenin-associated proteins into individual clusters. Proc. Natl. Acad. Sci. USA 2021, 118, e2105550118. [Google Scholar] [CrossRef]
- Bosseboeuf, E.; Chikh, A.; Chaker, A.B.; Mitchell, T.P.; Vignaraja, D.; Rajendrakumar, R.; Khambata, R.S.; Nightingale, T.D.; Mason, J.C.; Randi, A.M.; et al. Neuropilin-1 interacts with VE-cadherin and TGFBR2 to stabilize adherens junctions and prevent activation of endothelium under flow. Sci. Signal. 2023, 16, eabo4863. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primaries | Host Organism | Company | Catalog Number | RRID |
|---|---|---|---|---|
| Sdc4 | Rabbit | BioVision | Cat# 3644-100 | AB_2183016 |
| Sdc4 | Mouse | Santa Cruz Biotechnology | Cat# sc-12766 | AB_628314 |
| TMEM184A | Rabbit | Thermo Fisher Scientific | Cat# PA5-96834 | AB_2808636 |
| TMEM184A | Rat | GenScript | protein G-purified peptide sequence from the N-terminal region of bovine TMEM184A (N-PAGPQMDHMGNSSQC) | |
| VE-cadherin | Goat | Santa Cruz Biotechnology | Cat# sc-6458 | AB_2077955 |
| VE-cadherin | Rabbit | Cell Signaling Technology | Cat# 2500 | AB_10839118 |
| Rab4 | Rabbit | Abcam | Cat# ab13252 | AB_2269374 |
| Rab11a | Rabbit | Cell Signaling Technology | Cat# 5589 | AB_10693925 |
| HA | Goat | Novus | Cat# NB600362 | AB_10124937 |
| β-Actin | Rabbit | Cell Signaling Technology | Cat# 4970 | AB_2223172 |
| Tubulin | Mouse | Abcam | Cat# 7291 | AB_2241126 |
| Secondaries | Host Species | Company | Catalog number | RRID |
| Alexa 647 anti-mouse | Donkey | Jackson ImmunoResearch Labs | Cat# 715-605-151 | AB_2340863 |
| Alexa 647 anti-Rabbit | Donkey | Jackson ImmunoResearch Labs | Cat# 711-605-152 | AB_2492288 |
| Alexa 647 anti-rabbit, FC specific | Goat | Jackson ImmunoResearch Labs | Cat# 111-605-046 | AB_2338076 |
| Alexa 488 anti-rat | Donkey | Jackson ImmunoResearch Labs | Cat# 712-545-153 | AB_2340684 |
| Alexa 488 anti-rat, F(ab’)2 specific | Donkey | Jackson ImmunoResearch Labs | Cat# 112-545-072 | AB_2338359 |
| Alexa 488 anti-rabbit | Donkey | Jackson ImmunoResearch Labs | Cat# 711-545-152 | AB_2313584 |
| Alexa 488 anti-goat | Donkey | Jackson ImmunoResearch Labs | Cat# 705-545-147 | AB_2336933 |
| CY3 anti-mouse | Donkey | Jackson ImmunoResearch Labs | Cat# 715-165-150 | AB_2340813 |
| TRITC anti-rabbit | Donkey | Jackson ImmunoResearch Labs | Cat# 711-025-152 | AB_2340588 |
| TRITC anti-goat | Donkey | Jackson ImmunoResearch Labs | Cat# 705-025-147 | AB_2340389 |
| Figure | Primary1 | Primary2 | Secondary1 | Secondary2 |
|---|---|---|---|---|
| Figure 1A,B | Sdc4 rb (1:100) | TMEM NTD rat (1:50) | Alexa 647 α-rb (1:200) | Alexa 488 α-rat (1:500) |
| Figure S1C | TMEM CTD rb (1:50) | TMEM NTD rat (1:50) | TRITC α-rb (1:200) | Alexa 488 α-rat (1:500) |
| Figure S2C | Sdc4 rb (1:100) | TMEM NTD rat (1:50) | Alexa 647 α-rb (1:200) | Alexa 488 α-rat (1:500) |
| Figure S2A,B | Sdc4 mo (1:50) | TMEM CTD rb (1:50) | Alexa 488 α-mo (1:200) | TRITC α-rb (1:200) |
| Figure 3 and Figure 4 | VE cad gt(1:200) | TMEM CTD (1:100) | TRITC α-gt (1:200) | Alexa 647 α-rb (1:200) |
| Figure S3 | VE cad gt (1:200) | TMEM CTD (1:100) | Alexa 488 α-gt (1:200) | Alexa 647 α-rb (1:200) |
| Figure WB | Primary1 | Primary2 | Secondary1 | Secondary2 |
|---|---|---|---|---|
| Figures 3C and 4D | VE-cad rb (1:1000) | β-actin rb (1:1000) | Alexa-488 α-rb (1:10,000) | Alexa-488 α-rat F(ab’)2 specific (1:10,000) |
| Figure 4C | TMEM CTD rb (1:500) | Tubulin mo (1:10,000) | Alexa-488 α-rb (1:10,000) | Alexa 647 α-mo (1:10,000) |
| Figure S1A,B | TMEM CTD rb (1:500) | Tubulin mo (1:10,000) | Alexa-488 α-rb (1:10,000) | CY3 α- mo (1:10,000) |
| Figure S1D | TMEM CTD rb (1:200) | Sdc4 mo (1:200) | Alexa 647 α-rb (1:10,000) | CY3 α- mo (1:10,000) |
| Figure S1E | HA gt (1:1000) | Tubulin mo (1:10,000) | Alexa-488 α-gt (1:2000) | CY3 α- mo (1:10,000) |
| Figure S2D | Sdc4 rb (1:500) | TMEM NTD rat (1:200) | Alexa 647 α-rb FC site Specific (1:10,000) | Alexa-488 α-rat F(ab’)2 specific (1:10,000) |
| Figure S3A | VE-cad goat (1:5000) | Tubulin mo (1:10,000) | Alexa-488 α-goat (1:10,000) | Alexa 647 α-mo (1:10,000) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altenburg, L.M.; Wang, S.H.; Ciabattoni, G.O.; Kennedy, A.; O’Toole, R.L.; Farwell, S.L.N.; Iovine, M.K.; Lowe-Krentz, L.J. Transmembrane Protein-184A Interacts with Syndecan-4 and Rab GTPases and Is Required to Maintain VE-Cadherin Levels. Cells 2025, 14, 833. https://doi.org/10.3390/cells14110833
Altenburg LM, Wang SH, Ciabattoni GO, Kennedy A, O’Toole RL, Farwell SLN, Iovine MK, Lowe-Krentz LJ. Transmembrane Protein-184A Interacts with Syndecan-4 and Rab GTPases and Is Required to Maintain VE-Cadherin Levels. Cells. 2025; 14(11):833. https://doi.org/10.3390/cells14110833
Chicago/Turabian StyleAltenburg, Leanna M., Stephanie H. Wang, Grace O. Ciabattoni, Amelia Kennedy, Rachel L. O’Toole, Sara L. N. Farwell, M. Kathryn Iovine, and Linda J. Lowe-Krentz. 2025. "Transmembrane Protein-184A Interacts with Syndecan-4 and Rab GTPases and Is Required to Maintain VE-Cadherin Levels" Cells 14, no. 11: 833. https://doi.org/10.3390/cells14110833
APA StyleAltenburg, L. M., Wang, S. H., Ciabattoni, G. O., Kennedy, A., O’Toole, R. L., Farwell, S. L. N., Iovine, M. K., & Lowe-Krentz, L. J. (2025). Transmembrane Protein-184A Interacts with Syndecan-4 and Rab GTPases and Is Required to Maintain VE-Cadherin Levels. Cells, 14(11), 833. https://doi.org/10.3390/cells14110833