
The Aryl Hydrocarbon Receptor (AhR) Is a Novel Gene Involved in Proper Physiological Functions of Pancreatic β-Cells

 , , , ,
, , , ,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. RNA-Seq Expression Data
2.2. Cell Culturing and siRNA Transfection
2.3. RNA Extraction and Quantitative PCR (qPCR)
2.4. Insulin Secretion Assays
2.5. Glucose Uptake Assay
2.6. Intracellular Reactive Oxygen Species (ROS) Measurement
2.7. Apoptosis and Cell Viability Assay
2.8. Western Blot Analysis
2.9. Immunostaining
2.10. Statistical Analysis
3. Results
3.1. mRNA Expression of AHR in Human Pancreatic Islets
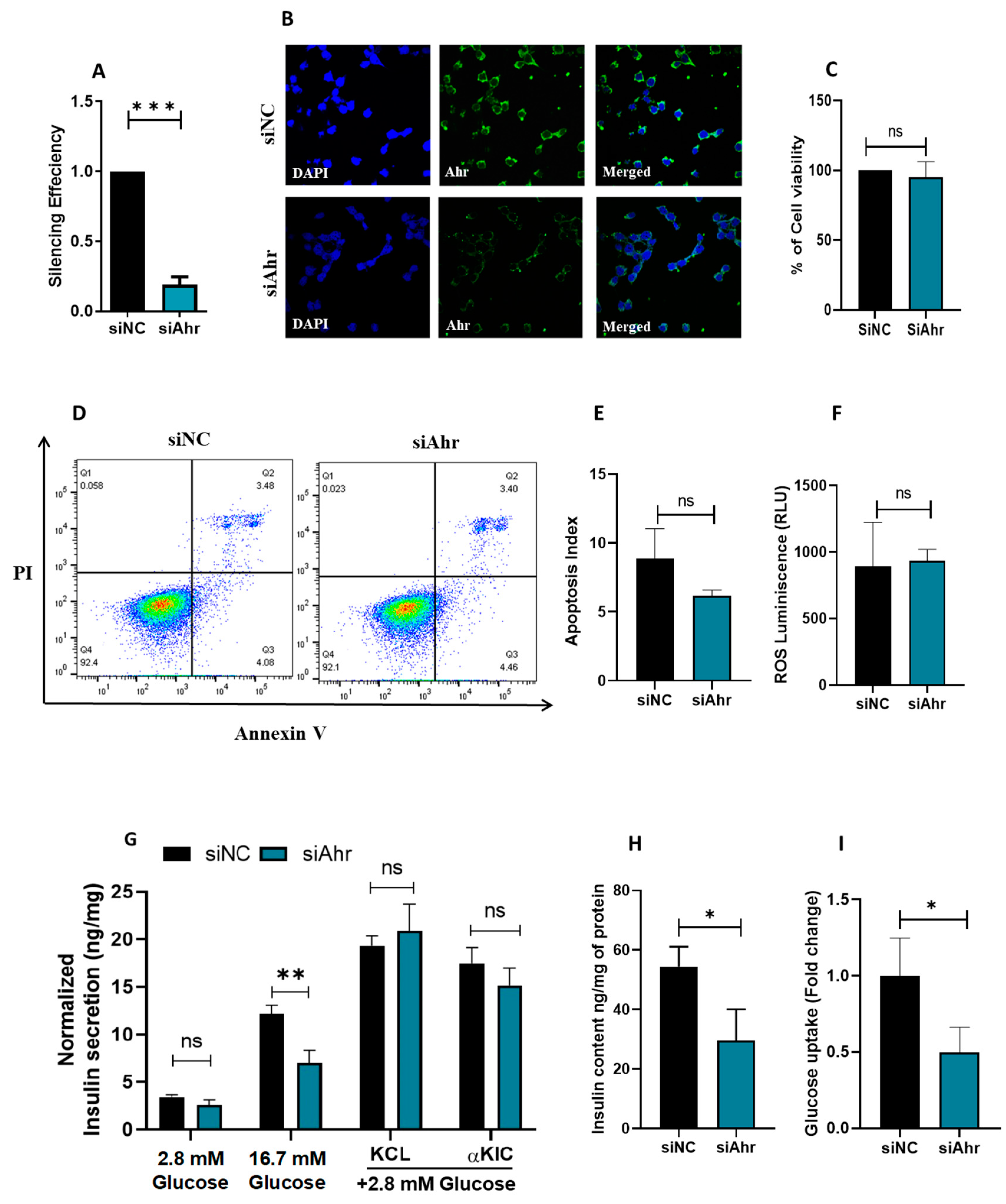
3.2. siRNA Silencing of Ahr in INS 1 Impairs Insulin Secretion and Glucose Uptake
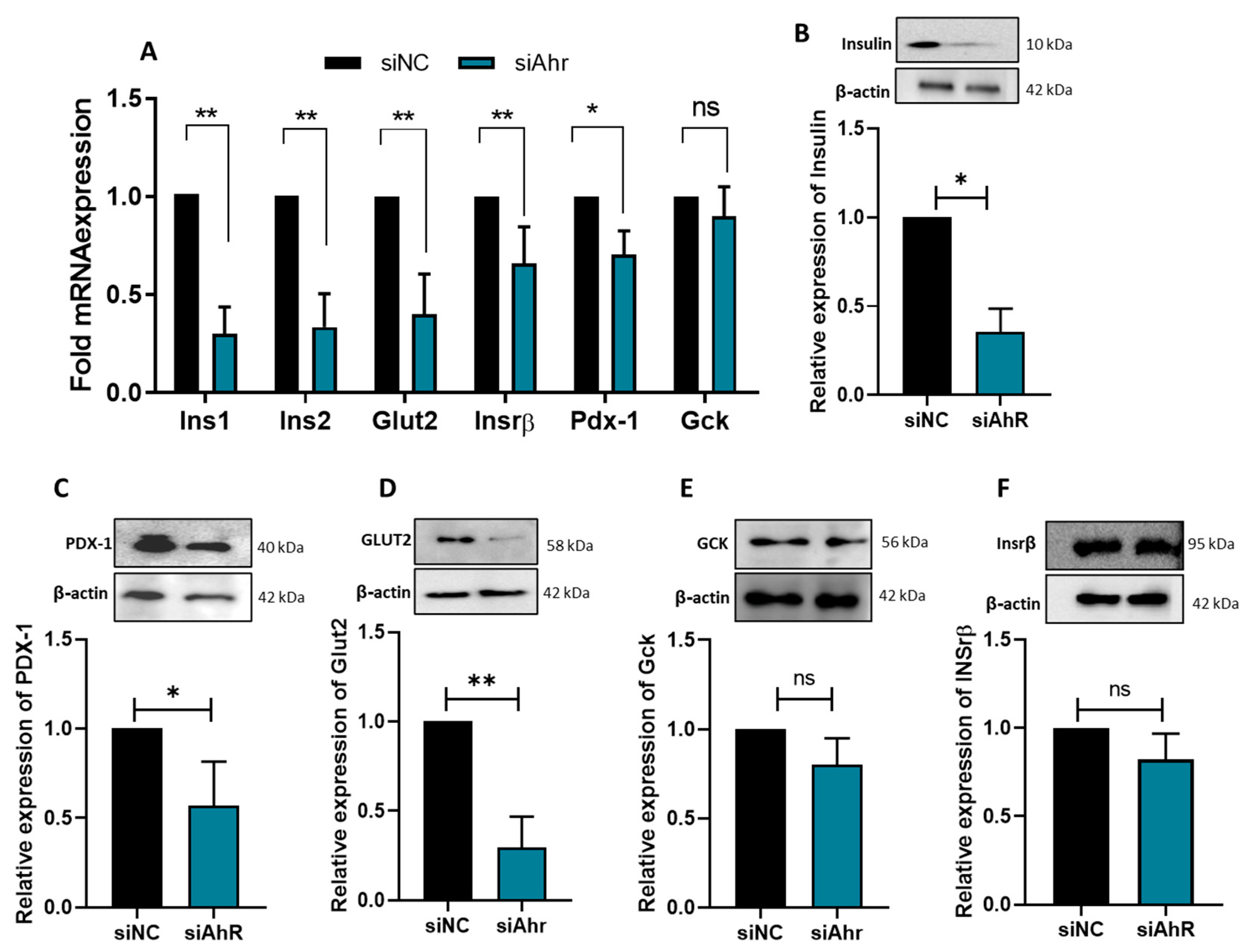
3.3. AhR Silencing Affects the Expression of β-Cell Functional Genes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Savitz, J. The kynurenine pathway: A finger in every pie. Mol. Psychiatry 2020, 25, 131–147. [Google Scholar] [CrossRef]
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [PubMed]
- Koziel, K.; Urbanska, E.M. Kynurenine Pathway in Diabetes Mellitus-Novel Pharmacological Target? Cells 2023, 12, 460. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Raynal, S.; Bailbe, D.; Gausseres, B.; Carbonne, C.; Autier, V.; Movassat, J.; Kergoat, M.; Portha, B. Expression of the kynurenine pathway enzymes in the pancreatic islet cells. Activation by cytokines and glucolipotoxicity. Biochim. Biophys. Acta 2015, 1852, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, W.H.; Whelan, S.A.; Lee, N. Tryptophan, kynurenine pathway, and diabetic ketoacidosis in type 1 diabetes. PLoS ONE 2021, 16, e0254116. [Google Scholar] [CrossRef]
- Kiluk, M.; Lewkowicz, J.; Kowalska, I.; Pawlak, D.; Lagoda, K.; Tankiewicz-Kwedlo, A. Alterations of the kynurenine pathway in patients with type 1 diabetes are associated with metabolic control of diabetes. Pol. Arch. Intern. Med. 2023, 133, 16581. [Google Scholar] [CrossRef]
- Vangipurapu, J.; Fernandes Silva, L.; Kuulasmaa, T.; Smith, U.; Laakso, M. Microbiota-Related Metabolites and the Risk of Type 2 Diabetes. Diabetes Care 2020, 43, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.; Papandreou, C.; Ruiz-Canela, M.; Guasch-Ferre, M.; Clish, C.B.; Dennis, C.; Liang, L.; Corella, D.; Fito, M.; Razquin, C.; et al. Association of Tryptophan Metabolites with Incident Type 2 Diabetes in the PREDIMED Trial: A Case-Cohort Study. Clin. Chem. 2018, 64, 1211–1220. [Google Scholar] [CrossRef]
- Trepci, A.; Imbeault, S.; Wyckelsma, V.L.; Westerblad, H.; Hermansson, S.; Andersson, D.C.; Piehl, F.; Venckunas, T.; Brazaitis, M.; Kamandulis, S.; et al. Quantification of Plasma Kynurenine Metabolites Following One Bout of Sprint Interval Exercise. Int. J. Tryptophan Res. 2020, 13, 1178646920978241. [Google Scholar] [CrossRef]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef]
- Kim, K. The Role of Endocrine Disruption Chemical-Regulated Aryl Hydrocarbon Receptor Activity in the Pathogenesis of Pancreatic Diseases and Cancer. Int. J. Mol. Sci. 2024, 25, 3818. [Google Scholar] [CrossRef]
- Rejano-Gordillo, C.M.; Marín-Díaz, B.; Ordiales-Talavero, A.; Merino, J.M.; González-Rico, F.J.; Fernández-Salguero, P.M. From nucleus to organs: Insights of aryl hydrocarbon receptor molecular mechanisms. Int. J. Mol. Sci. 2022, 23, 14919. [Google Scholar] [CrossRef] [PubMed]
- Thackaberry, E.A.; Bedrick, E.J.; Goens, M.B.; Danielson, L.; Lund, A.K.; Gabaldon, D.; Smith, S.M.; Walker, M.K. Insulin regulation in AhR-null mice: Embryonic cardiac enlargement, neonatal macrosomia, and altered insulin regulation and response in pregnant and aging AhR-null females. Toxicol. Sci. 2003, 76, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Fadista, J.; Vikman, P.; Laakso, E.O.; Mollet, I.G.; Esguerra, J.L.; Taneera, J.; Storm, P.; Osmark, P.; Ladenvall, C.; Prasad, R.B.; et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 13924–13929. [Google Scholar] [CrossRef]
- Hamad, M.; Mohammed, A.K.; Hachim, M.Y.; Mukhopadhy, D.; Khalique, A.; Laham, A.; Dhaiban, S.; Bajbouj, K.; Taneera, J. Heme Oxygenase-1 (HMOX-1) and inhibitor of differentiation proteins (ID1, ID3) are key response mechanisms against iron-overload in pancreatic beta-cells. Mol. Cell Endocrinol. 2021, 538, 111462. [Google Scholar] [CrossRef] [PubMed]
- El-Huneidi, W.; Anjum, S.; Mohammed, A.K.; Unnikannan, H.; Saeed, R.; Bajbouj, K.; Abu-Gharbieh, E.; Taneera, J. Copine 3 “CPNE3” is a novel regulator for insulin secretion and glucose uptake in pancreatic beta-cells. Sci. Rep. 2021, 11, 20692. [Google Scholar] [CrossRef]
- Murray, I.A.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Okino, S.T.; Pookot, D.; Basak, S.; Dahiya, R. Toxic and chemopreventive ligands preferentially activate distinct aryl hydrocarbon receptor pathways: Implications for cancer prevention. Cancer Prev. Res. 2009, 2, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Zhang, L. The Role of the Aryl Hydrocarbon Receptor (AhR) and Its Ligands in Breast Cancer. Cancers 2022, 14, 5574. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, T.B.; Morris, G.Z.; Tomblin, J.K.; Chaudhry, A.R.; Cook, C.R.; Santanam, N. Aryl hydrocarbon receptor ligands inhibit igf-ii and adipokine stimulated breast cancer cell proliferation. ISRN Endocrinol. 2013, 2013, 104850. [Google Scholar] [CrossRef] [PubMed]
- Walczak, K.; Langner, E.; Makuch-Kocka, A.; Szelest, M.; Szalast, K.; Marciniak, S.; Plech, T. Effect of Tryptophan-Derived AhR Ligands, Kynurenine, Kynurenic Acid and FICZ, on Proliferation, Cell Cycle Regulation and Cell Death of Melanoma Cells-In Vitro Studies. Int. J. Mol. Sci. 2020, 21, 7946. [Google Scholar] [CrossRef]
- Zarate, L.V.; Pontillo, C.A.; Espanol, A.; Miret, N.V.; Chiappini, F.; Cocca, C.; Alvarez, L.; de Pisarev, D.K.; Sales, M.E.; Randi, A.S. Angiogenesis signaling in breast cancer models is induced by hexachlorobenzene and chlorpyrifos, pesticide ligands of the aryl hydrocarbon receptor. Toxicol. Appl. Pharmacol. 2020, 401, 115093. [Google Scholar] [CrossRef] [PubMed]
- de Las Heras, N.; Lahera, V. Relevance of mitochondrial dysfunction in heart disease associated with insulin resistance conditions. Pflugers Arch. 2022, 474, 21–31. [Google Scholar] [CrossRef]
- Mone, P.; Morgante, M.; Pansini, A.; Jankauskas, S.S.; Rizzo, M.; Lombardi, A.; Frullone, S.; Santulli, G. Effects of insulin resistance on mitochondrial (dys)function. Atherosclerosis 2022, 341, 52–54. [Google Scholar] [CrossRef]
- Villarreal-Martinez, A.; Martinez-de-Villarreal, L.E.; Gomez-Flores, M.; Chavez-Alvarez, S.; Cerda-Flores, R.; Ocampo-Candiani, J.; Ruiz-Herrera, C.; Rodriguez-Rivera, M.R.; Villarreal-Perez, J.Z.; Gonzalez-Gonzalez, J.G.; et al. Mitochondrial dysfunction: The pathological link between psoriasis and insulin resistance? J. Eur. Acad. Dermatol. Venereol. 2023, 37, 340–347. [Google Scholar] [CrossRef]
- Zhao, R.X.; He, Q.; Sha, S.; Song, J.; Qin, J.; Liu, P.; Sun, Y.J.; Sun, L.; Hou, X.G.; Chen, L. Increased AHR Transcripts Correlate with Pro-inflammatory T-Helper Lymphocytes Polarization in Both Metabolically Healthy Obesity and Type 2 Diabetic Patients. Front. Immunol. 2020, 11, 1644. [Google Scholar] [CrossRef]
- Ottosson-Laakso, E.; Krus, U.; Storm, P.; Prasad, R.B.; Oskolkov, N.; Ahlqvist, E.; Fadista, J.; Hansson, O.; Groop, L.; Vikman, P. Glucose-Induced Changes in Gene Expression in Human Pancreatic Islets: Causes or Consequences of Chronic Hyperglycemia. Diabetes 2017, 66, 3013–3028. [Google Scholar] [CrossRef] [PubMed]
- Nakajima-Nagata, N.; Sugai, M.; Sakurai, T.; Miyazaki, J.; Tabata, Y.; Shimizu, A. Pdx-1 enables insulin secretion by regulating synaptotagmin 1 gene expression. Biochem. Biophys. Res. Commun. 2004, 318, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Iezzi, M.; Theander, S.; Antinozzi, P.A.; Gauthier, B.R.; Halban, P.A.; Wollheim, C.B. Suppression of Pdx-1 perturbs proinsulin processing, insulin secretion and GLP-1 signalling in INS-1 cells. Diabetologia 2005, 48, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.; MacFarlane, E.M.; Matteo, G.; Hoyeck, M.P.; Rick, K.R.C.; Farokhi, S.; Copley, C.M.; O’Dwyer, S.; Bruin, J.E. Functional cytochrome P450 1A enzymes are induced in mouse and human islets following pollutant exposure. Diabetologia 2020, 63, 162–178. [Google Scholar] [CrossRef]
- MacDonald, P.E.; Joseph, J.W.; Rorsman, P. Glucose-sensing mechanisms in pancreatic beta-cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2211–2225. [Google Scholar] [CrossRef] [PubMed]
- Maiztegui, B.; Roman, C.L.; Barbosa-Sampaio, H.C.; Boschero, A.C.; Gagliardino, J.J. Role of Islet Glucokinase, Glucose Metabolism, and Insulin Pathway in the Enhancing Effect of Islet Neogenesis-Associated Protein on Glucose-Induced Insulin Secretion. Pancreas 2015, 44, 959–966. [Google Scholar] [CrossRef]
- Schuit, F.C.; Huypens, P.; Heimberg, H.; Pipeleers, D.G. Glucose sensing in pancreatic beta-cells: A model for the study of other glucose-regulated cells in gut, pancreas, and hypothalamus. Diabetes 2001, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Whitticar, N.B.; Nunemaker, C.S. Reducing Glucokinase Activity to Enhance Insulin Secretion: A Counterintuitive Theory to Preserve Cellular Function and Glucose Homeostasis. Front. Endocrinol. 2020, 11, 378. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.C.; Williams, D.; Vicogne, J.; Pessin, J.E. The glucose transporter 2 undergoes plasma membrane endocytosis and lysosomal degradation in a secretagogue-dependent manner. Endocrinology 2009, 150, 4056–4064. [Google Scholar] [CrossRef] [PubMed]
- Lalwani, A.; Stokes, R.A.; Lau, S.M.; Gunton, J.E. Deletion of ARNT (Aryl hydrocarbon receptor nuclear translocator) in beta-cells causes islet transplant failure with impaired beta-cell function. PLoS ONE 2014, 9, e98435. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.; Huypens, P.; Huang, M.; Schaefer, S.; Sheinin, T.; Wettig, S.D.; Joseph, J.W. Aryl hydrocarbon receptor nuclear translocator/hypoxia-inducible factor-1β plays a critical role in maintaining glucose-stimulated anaplerosis and insulin release from pancreatic β-cells. J. Biol. Chem. 2011, 286, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Nishiumi, S.; Yoshida, M.; Azuma, T.; Yoshida, K.-i.; Ashida, H. 2,3,7,8-Tetrachlorodibenzo-p-dioxin impairs an insulin signaling pathway through the induction of tumor necrosis factor-α in adipocytes. Toxicol. Sci. 2010, 115, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; He, J.; Liang, H.; Hu, K.; Jiang, S.; Yang, L.; Mei, S.; Zhu, X.; Yu, J.; Kijlstra, A.; et al. Aryl Hydrocarbon Receptor Regulates Apoptosis and Inflammation in a Murine Model of Experimental Autoimmune Uveitis. Front. Immunol. 2018, 9, 1713. [Google Scholar] [CrossRef]
- Denison, M.S.; Faber, S.C. And Now for Something Completely Different: Diversity in Ligand-Dependent Activation of Ah Receptor Responses. Curr. Opin. Toxicol. 2017, 2, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Grishanova, A.Y.; Perepechaeva, M.L. Aryl hydrocarbon receptor in oxidative stress as a double agent and its biological and therapeutic significance. Int. J. Mol. Sci. 2022, 23, 6719. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5’-3’) | Reverse (5’-3’) |
|---|---|---|
| Hprt | TTGTGTCATCAGCGAAAGTGG | CACAGGACTAGAACGTCTGCT |
| Insr | GAGTCCAGAGCTCACCAAGG | TCTCAGACTCCACACAACGC |
| Gck | CCTGGGAGGAACCAACTTCA | TCTTGTGCTTCATCATCTCGGC |
| Pdx1 | TTCAGTGCTAATCCCCCTGC | ACTTCCCTGTTCCAGCGTTC |
| Glut2 | GCAACATGTCAGAAGACAAGATCA | TGTCATATCCGAACTGGAAGGA |
| Ahr | GCAAGGTAAGGGCTTTCTACAG | GTCTGAAGGTGGCCAATGCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bin Eshaq, S.; Taneera, J.; Anjum, S.; Mohammed, A.K.; Semreen, M.H.; Alzoubi, K.H.; Eladl, M.; Bustanji, Y.; Abu-Gharbieh, E.; El-Huneidi, W. The Aryl Hydrocarbon Receptor (AhR) Is a Novel Gene Involved in Proper Physiological Functions of Pancreatic β-Cells. Cells 2025, 14, 57. https://doi.org/10.3390/cells14010057
Bin Eshaq S, Taneera J, Anjum S, Mohammed AK, Semreen MH, Alzoubi KH, Eladl M, Bustanji Y, Abu-Gharbieh E, El-Huneidi W. The Aryl Hydrocarbon Receptor (AhR) Is a Novel Gene Involved in Proper Physiological Functions of Pancreatic β-Cells. Cells. 2025; 14(1):57. https://doi.org/10.3390/cells14010057
Chicago/Turabian StyleBin Eshaq, Shuhd, Jalal Taneera, Shabana Anjum, Abdul Khader Mohammed, Mohammad H. Semreen, Karem H. Alzoubi, Mohamed Eladl, Yasser Bustanji, Eman Abu-Gharbieh, and Waseem El-Huneidi. 2025. "The Aryl Hydrocarbon Receptor (AhR) Is a Novel Gene Involved in Proper Physiological Functions of Pancreatic β-Cells" Cells 14, no. 1: 57. https://doi.org/10.3390/cells14010057
APA StyleBin Eshaq, S., Taneera, J., Anjum, S., Mohammed, A. K., Semreen, M. H., Alzoubi, K. H., Eladl, M., Bustanji, Y., Abu-Gharbieh, E., & El-Huneidi, W. (2025). The Aryl Hydrocarbon Receptor (AhR) Is a Novel Gene Involved in Proper Physiological Functions of Pancreatic β-Cells. Cells, 14(1), 57. https://doi.org/10.3390/cells14010057