
Expression Profiling and Function Analysis Identified New Cumulus Cells-Expressed Genes and miRNAs Predictive of Oocyte Developmental Potential

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
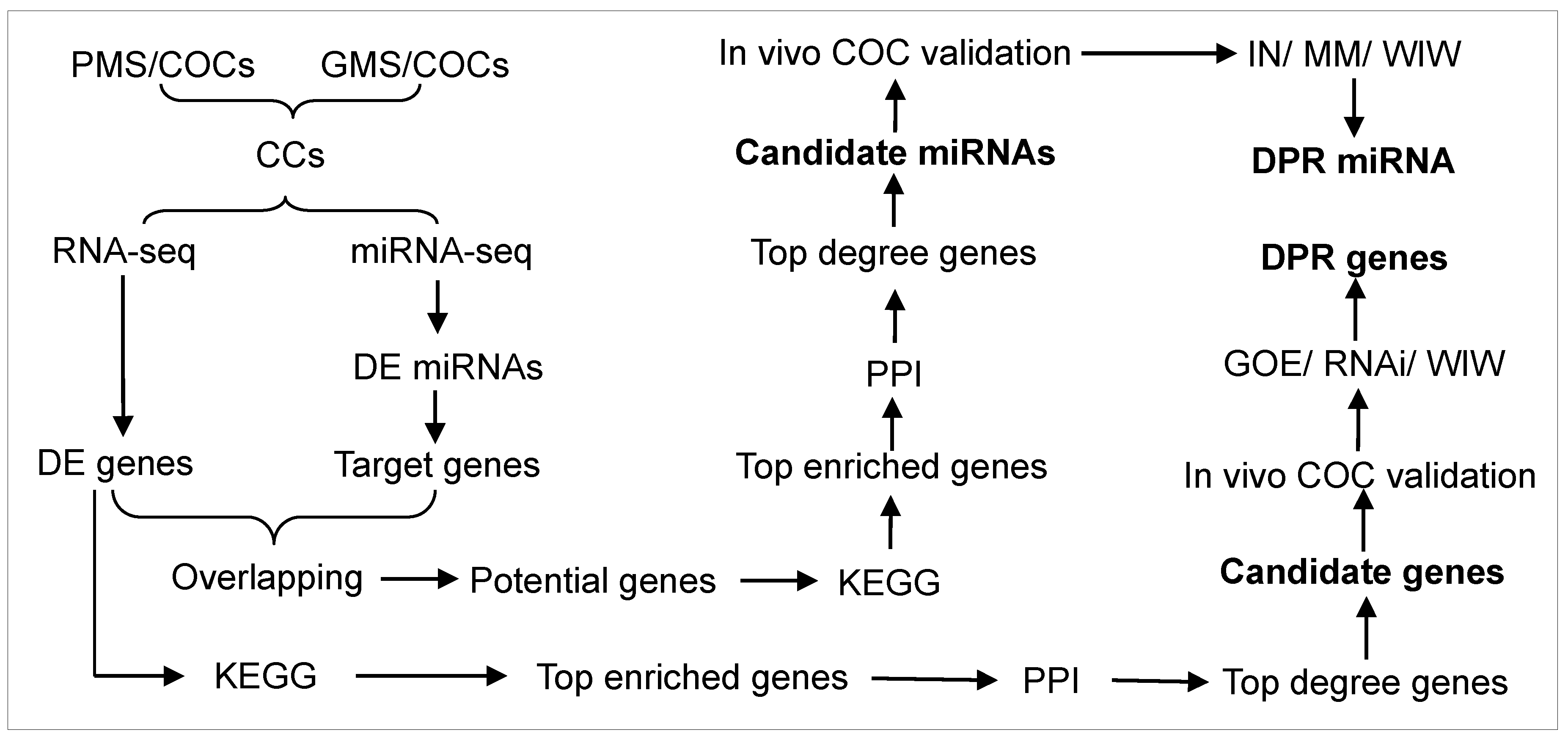
2.1. Experimental Design
2.2. Oocyte Collection
2.3. Oocyte In Vitro Maturation
2.4. Oocyte Activation and Embryo Culture
2.5. Glutathione Measurement
2.6. RT-qPCR Measurement of mRNA
2.7. RT-qPCR Measurement of miRNA
2.8. Measurement of Mitochondrial Membrane Potential (MMP)
2.9. Measurement for Cumulus Expansion Area
2.10. Flow Cytometry Detection for Apoptosis of CCs
2.11. Illumina HiSeq Sequencing of mRNA and miRNA in CCs
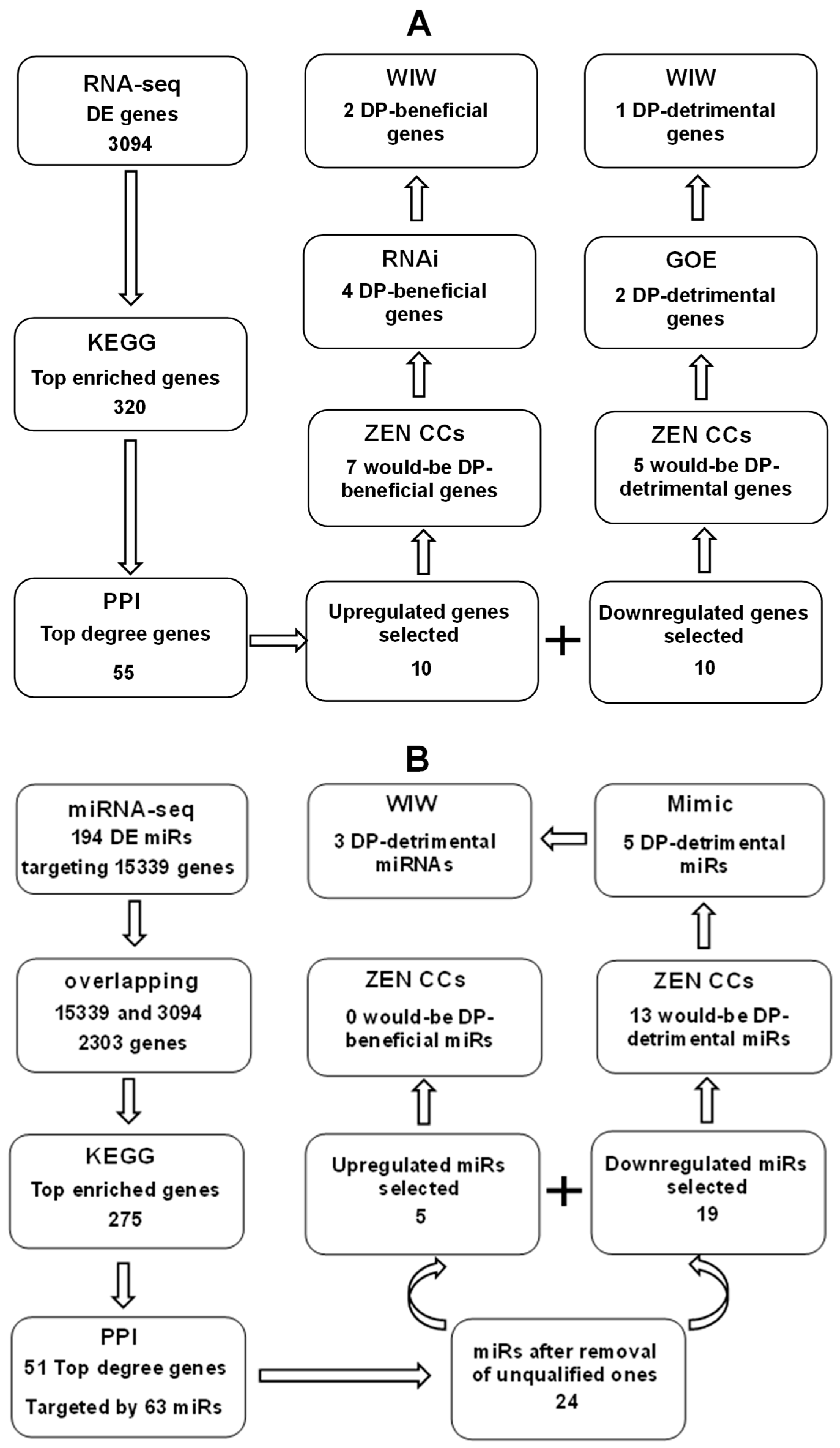
2.12. Bioinformatic Analysis
2.13. Transfection of COCs with siRNA or Plasmids
2.14. Transfection of COCs with miRNA Mimics
2.15. Activation and Embryo Culture in the WIW System
2.16. Data Analysis
3. Results
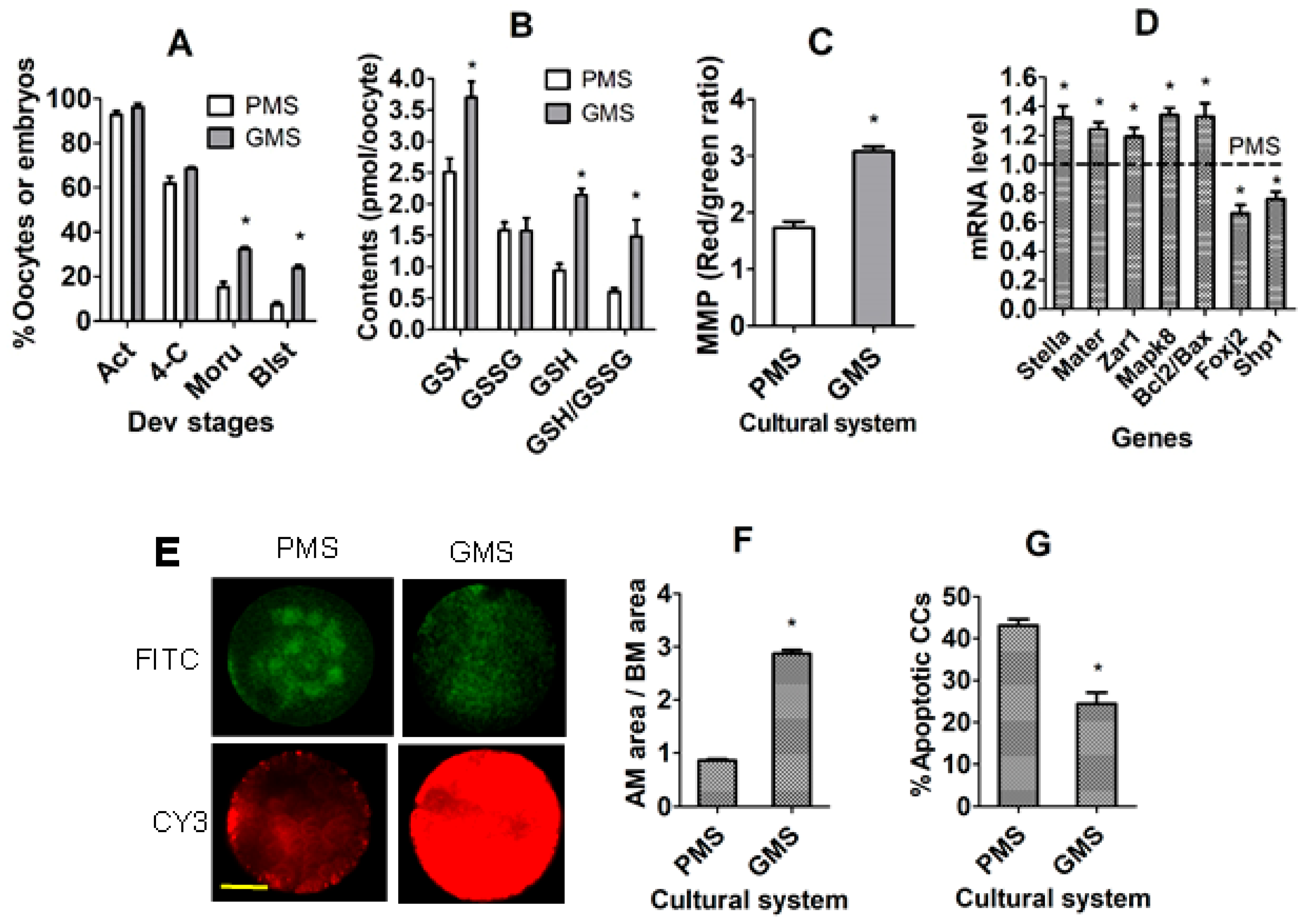
3.1. The Establishment of In Vitro CC Models Using In Vitro Matured COCs with Significantly Different DP
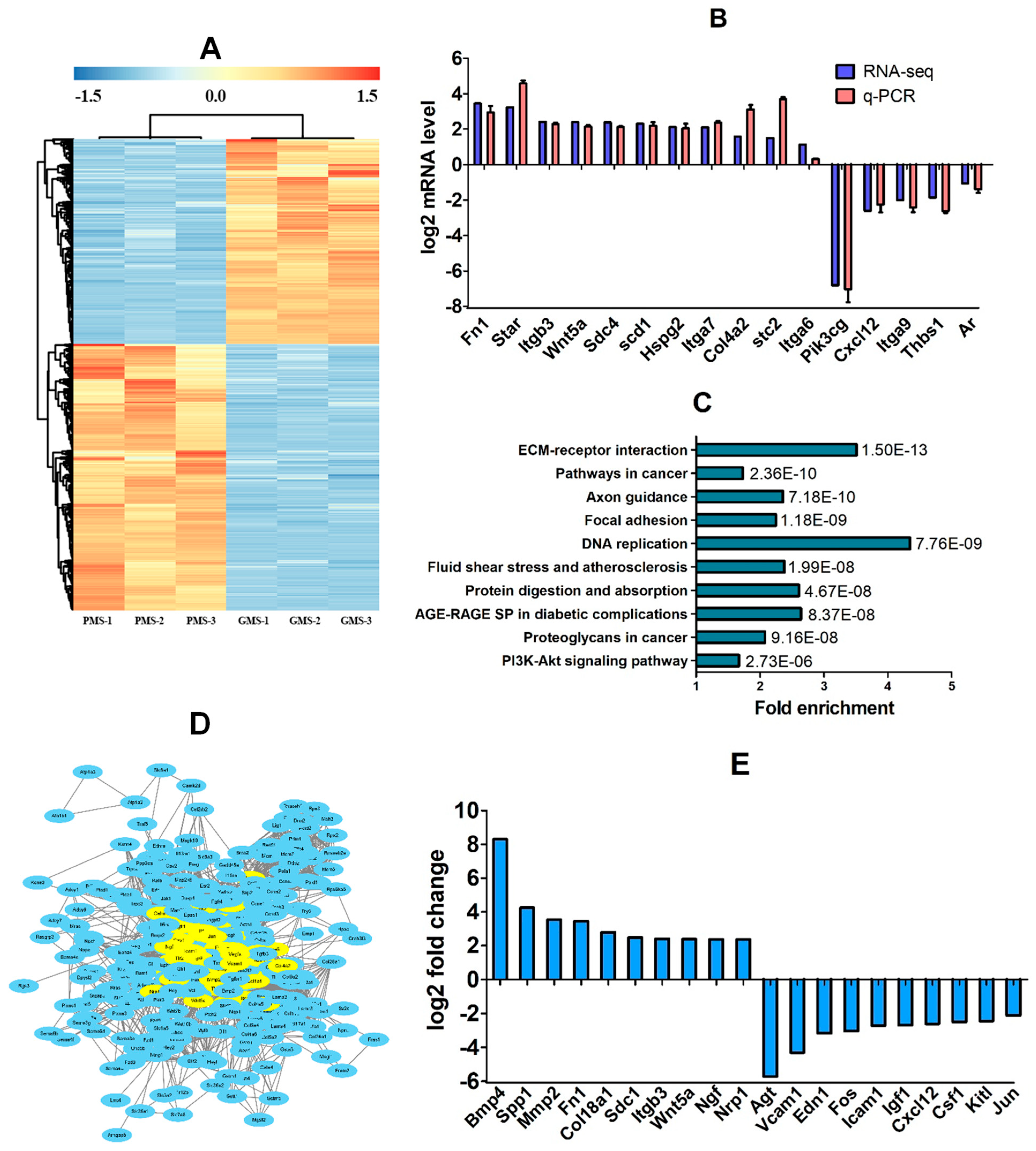
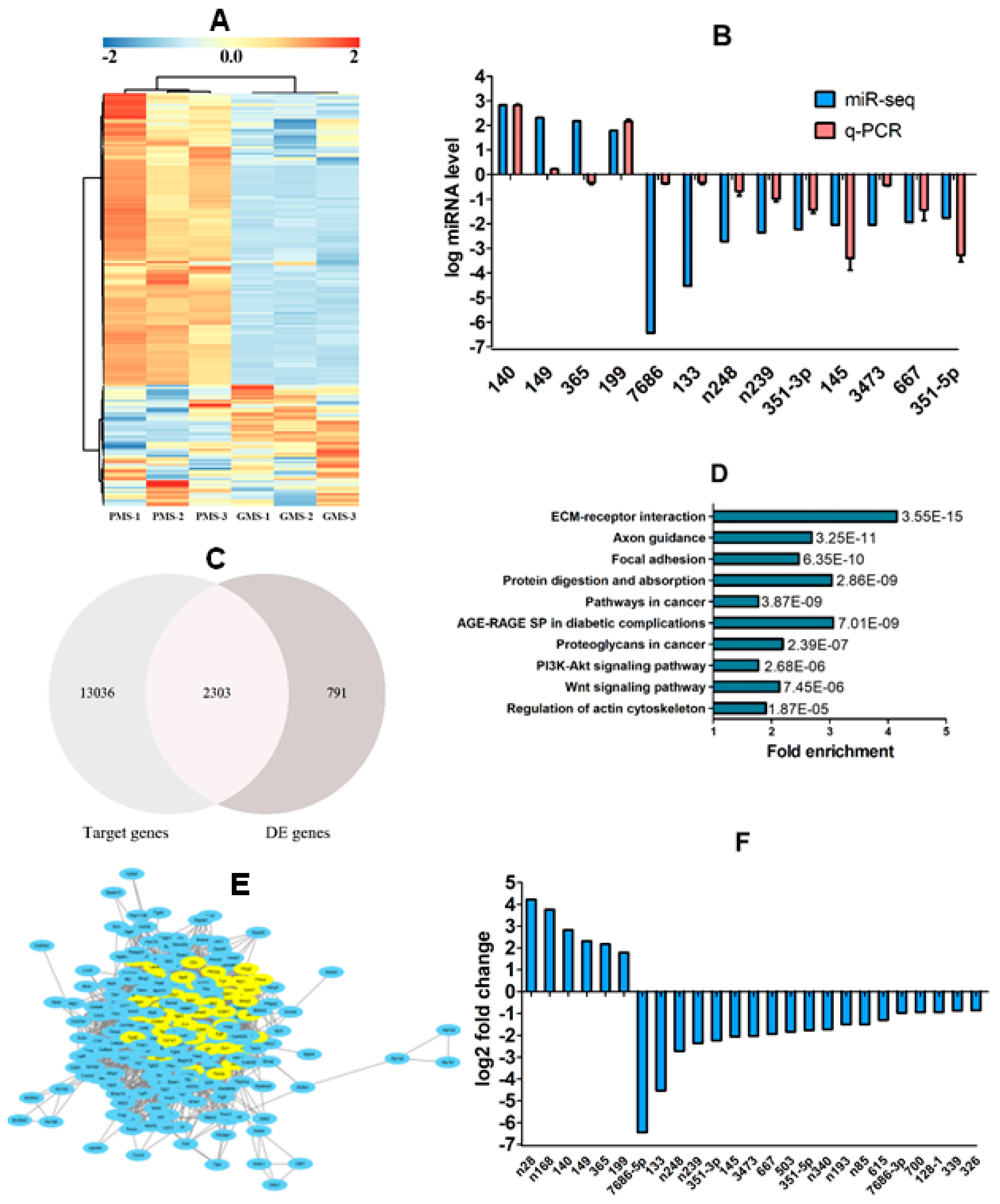
3.2. Identification of DE Genes in CCs Between PMS- and GMS-Matured Oocytes
3.3. KEGG and PPI Analyses on DE Genes
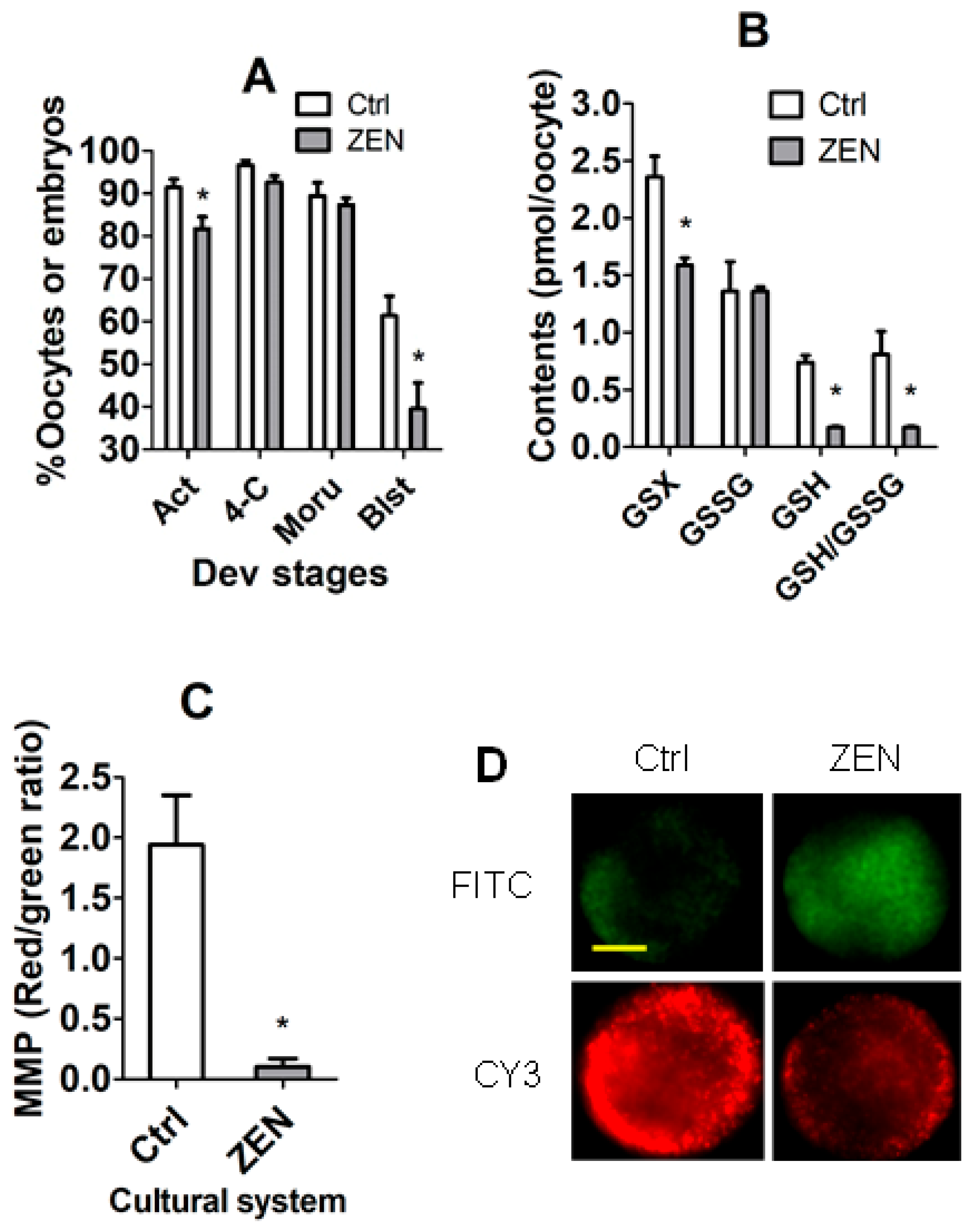
3.4. The Establishment of In Vivo CC Models Using In Vivo Matured COCs with Significantly Different DP
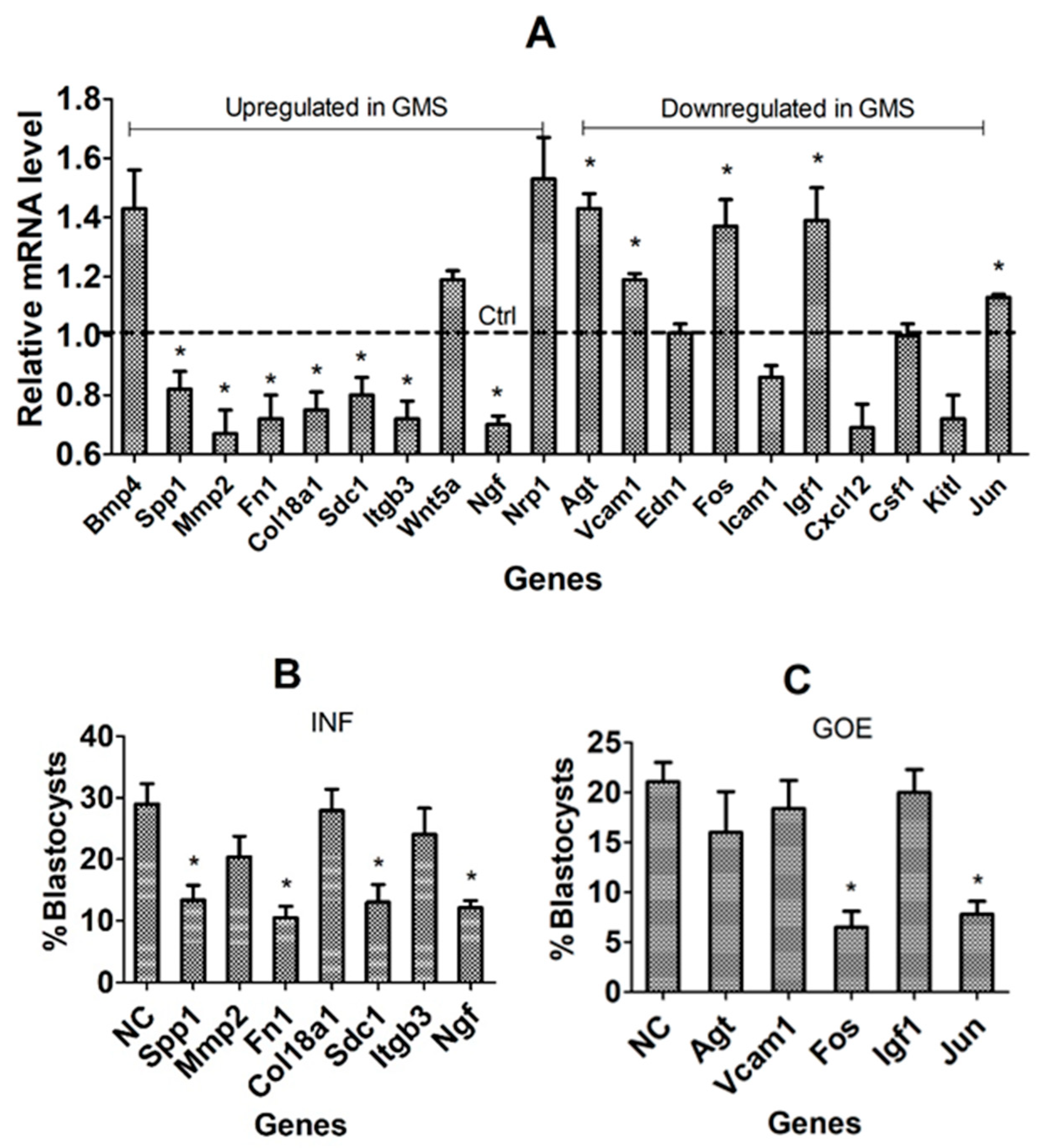
3.5. Validation of Candidate Genes Using In Vivo COCs and by RNAi or Gene Overexpression (GOE)
3.6. Identification of DE miRNAs in CCs Between PMS- and GMS-Matured Oocytes
3.7. Bioinformatic Analysis on the DE miRNAs
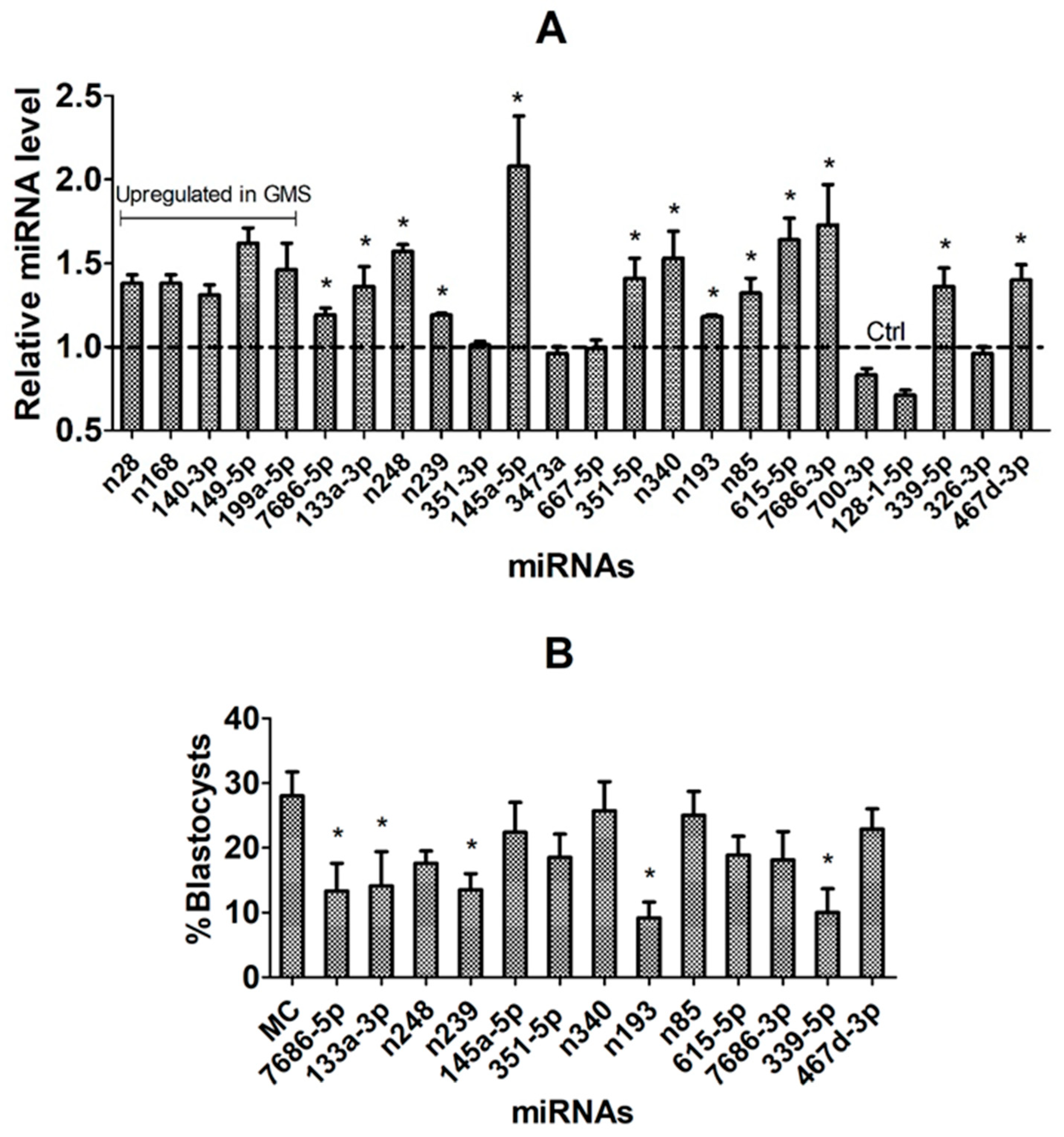
3.8. Validation of Candidate miRNAs Using In Vivo COC Models and by miR Mimics
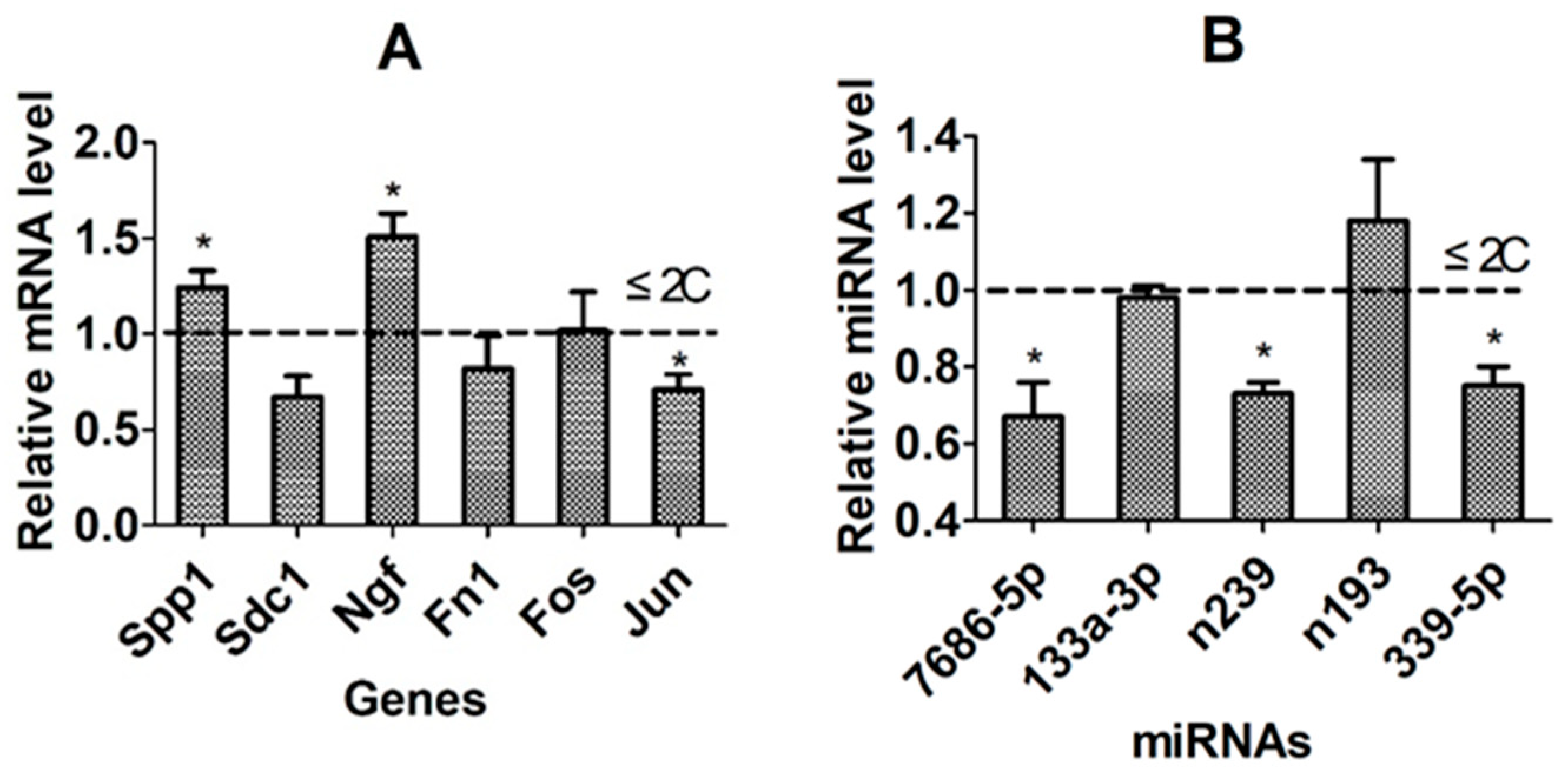
3.9. Further Validation of Candidate Genes/miRNAs Using the WIW System of Activation and Embryo Culture
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Trounson, A.; Wood, C.; Kausche, A. In Vitro Maturation and the Fertilization and Developmental Competence of Oocytes Recovered from Untreated Polycystic Ovarian Patients. Fertil. Steril. 1994, 62, 353–362. [Google Scholar] [CrossRef]
- Barnes, F.L.; Kausche, A.; Tiglias, J.; Wood, C.; Wilton, L.; Trounson, A. Production of Embryos from in Vitro-Matured Primary Human Oocytes. Fertil. Steril. 1996, 65, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Cha, K.Y.; Han, S.Y.; Chung, H.M.; Choi, D.H.; Lim, J.M.; Lee, W.S.; Ko, J.J.; Yoon, T.K. Pregnancies and Deliveries after in Vitro Maturation Culture Followed by in Vitro Fertilization and Embryo Transfer without Stimulation in Women with Polycystic Ovary Syndrome. Fertil. Steril. 2000, 73, 978–983. [Google Scholar] [CrossRef]
- Aguila, L.; Treulen, F.; Therrien, J.; Felmer, R.; Valdivia, M.; Smith, L.C. Oocyte Selection for in Vitro Embryo Production in Bovine Species: Noninvasive Approaches for New Challenges of Oocyte Competence. Animals 2020, 10, 2196. [Google Scholar] [CrossRef]
- Assou, S.; Haouzi, D.; De Vos, J.; Hamamah, S. Human Cumulus Cells as Biomarkers for Embryo and Pregnancy Outcomes. Mol. Hum. Reprod. 2010, 16, 531–538. [Google Scholar] [CrossRef]
- Racowsky, C.; Needleman, D.J. Cumulus Cell Gene Expression as a Potential Biomarker for Oocyte Quality. Fertil. Steril. 2018, 109, 438–439. [Google Scholar] [CrossRef]
- Bosco, L.; Chiarelli, R.; Roccheri, M.C.; Matranga, D.; Ruvolo, G. Relationship between Apoptosis and Survival Molecules in Human Cumulus Cells as Markers of Oocyte Competence. Zygote 2017, 25, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Moulavi, F.; Hosseini, S.M. Diverse Patterns of Cumulus Cell Expansion During In vitro Maturation Reveal Heterogeneous Cellular and Molecular Features of Oocyte Competence in Dromedary Camel. Theriogenology 2018, 119, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, R.B.; Ritter, L.J.; Armstrong, D.T. Oocyte-Somatic Cell Interactions During Follicle Development in Mammals. Anim. Reprod. Sci. 2004, 82–83, 431–446. [Google Scholar] [CrossRef]
- Shepel, E.A.; Voznesenskaya, T.V.; Blashkiv, T.V.; Yanchii, R.I. umulus Cell Genes as Potential Biomarkers of Oocyte and Embryo Developmental Competence. Fiziolohichnyi Zhurnal 2016, 62, 107–113. [Google Scholar] [CrossRef]
- Uyar, A.; Torrealday, S.; Seli, E. Cumulus and Granulosa Cell Markers of Oocyte and Embryo Quality. Fertil. Steril. 2013, 99, 979–997. [Google Scholar] [CrossRef] [PubMed]
- Assidi, M.; Dufort, I.; Ali, A.; Hamel, M.; Algriany, O.; Dielemann, S.; Sirard, M.A. Identification of Potential Markers of Oocyte Competence Expressed in Bovine Cumulus Cells Matured with Folli-cle-Stimulating Hormone and/or Phorbol Myristate Acetate in Vitro. Biol. Reprod. 2008, 79, 209–222. [Google Scholar] [CrossRef]
- Bunel, A.; Jorssen, E.P.; Merckx, E.; Leroy, J.L.; Bols, P.E.; Sirard, M.A. Individual Bovine In vitro Embryo Production and Cumulus Cell Transcriptomic Analysis to Distinguish Cumulus-Oocyte Complexes with High or Low Developmental Potential. Theriogenology 2015, 83, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Moro, Á.; González-Brusi, L.; Lamas-Toranzo, I.; O’Callaghan, E.; Esteve-Codina, A.; Lonergan, P.; Bermejo-Álvarez, P. Rna-Sequencing Reveals Genes Linked with Oocyte Developmental Potential in Bovine Cumulus Cells. Mol. Reprod. Dev. 2022, 89, 399–412. [Google Scholar] [CrossRef]
- Burnik Papler, T.; Vrtacnik Bokal, E.; Lovrecic, L.; Kopitar, A.N.; Maver, A. No Specific Gene Expression Signature in Human Granulosa and Cumulus Cells for Prediction of Oocyte Fertilisation and Embryo Implantation. PLoS ONE 2015, 10, e0115865. [Google Scholar] [CrossRef] [PubMed]
- Green, K.A.; Franasiak, J.M.; Werner, M.D.; Tao, X.; Landis, J.N.; Scott, R.T., Jr.; Treff, N.R. Cumulus Cell Transcriptome Profiling Is Not Predictive of Live Birth after In vitro Fertilization: A Paired Analysis of Euploid Sibling Blastocysts. Fertil. Steril. 2018, 109, 460–466. [Google Scholar] [CrossRef]
- Martínez-Moro, Á.; González-Brusi, L.; Lamas-Toranzo, I.; González-Dosal, P.; Rodríguez-Juárez, F.; Bermejo-Álvarez, P. The Human Cumulus Cell Transcriptome Provides Poor Predictive Value for Embryo Transfer Outcome. Reprod. BioMed. Online 2023, 46, 783–791. [Google Scholar] [CrossRef]
- Kumar, S.; Vijayan, M.; Bhatti, J.S.; Reddy, P.H. Micrornas as Peripheral Biomarkers in Aging and Age-Related Diseases. Prog. Mol. Biol. Transl. Sci. 2017, 146, 47–94. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Ochiya, T. Circulating Micrornas and Extracellular Vesicles as Potential Cancer Biomarkers: A Systematic Review. Int. J. Clin. Oncol. 2017, 22, 413–420. [Google Scholar] [CrossRef]
- Rosenbluth, E.M.; Shelton, D.N.; Wells, L.M.; Sparks, A.E.; Van Voorhis, B.J. Human Embryos Secrete Micrornas into Culture Media--a Potential Biomarker for Implantation. Fertil. Steril. 2014, 101, 1493–1500. [Google Scholar] [CrossRef]
- Capalbo, A.; Ubaldi, F.M.; Cimadomo, D.; Noli, L.; Khalaf, Y.; Farcomeni, A.; Ilic, D.; Rienzi, L. Micrornas in Spent Blastocyst Culture Medium Are Derived from Trophectoderm Cells and Can Be Explored for Human Embryo Reproductive Competence Assessment. Fertil. Steril. 2016, 105, e221–e223. [Google Scholar] [CrossRef] [PubMed]
- Assou, S.; Al-edani, T.; Haouzi, D.; Philippe, N.; Lecellier, C.H.; Piquemal, D.; Commes, T.; Aït-Ahmed, O.; Dechaud, H.; Hamamah, S. Micrornas: New Candidates for the Regulation of the Human Cumulus-Oocyte Complex. Hum. Reprod. 2013, 28, 3038–3049. [Google Scholar] [CrossRef] [PubMed]
- Abd El Naby, W.S.; Hagos, T.H.; Hossain, M.M.; Salilew-Wondim, D.; Gad, A.Y.; Rings, F.; Cinar, M.U.; Tholen, E.; Looft, C.; Schellander, K.; et al. Expression Analysis of Regulatory Micrornas in Bovine Cumulus Oocyte Complex and Preimplantation Embryos. Zygote 2013, 21, 31–51. [Google Scholar] [CrossRef]
- Yao, G.; Liang, M.; Liang, N.; Yin, M.; Lü, M.; Lian, J.; Wang, Y.; Sun, F. Microrna-224 Is Involved in the Regulation of Mouse Cumulus Expansion by Targeting Ptx3. Mol. Cell. Endocrinol. 2014, 382, 244–253. [Google Scholar] [CrossRef]
- Han, X.; Zhang, M.; Yuan, H.J.; Wang, G.L.; Zhao, X.Y.; Li, Z.B.; Gong, S.; Tan, J.H. Expression Profiling and Function Analysis Identified New Micrornas Regulating Cumulus Expansion and Apoptosis in Cu-mulus Cells. Mol. Hum. Reprod. 2022, 28, gaab070. [Google Scholar] [CrossRef]
- Pan, B.; Toms, D.; Shen, W.; Li, J. Microrna-378 Regulates Oocyte Maturation Via the Suppression of Aromatase in Porcine Cumulus Cells. Am. J. Physiol.-Endocrinol. Metab. 2015, 308, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.; Sheng, Y.; Wang, Z. Microrna-224 Delays Oocyte Maturation through Targeting Ptx3 in Cumulus Cells. Mech. Dev. 2017, 143, 20–25. [Google Scholar] [CrossRef]
- Han, X.; Xue, R.; Yuan, H.J.; Wang, T.Y.; Lin, J.; Zhang, J.; Liang, B.; Tan, J.H. Microrna-21 Plays a Pivotal Role in the Oocyte-Secreted Factor-Induced Suppression of Cumulus Cell Apoptosis. Biol. Reprod. 2017, 96, 1167–1180. [Google Scholar] [CrossRef]
- Liu, C.; Yuan, B.; Chen, H.; Xu, M.; Sun, X.; Xu, J.; Gao, Y.; Chen, C.; Jiang, H.; Zhang, J. Effects of Mir-375-Bmpr2 as a Key Factor Downstream of Bmp15/Gdf9 on the Smad1/5/8 and Smad2/3 Signaling Pathways. Cell. Physiol. Biochem. 2018, 46, 213–225. [Google Scholar] [CrossRef]
- Cui, L.; Fang, L.; Mao, X.; Chang, H.M.; Leung, P.C.K.; Ye, Y. Gdnf-Induced Downregulation of Mir-145-5p Enhances Human Oocyte Maturation and Cumulus Cell Viability. J. Clin. Endocrinol. Metab. 2018, 103, 2510–2521. [Google Scholar] [CrossRef]
- Sinha, P.B.; Tesfaye, D.; Rings, F.; Hossien, M.; Hoelker, M.; Held, E.; Neuhoff, C.; Tholen, E.; Schellander, K.; Salilew-Wondim, D. Microrna-130b Is Involved in Bovine Granulosa and Cumulus Cells Function, Oocyte Maturation and Blastocyst Formation. J. Ovarian Res. 2017, 10, 37. [Google Scholar] [CrossRef]
- Sabry, R.; Saleh, A.C.; Stalker, L.; LaMarre, J.; Favetta, L.A. Effects of Bisphenol a and Bisphenol S on Microrna Expression During Bovine (Bos Taurus) Oocyte Maturation and Early Embryo Development. Reprod. Toxicol. 2021, 99, 96–108. [Google Scholar] [CrossRef]
- Uhde, K.; van Tol, H.T.A.; Stout, T.A.E.; Roelen, B.A.J. Microrna Expression in Bovine Cumulus Cells in Relation to Oocyte Quality. Non-Coding RNA 2017, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wu, J.S.; Han, X.; Ma, R.J.; Xu, J.L.; Xu, M.T.; Yuan, H.J.; Luo, M.J.; Tan, J.H. Expression Profiling and Function Analysis Identified New Genes Regulating Cumulus Expansion and Cumulus Cell Apoptosis in Mouse Oocytes. Reproduction 2024, 168, 3. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Sui, H.S.; Lan, G.C.; Liu, N.; Wang, J.Z.; Tan, J.H. Coculture with Cumulus Cells Improves Maturation of Mouse Oocytes Denuded of the Cumulus Oophorus: Observations of Nuclear and Cytoplasmic Events. Fertil. Steril. 2008, 90, 2376–2388. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.Z.; Cheng, H.; Zhang, J.; Gong, S.; Tian, X.D.; Pan, C.J.; Luo, M.J.; Tan, J.H. Invivo Zearalenone Exposure Dose-Dependently Compromises Mouse Oocyte Competence by Impairing Chromatin Configuration and Gene Transcription. Reprod. Fertil. Dev. 2021, 33, 229–238. [Google Scholar] [CrossRef]
- Han, Z.B.; Lan, G.C.; Wu, Y.G.; Han, D.; Feng, W.G.; Wang, J.Z.; Tan, J.H. Interactive Effects of Granulosa Cell Apoptosis, Follicle Size, Cumulus-Oocyte Complex Morphology, and Cumulus Expansion on the Developmental Competence of Goat Oocytes: A Study Using the Well-in-Drop Culture System. Reproduction 2006, 132, 749–758. [Google Scholar] [CrossRef]
- Wu, J.S.; Gong, S.; Zhang, M.; Ma, R.J.; Wang, H.L.; Luo, M.J.; He, N.; Tan, J.H. Role and Action Mechanisms of Mir-149 and Mir-31 in Regulating Function of Pig Cumulus Cells and Oocytes. Theriogenology 2024, 220, 84–95. [Google Scholar] [CrossRef]
- Park, J.E.; Kim, M.; Lee, J.; Chai, C.; Lee, S.T.; Lee, E. In vitro Maturation on an Agarose Matrix Improves the Developmental Competence of Porcine Oocytes. Theriogenology 2020, 157, 7–17. [Google Scholar] [CrossRef]
- Xu, Y.; Zhou, T.; Shao, L.; Zhang, B.; Liu, K.; Gao, C.; Gao, L.; Liu, J.; Cui, Y.; Chian, R.C. Gene Expression Profiles in Mouse Cumulus Cells Derived from In vitro Matured Oocytes with and without Blastocyst For-mation. Gene Expr. Patterns 2017, 25–26, 46–58. [Google Scholar] [CrossRef]
- Chermuła, B.; Kranc, W.; Jopek, K.; Budna-Tukan, J.; Hutchings, G.; Dompe, C.; Moncrieff, L.; Janowicz, K.; Józkowiak, M.; Jeseta, M.; et al. Human Cumulus Cells in Long-Term in Vitro Culture Reflect Differential Expression Profile of Genes Responsible for Planned Cell Death and Aging-a Study of New Molecular Markers. Cells 2020, 9, 1265. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, J.; Wen, H.; Feng, Y.; Zhang, X.; Xiang, H.; Cao, Y.; Tong, X.; Ji, Y.; Xue, Z. Analyzing the Transcriptome Profile of Human Cumulus Cells Related to Embryo Quality Via Rna Sequencing. BioMed Res. Int. 2018, 24, 9846274. [Google Scholar] [CrossRef]
- Thomas, R.E.; Armstrong, D.T.; Gilchrist, R.B. Bovine Cumulus Cell-Oocyte Gap Junctional Communication During in Vitro Maturation in Response to Manipulation of Cell-Specific Cyclic Adenosine 3′,5′-Monophosophate Levels. Biol. Reprod. 2004, 70, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.L.; Salustri, A. Extracellular Matrix of the Cumulus-Oocyte Complex. Semin. Reprod. Med. 2006, 24, 217–227. [Google Scholar] [CrossRef]
- Gilmore, A.P.; Owens, T.W.; Foster, F.M.; Lindsay, J. How Adhesion Signals Reach a Mitochondrial Conclusion--Ecm Regulation of Apoptosis. Curr. Opin. Cell Biol. 2009, 21, 654–661. [Google Scholar] [CrossRef]
- Chen, X.; Gong, L.; Li, Q.; Hu, J.; Liu, X.; Wang, Y.; Bai, J.; Ran, X.; Wu, J.; Ge, Q.; et al. The Appropriate Remodeling of Extracellular Matrix Is the Key Molecular Signature in Subcutaneous Adipose Tissue Following Roux-En-Y Gastric Bypass. Life Sci. 2019, 218, 265–273. [Google Scholar] [CrossRef]
- He, Y.; Liu, Y.; Gong, J.; Liu, C.; Zhang, H.; Wu, H. Identification of Key Pathways and Candidate Genes in Pancreatic Ductal Adenocarcinoma Using Bioinformatics Analysis. Oncol. Lett. 2019, 17, 3751–3764. [Google Scholar] [CrossRef]
- Masu, M. Proteoglycans and Axon Guidance: A New Relationship between Old Partners. J. Neurochem. 2016, 139, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Saied-Santiago, K.; Bülow, H.E. Diverse Roles for Glycosaminoglycans in Neural Patterning. Dev. Dyn. 2018, 247, 54–74. [Google Scholar] [CrossRef]
- Nagyova, E. The Biological Role of Hyaluronan-Rich Oocyte-Cumulus Extracellular Matrix in Female Reproduction. Int. J. Mol. Sci. 2018, 19, 283. [Google Scholar] [CrossRef]
- Kaneko, T.; Saito, H.; Toya, M.; Satio, T.; Nakahara, K.; Hiroi, M. Hyaluronic Acid Inhibits Apoptosis in Granulosa Cells Via Cd44. J. Assist. Reprod. Genet. 2000, 17, 162–167. [Google Scholar] [CrossRef]
- Arakawa, H. P53, Apoptosis and Axon-Guidance Molecules. Cell Death Differ. 2005, 12, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A Model for P53-Induced Apoptosis. Nature 1997, 389, 300–305. [Google Scholar] [CrossRef]
- Kitasaka, H.; Kawai, T.; Hoque, S.A.M.; Umehara, T.; Fujita, Y.; Shimada, M. Inductions of Granulosa Cell Luteinization and Cumulus Expansion Are Dependent on the Fibronectin-Integrin Pathway During Ovulation Process in Mice. PLoS ONE 2018, 13, e0192458. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Gross, S.; Kaur, A.S.; Wilson, R.I.; Xu, L.H.; Yang, X.H.; Cance, W.G. Simultaneous Inhibition of Focal Adhesion Kinase and Src Enhances Detachment and Apoptosis in Colon Cancer Cell Lines. Mol. Cancer Res. 2003, 1, 755–764. [Google Scholar] [PubMed]
- Peng, N.; Meng, N.; Wang, S.; Zhao, F.; Zhao, J.; Su, L.; Zhang, S.; Zhang, Y.; Zhao, B.; Miao, J. An Activator of Mtor Inhibits Oxldl-Induced Autophagy and Apoptosis in Vascular Endothelial Cells and Restricts Atheroscle-rosis in Apolipoprotein E⁻/⁻ Mice. Sci. Rep. 2014, 4, 5519. [Google Scholar] [CrossRef]
- Pandurangan, A.K.; Divya, T.; Kumar, K.; Dineshbabu, V.; Velavan, B.; Sudhandiran, G. Colorectal Carcinogenesis: Insights into the Cell Death and Signal Transduction Pathways: A Review. World J. Gastrointest. Oncol. 2018, 10, 244–259. [Google Scholar] [CrossRef]
- Dragovic, R.A.; Ritter, L.J.; Schulz, S.J.; Amato, F.; Armstrong, D.T.; Gilchrist, R.B. Role of Oocyte-Secreted Growth Differentiation Factor 9 in the Regulation of Mouse Cumulus Expansion. Endocrinology 2005, 146, 2798–2806. [Google Scholar] [CrossRef]
- Abdelkader, N.F.; Ibrahim, S.M.; Moustafa, P.E.; Elbaset, M.A. Inosine Mitigated Diabetic Peripheral Neuropathy Via Modulating Glo1/Ages/Rage/Nf-Κb/Nrf2 and Tgf-Β/Pkc/Trpv1 Signaling Pathways. Biomed. Pharmacother. 2022, 145, 112395. [Google Scholar] [CrossRef]
- Park, H.J.; Chae, S.K.; Kim, J.W.; Yang, S.G.; Jung, J.M.; Kim, M.J.; Wee, G.; Lee, D.S.; Kim, S.U.; Koo, D.B. Ganglioside Gm3 Induces Cumulus Cell Apoptosis through Inhibition of Epidermal Growth Factor Receptor-Mediated Pi3k/Akt Signaling Pathways During in Vitro Maturation of Pig Oocytes. Mol. Reprod. Dev. 2017, 84, 702–711. [Google Scholar] [CrossRef]
- Jiao, Y.; Zhu, S.; Li, J.; Jam Zaheer, A.; Li, M.; Huang, B. Ps48 Promotes in Vitro Maturation and Developmental Competence of Porcine Oocytes through Activating Pi3k/Akt Signal-ling Pathway. Reprod. Domest. Anim. 2020, 55, 1678–1687. [Google Scholar] [CrossRef] [PubMed]
- Thoumine, O.; Nerem, R.M.; Girard, P.R. Changes in Organization and Composition of the Extracellular Matrix Underlying Cultured Endothelial Cells Exposed to Laminar Steady Shear Stress. Lab. Investig. 1995, 73, 565–576. [Google Scholar]
- Cheng, H.; Zhong, W.; Wang, L.; Zhang, Q.; Ma, X.; Wang, Y.; Wang, S.; He, C.; Wei, Q.; Fu, C. Effects of Shear Stress on Vascular Endothelial Functions in Atherosclerosis and Potential Therapeutic Approaches. Biomed. Pharmacother. 2023, 158, 114198. [Google Scholar] [CrossRef] [PubMed]
- Munakata, Y.; Kawahara-Miki, R.; Shiratsuki, S.; Tasaki, H.; Itami, N.; Shirasuna, K.; Kuwayama, T.; Iwata, H. Gene Expression Patterns in Granulosa Cells and Oocytes at Various Stages of Follicle Development as Well as in in Vitro Grown Oocyte-and-Granulosa Cell Complexes. J. Reprod. Dev. 2016, 62, 359–366. [Google Scholar] [CrossRef]
- Li, Z.; Liu, Y.; Ma, T.; Lv, C.; Li, Y.; Duan, H.; Zhao, X.; Wang, J.; Zhang, Y. Smart-Seq2 Technology Reveals a Novel Mechanism That Zearalenone Inhibits the in Vitro Maturation of Ovine Oocytes by Influencing Tnfaip6 Expression. Toxins 2023, 15, 617. [Google Scholar] [CrossRef] [PubMed]
- Chaves, R.N.; Alves, A.M.; Lima, L.F.; Matos, H.M.; Rodrigues, A.P.; Figueiredo, J.R. Role of Nerve Growth Factor (Ngf) and Its Receptors in Folliculogenesis. Zygote 2013, 21, 187–197. [Google Scholar] [CrossRef]
- Wang, D.H.; Ren, J.; Zhou, C.J.; Han, Z.; Wang, L.; Liang, C.G. Supplementation with Ctgf, Sdf1, Ngf, and Hgf Promotes Ovine In vitro Oocyte Maturation and Early Embryo Development. Domest. Anim. Endocrinol. 2018, 65, 38–48. [Google Scholar] [CrossRef]
- Sun, M.; Wang, X.; Bi, F.; Xiang, H.; Wang, N.; Gao, W.; Liu, Y.; Lv, Z.; Li, Y.; Huan, Y. Fibronectin 1 Supports Oocyte in Vitro Maturation in Pigs. Int. J. Biol. Macromol. 2024, 264, 130590. [Google Scholar] [CrossRef]
- Hassani, F.; Oryan, S.; Eftekhari-Yazdi, P.; Bazrgar, M.; Moini, A.; Nasiri, N.; Sharifi-Zarchi, A. Downregulation of Extracellular Matrix and Cell Adhesion Molecules in Cumulus Cells of Infertile Polycystic Ovary Syndrome Women with and without Insulin Resistance. Cell J. 2019, 21, 35–42. [Google Scholar] [CrossRef]
- Liu, C.; Li, S.; Li, Y.; Tian, J.; Sun, X.; Song, T.; Yan, G.; Ding, L.; Sun, H. Growth Hormone Ameliorates the Age-Associated Depletion of Ovarian Reserve and Decline of Oocyte Quality Via Inhibiting the Activation of Fos and Jun Signaling. Aging 2021, 13, 6765–6781. [Google Scholar] [CrossRef]
- Deb, G.K.; Dey, S.R.; Bang, J.I.; Lee, J.G.; Kong, I.K. 9-Cis Retinoic Acid Inhibits Cumulus Cell Apoptosis During the Maturation of Bovine Cumulus-Oocyte-Complexes. J. Anim. Sci. 2012, 90, 1798–1806. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bi, X.; Wu, X.; Chen, Z.; Cao, Y.; Zhao, G. Improving Vitrification Efficiency of Human in Vitro Matured Oocytes by the Addition of Lea Proteins. Hum. Reprod. 2024, 39, 1275–1290. [Google Scholar] [CrossRef] [PubMed]
- Buñay, J.; Larriba, E.; Moreno, R.D.; Del Mazo, J. Chronic Low-Dose Exposure to a Mixture of Environmental Endocrine Disruptors Induces Micrornas/Isomirs Deregulation in Mouse Concomitant with Intratesticular Estradiol Reduction. Sci. Rep. 2017, 7, 3373. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhang, J.; Liang, W.; Lv, Y.; Luo, X.; Li, C.; Qu, X.; Zhang, Y.; Gu, W.; Chen, X.; et al. Follicular Fluid Exosome-Derived Mir-339-5p Enhances in Vitro Maturation of Porcine Oocytes Via Targeting Sfpq, a Regulator of the Erk1/2 Pathway. Theriogenology 2024, 225, 107–118. [Google Scholar] [CrossRef]
- Tian, X.; Li, L.; Fu, G.; Wang, J.; He, Q.; Zhang, C.; Qin, B.; Wang, J. Mir-133a-3p Regulates the Proliferation and Apoptosis of Intestinal Epithelial Cells by Modulating the Expression of Tagln2. Exp. Ther. Med. 2021, 22, 824. [Google Scholar] [CrossRef]
- Yin, Y.; Du, L.; Li, X.; Zhang, X.; Gao, Y. Mir-133a-3p Suppresses Cell Proliferation, Migration, and Invasion and Promotes Apoptosis in Esophageal Squamous Cell Carcinoma. J. Cell. Physiol. 2019, 234, 12757–12770. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mRNAs | Forward | Reverse |
|---|---|---|
| β-actin | AGTGTGACGTTGACATCCGT | GCAGCTCAGTAACAGTCCGC |
| Stella | AGGGTCCGCACTTTGTTGTC | GGCTCACTGTCCCGTTCAAA |
| Mater | TCAAGAACTGGAACTAGTGGAC | GCTTGGTTGTTGTGATCATACA |
| Zar1 | GACGCCTCGGTGCAGTGTTC | CACAGAAGGTCACGGACGAGAAC |
| Mapk8 | AAACTGTTCCCCGATGTGCTT | CGTTGATGTATGGGTGCTGGA |
| Bcl2 | GAGCGTCAACAGGGAGATG | GGGCCATATAGTTCCACAAAGG |
| Bax | TGCAGAGGATGATTGCTGAC | GATCAGCTCGGGCACTTTAG |
| Foxj2 | TCAGCAAAGATGAGGCAGCG | ACCGATGCCAGCGTTCTTGTA |
| Shp1 | GGGCTAGACTGTGACATTGATA | TTTCTTCTTGGTCGTTTCGATG |
| Fn1 | ATCATAGTGGAGGCACTGCAGAA | GGTCAAAGCATGAGTCATCTGTAGG |
| Star | ACTCACTTGGCTGCTCAGTATTG | CAGGTGGTTGGCGAACTCTAT |
| Itgb3 | GGCGTTGTTGTTGGAGAGTC | CTTCAGGTTACATCGGGGTGA |
| t5a | GACGCTAGAGAAAGGGAACGAATC | GCCAGACACTCCATGACACTTAC |
| c4 | AGGGCAGCAACATCTTTGAGAGAA | CACCAGCAGCAGGATCAGGAA |
| Scd1 | TACACCTGCCTCTTCGGGATT | CAGCCGTGCCTTGTAAGTTCT |
| Hspg2 | ATCTGTAACGCCACCAACTCT | GCCGAGGTATGTTCTGGAATTATG |
| Itga7 | TACCTCATCCTCAGCACCTCT | GACAGTGGCAATTCAATGAAGACA |
| Col4a2 | CAGCCTGGTGTACTCGGTCTTC | TTGGTCGCCTTTGGGTCCTTT |
| Stc2 | AGTTTGTGACCCTGGCTTTGG | CTGGATCTCCGCTGTGTTCTG |
| Itga6 | TCTCGTTCTTCGTTCCAGGTTGT | AGCAGCAGCGGTGACATCTAT |
| Pik3cg | TGACAGGCACAACGACAACATT | TTAGGACGAAGGGCACTCTCTC |
| Cxcl12 | TGCATCAGTGACGGTAAACCA | TTCTTCAGCCGTGCAACAATC |
| Itga9 | CCATCAACATCACAGCACCTCAG | TCAGCCGTCAGATTGTAGTTCAGA |
| Thbs1 | GACTATGACAAGGACGGGATTGG | ACTGGGCTGGGTTGTAATGGAA |
| Ar | TTCCTGGATGGGACTGATGGTA | CGAGACTTGTGCATGCGGTA |
| Bmp4 | GGGATCTTTACCGGCTCCAG | GCTGCTGAGGTTGAAGAGGA |
| Spp1 | TACGACCATGAGATTGGCAGTGA | TATAGGATCTGGGTGCAGGCTGTAA |
| Mmp2 | CAGGGCACCTCCTACAACAG | CAGTGGACATAGCGGTCTCG |
| Col18a1 | GCCAGAAAGGCAGTGTTGGTG | GAATCCAGCAGCAAATCCTGG |
| Ngf | GCGTTTTTGATCGGCGTACA | AGGGCTGTGTCAAGGGAATG |
| Nrp1 | GGAGGAATGTTCTGTCGCTATG | GCACACTGTAGTTGGCTGAG |
| Agt | TCTCCTTTACCACAACAAGAGCA | CTTCTCATTCACAGGGGAGGT |
| Vcam1 | TCAGGAAATGCCACCCTCAC | CAGCACACGTCAGAACAACC |
| Edn1 | TCTGCCACCTGGACATCATCT | AACGCTTGGACCTGGAAGAAC |
| Fos | ACCGTGTCAGGAGGCAGA | GCAGCCATCTTATTCCGTTCC |
| Icam1 | GTGGGTCGAAGGTGGTTCTT | CCGAGGACCATACAGCACG |
| Igf1 | CAGTTCGTGTGTGGACCGAG | AGTGGGGCACAGTACATCTC |
| Csf1 | CGCTGCCCTTCTTCGACA | AGGCAATCTGGCATGAAGTCT |
| Kitl | TGATAACCCTCAACTATGTCGCC | TGTCCAGAAGAGTAGTCAAGCTG |
| Jun | ACGACCTTCTACGACGATGC | GCCAGGTTCAAGGTCATGCT |
| miRNAs | Forward | Reverse |
|---|---|---|
| miR-140-3p | TACCACAGGGTAGAACCACGG | mRQ 3′ Primer from TAKARA miRNA RT-qPCR kit (TaKaRa Bio Inc., Kusatsu, Japan) |
| miR-149-5p | TCTGGCTCCGTGTCTTCACTCCC | |
| miR-365-2-5p | GGACTTTCAGGGGCAGCTAAA | |
| miR-199a-5p | TCTCCCAGTGTTCAGACTACCT | |
| miR-7686-5p | GACCTGGGGCTGGGCAAAA | |
| miR-133a-3p | CTCCCCTTGAACCAGCTGAAA | |
| novel-miR-248 | GCTGGCCTTGAACTCACAGAA | |
| novel-miR-239 | AGTTCCTTGGCTGTGTCTGAG | |
| miR-351-3p | TTCAAGATGCGCCTGGGAAC | |
| miR-145a-5p | CAGTTTTCCCAGGAATCCCT | |
| miR-3473a | GGAGAGATGGCTCAGCAAAA | |
| miR-667-5p | GTGGAGCAGTGAGCACGAAA | |
| miR-351-5p | TCCCTGAGGAGCCCTTTGAGCCTG | |
| novel-miR-28 | CGAGATCGTGGGTTCGAGTC | |
| novel-miR-168 | CGAGATCGTGGGTTCGAGTC | |
| novel-miR-340 | CCTGAGTGTGTGTGTGTGTATTA | |
| novel-miR-193 | TGGACAACCCAGGAGGTCAA | |
| novel-miR-85 | GGGAATTGAACTCAGGACCTAA | |
| miR-615-5p | CCCGGTGCTCGGATCAAAA | |
| miR-7686-3p | CTCGGGGCACTGTAAGAGAA | |
| miR-700-3p | GGGAACCGAGTCCACCAAA | |
| miR-128-1-5p | GGCCGTAGCACTGTCTGAAA | |
| miR-339-5p | CCTCCAGGAGCTCACGAAAA | |
| miR-326-3p | TGGGCCCTTCCTCCAGTAAA | |
| miR-467d-3p | GCATACATACACACACCTACACAA | |
| U6 | GGAACGATACAGAGAAGATTAGC | TGGAACGCTTCACGAATTTGCG |
| KEGG Pathways | Exp 1 | Exp 2 | Exp 3 | Exp 4 |
|---|---|---|---|---|
| ECM-receptor interaction # $ | 1 * | 1 | 5 | 6 |
| Pathways in cancer | 2 | 5 | 3 | 7 |
| Axon guidance | 3 | 2 | 1 | |
| Focal adhesion | 4 | 3 | 2 | 3 |
| DNA replication | 5 | |||
| Fluid shear stress and atherosclerosis | 6 | 6 | ||
| Protein digestion and absorption | 7 | 4 | 1 | 9 |
| AGE-RAGE SP in diabetic complications | 8 | 6 | ||
| Proteoglycans in cancer | 9 | 7 | 4 | |
| PI3K-Akt signaling pathway | 10 | 8 | 4 | 5 |
| Metabolic pathway | 8 | |||
| Cushing syndrome | 9 | |||
| EGFR tyrosine kinase inhibitor resistance | 10 | |||
| Wnt signaling pathway | 9 | 7 | ||
| Regulation of actin cytoskeleton | 10 | |||
| Rap1 signaling | 2 | |||
| Regulation lipolysis adipocytes | 8 | |||
| Ras signaling | 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Wang, M.-M.; Zhang, F.-L.; He, N.; Luo, M.-J.; Gong, S.; Fu, F.-Y.; Yuan, H.-J.; Zhang, J.; Tan, J.-H. Expression Profiling and Function Analysis Identified New Cumulus Cells-Expressed Genes and miRNAs Predictive of Oocyte Developmental Potential. Cells 2025, 14, 791. https://doi.org/10.3390/cells14110791
Zhang M, Wang M-M, Zhang F-L, He N, Luo M-J, Gong S, Fu F-Y, Yuan H-J, Zhang J, Tan J-H. Expression Profiling and Function Analysis Identified New Cumulus Cells-Expressed Genes and miRNAs Predictive of Oocyte Developmental Potential. Cells. 2025; 14(11):791. https://doi.org/10.3390/cells14110791
Chicago/Turabian StyleZhang, Min, Meng-Meng Wang, Fa-Li Zhang, Nan He, Ming-Jiu Luo, Shuai Gong, Fu-Yin Fu, Hong-Jie Yuan, Jie Zhang, and Jing-He Tan. 2025. "Expression Profiling and Function Analysis Identified New Cumulus Cells-Expressed Genes and miRNAs Predictive of Oocyte Developmental Potential" Cells 14, no. 11: 791. https://doi.org/10.3390/cells14110791
APA StyleZhang, M., Wang, M.-M., Zhang, F.-L., He, N., Luo, M.-J., Gong, S., Fu, F.-Y., Yuan, H.-J., Zhang, J., & Tan, J.-H. (2025). Expression Profiling and Function Analysis Identified New Cumulus Cells-Expressed Genes and miRNAs Predictive of Oocyte Developmental Potential. Cells, 14(11), 791. https://doi.org/10.3390/cells14110791