
TurboID-Based IRE1 Interactome Reveals Participants of the Endoplasmic Reticulum-Associated Protein Degradation Machinery in the Human Mast Cell Leukemia Cell Line HMC-1.2


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmid Constructs
2.3. Mammalian Cell Transfections
2.4. Pharmacological Inhibitors and ER Stress Inducers
2.5. RNA Preparation and Quantitative RT-PCR
2.6. TurboID, Biotinylation Assay, and Mass Spectrometry
2.7. Immunoprecipitation, Immunoblotting, and Antibodies
2.8. Densitometric Analysis
2.9. Immunofluorescence (IF)
2.10. Statistical and Bioinformatic Analysis
3. Results
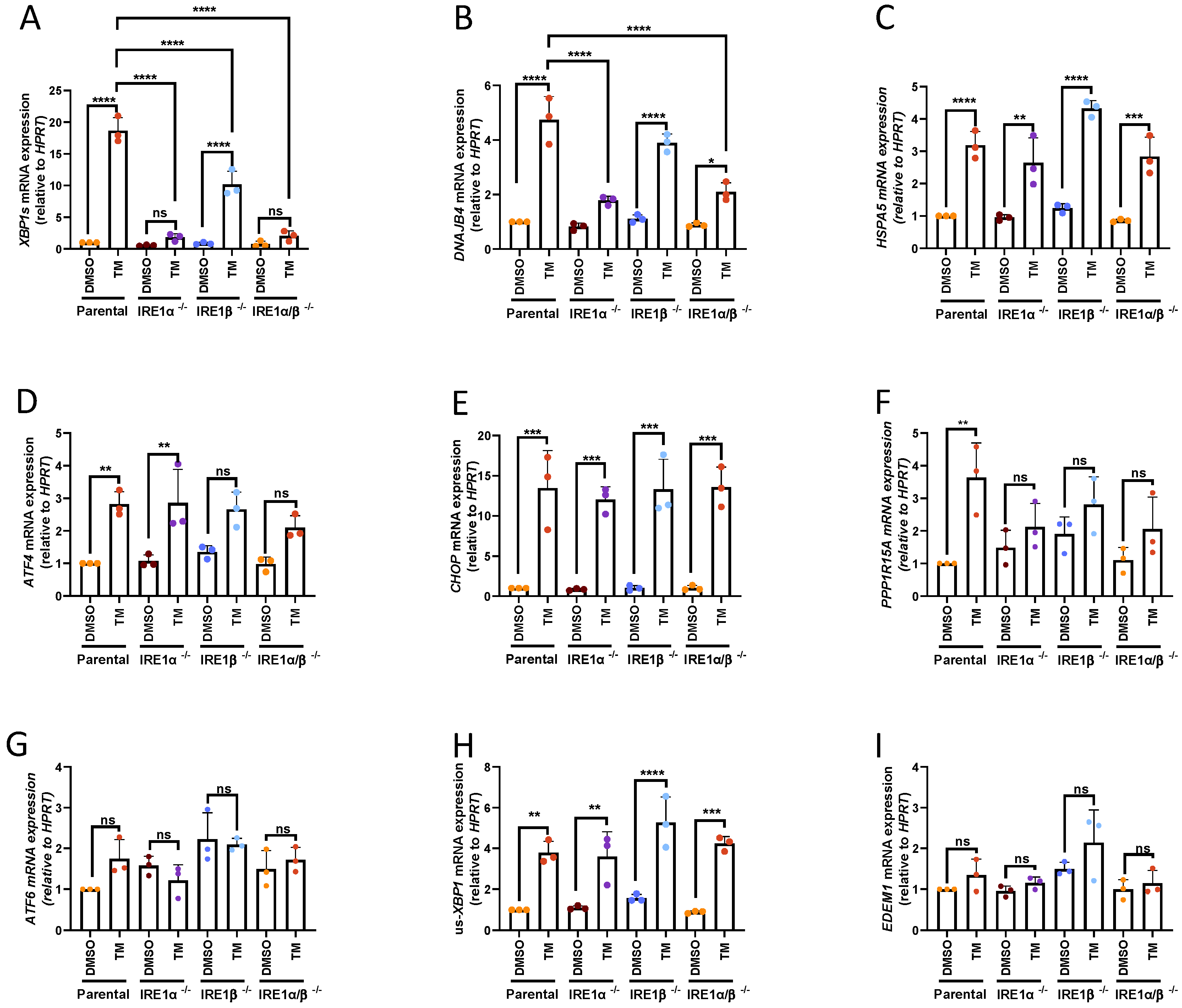
3.1. UPR Activation in IRE1α- and IRE1β-Deficient HMC-1.2 Cell Lines
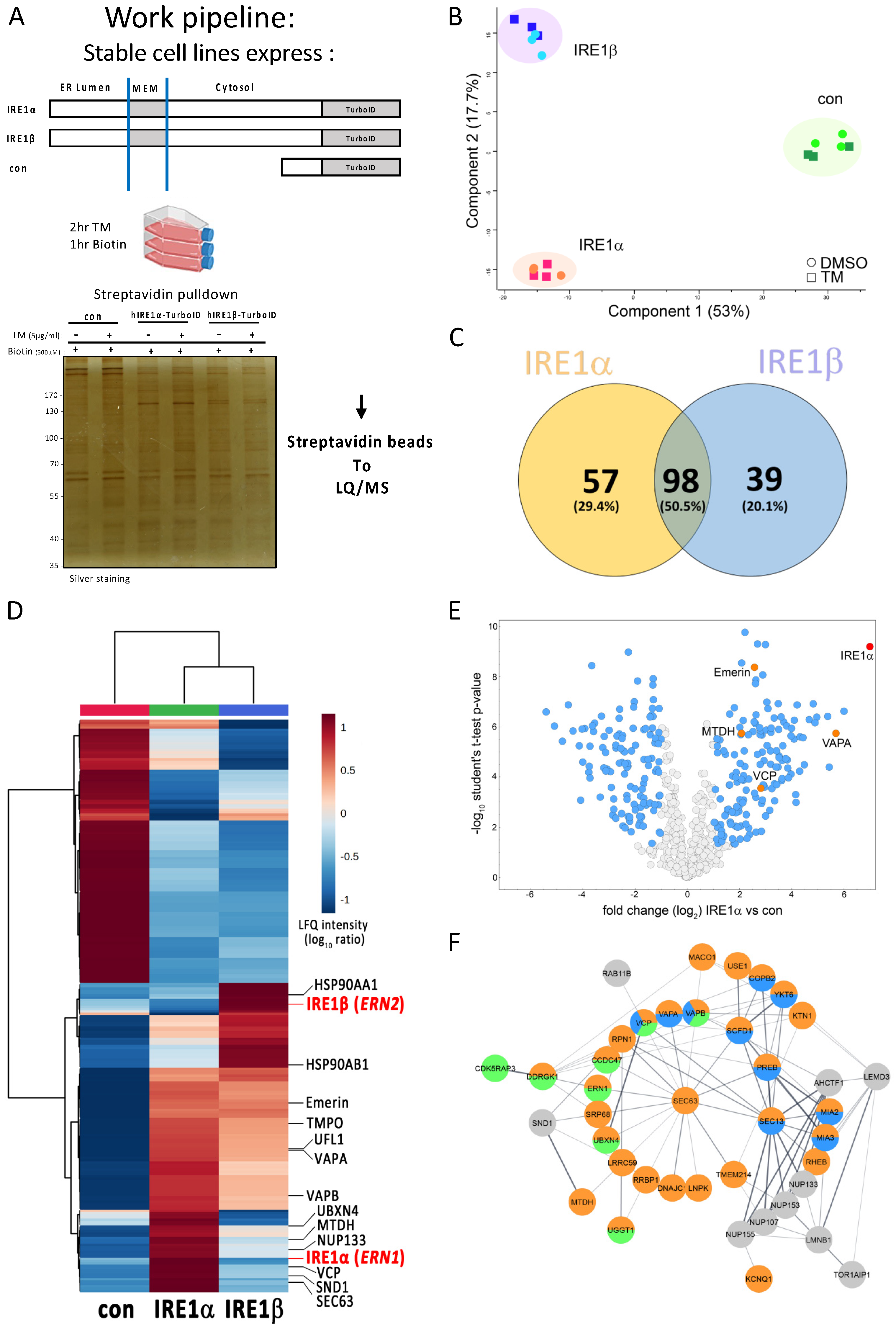
3.2. Identification of IRE1 Interacting Proteins Using a TurboID Approach
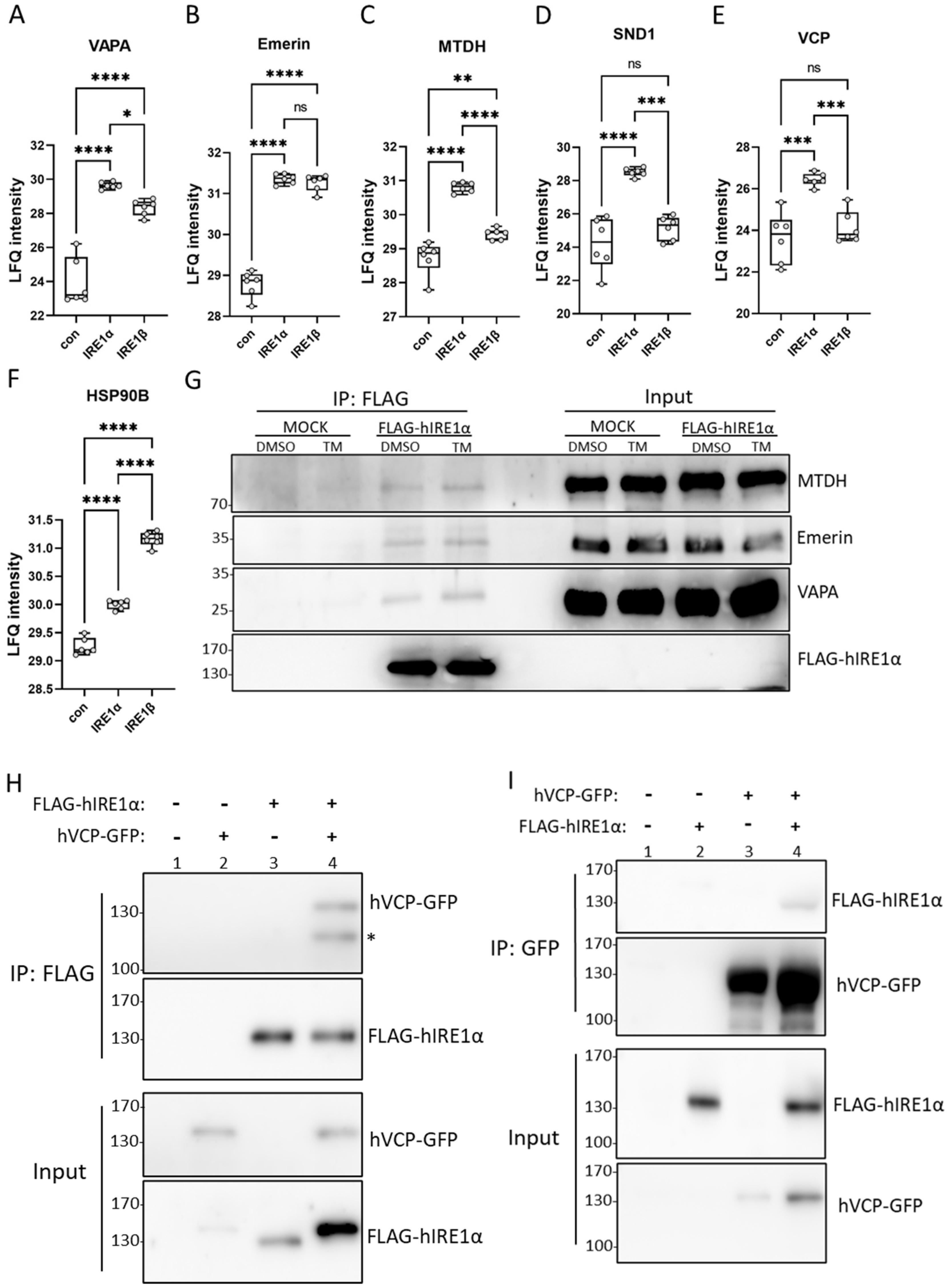
3.3. IRE1α Interacts with Proteins Involved in Vesicle Transport and Protein Stabilization
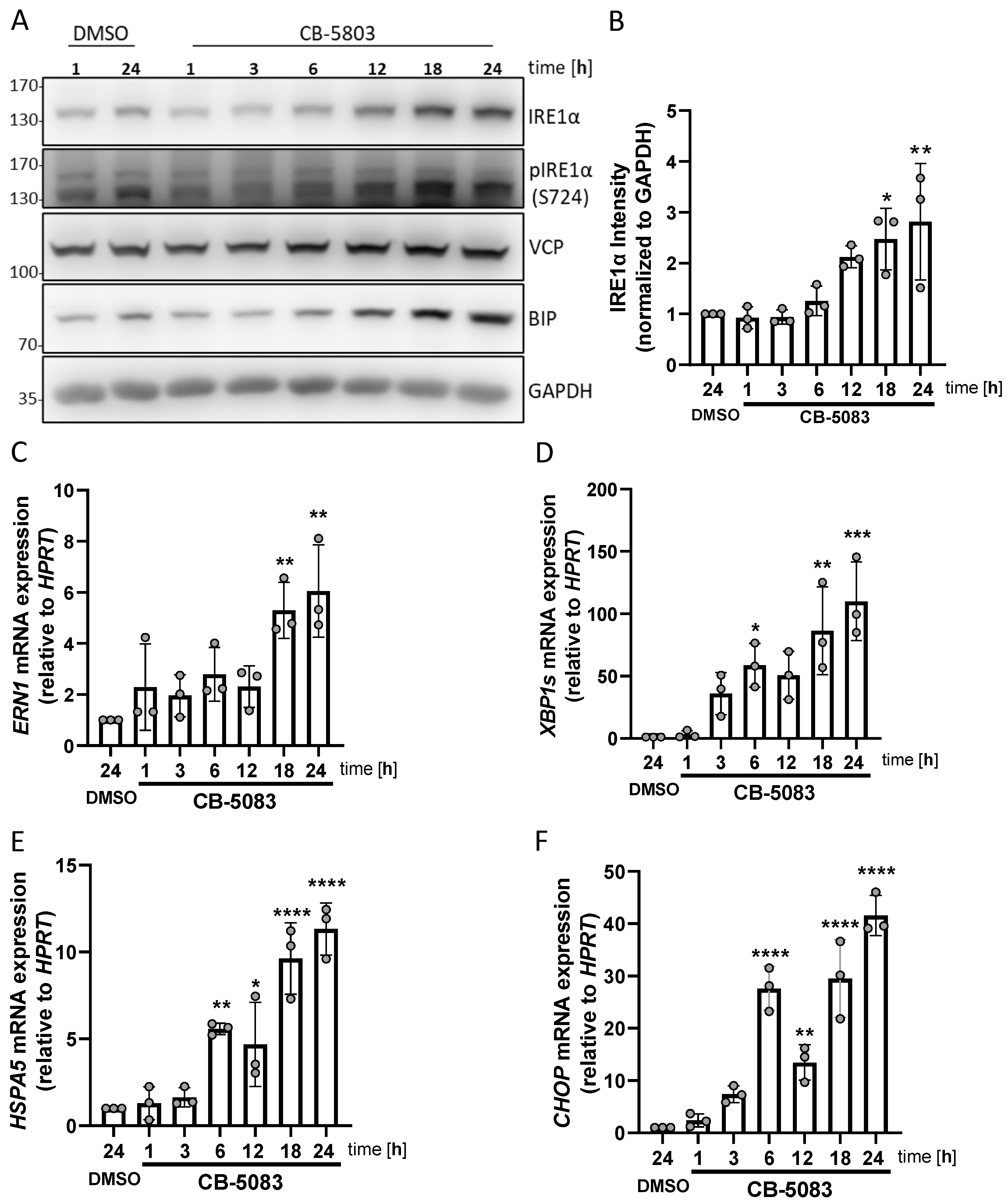
3.4. VCP Inhibition Stimulates UPR Activation and Enhancement of IRE1α Expression
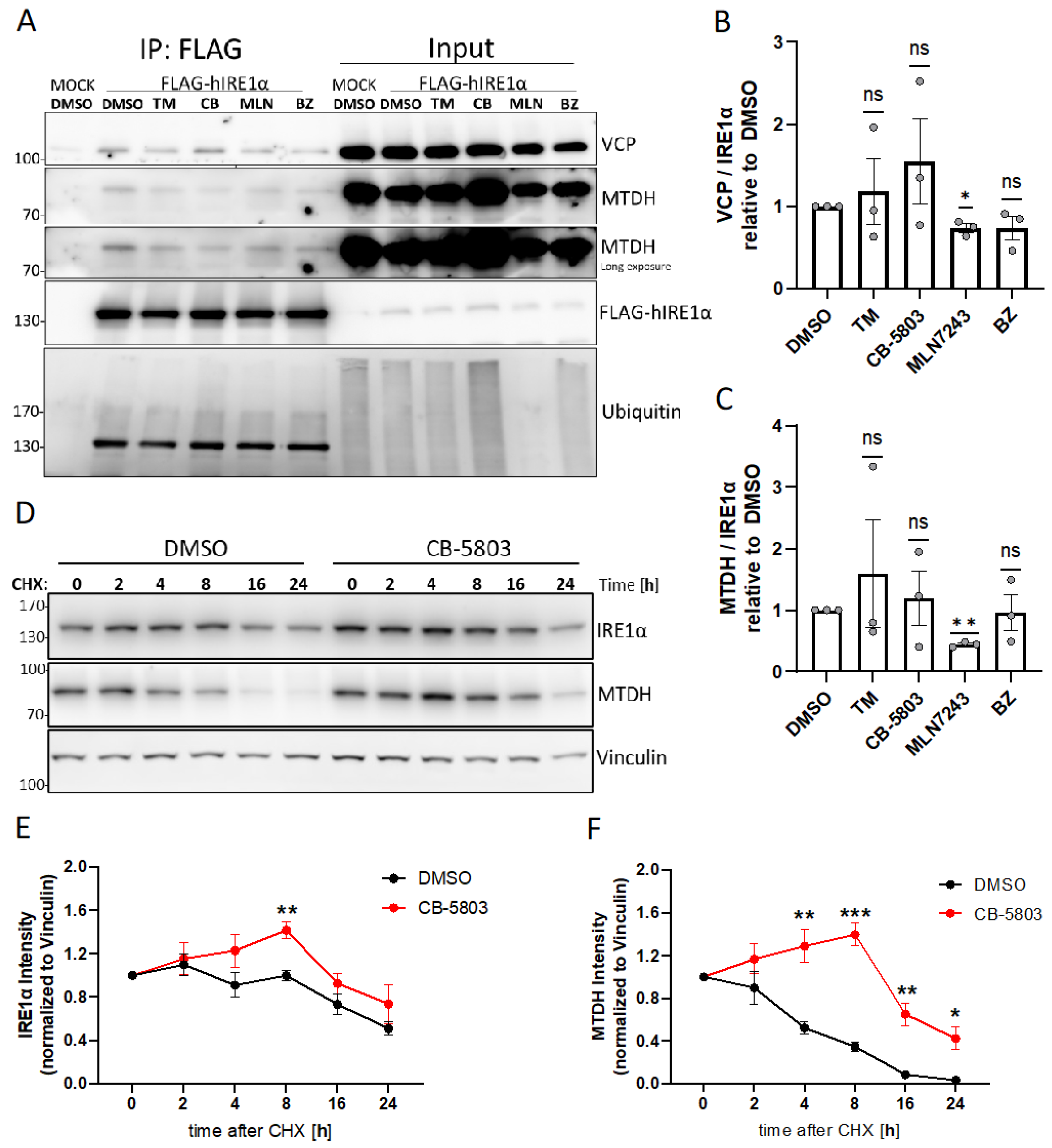
3.5. The Interaction between IRE1α, VCP, and MTDH Is Dependent on Ubiquitination
3.6. VCP Inhibition Stabilizes Expression of IRE1α and MTDH
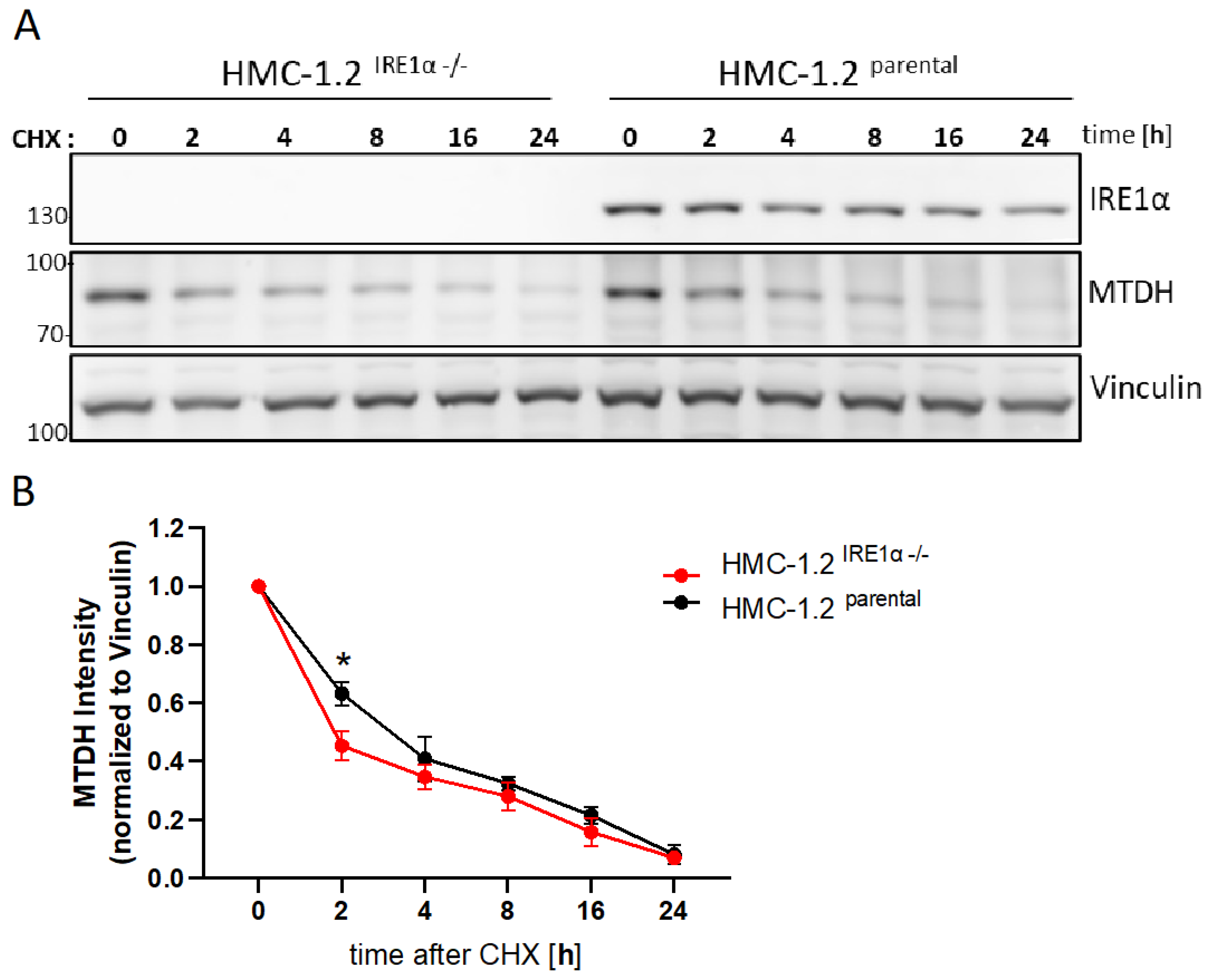
3.7. IRE1α Increases the Stability of MTDH Protein
4. Discussion
5. Conclusions
6. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huber, M.; Cato, A.C.B.; Ainooson, G.K.; Freichel, M.; Tsvilovskyy, V.; Jessberger, R.; Riedlinger, E.; Sommerhoff, C.P.; Bischoff, S.C. Regulation of the pleiotropic effects of tissue-resident mast cells. J. Allergy Clin. Immun. 2019, 144, S31–S45. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, D.D. Mast cells and mastocytosis. Blood 2008, 112, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Ustun, C.; Arock, M.; Kluin-Nelemans, H.C.; Reiter, A.; Sperr, W.R.; George, T.; Horny, H.-P.; Hartmann, K.; Sotlar, K.; Damaj, G.; et al. Advanced systemic mastocytosis: From molecular and genetic progress to clinical practice. Haematologica 2016, 101, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Akin, C.; Kirshenbaum, A.S.; Semere, T.; Worobec, A.S.; Scott, L.M.; Metcalfe, D.D. Analysis of the surface expression of c-kit and occurrence of the c-kit Asp816Val activating mutation in T cells, B cells, and myelomonocytic cells in patients with mastocytosis. Exp. Hematol. 2000, 28, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Fritsche-Polanz, R.; Jordan, J.; Feix, A.; Sperr, W.R.; Sunder-Plassmann, G.; Valent, P.; Födinger, M. Mutation analysis of C-KIT in patients with myelodysplastic syndromes without mastocytosis and cases of systemic mastocytosis. Br. J. Haematol. 2001, 113, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Longley, B.J.; Metcalfe, D.D.; Tharp, M.; Wang, X.; Tyrrell, L.; Lu, S.; Heitjan, D.; Ma, Y. Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc. Natl. Acad. Sci. USA 1999, 96, 1609–1614. [Google Scholar] [CrossRef] [PubMed]
- Gleixner, K.V.; Peter, B.; Blatt, K.; Suppan, V.; Reiter, A.; Radia, D.; Hadzijusufovic, E.; Valent, P.; Gleixner, K.V. Synergistic growth-inhibitory effects of ponatinib and midostaurin (PKC412) on neoplastic mast cells carrying KIT D816V. Haematologica 2013, 98, 1450–1457. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; Bick, F.; Peters, K.; Mohta, V.; Tirosh, B.; Patterson, J.B.; Kharabi-Masouleh, B.; Huber, M. Infliction of proteotoxic stresses by impairment of the unfolded protein response or proteasomal inhibition as a therapeutic strategy for mast cell leukemia. Oncotarget 2017, 9, 2984–3000. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional Induction of Mammalian ER Quality Control Proteins Is Mediated by Single or Combined Action of ATF6α and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Christianson, J.C.; Jarosch, E.; Sommer, T. Mechanisms of substrate processing during ER-associated protein degradation. Nat. Rev. Mol. Cell Biol. 2023, 24, 777–796. [Google Scholar] [CrossRef]
- Braxton, J.R.; Southworth, D.R. Structural insights of the p97/VCP AAA+ ATPase: How adapter interactions coordinate diverse cellular functionality. J. Biol. Chem. 2023, 299, 105182. [Google Scholar] [CrossRef] [PubMed]
- Emdad, L.; Das, S.K.; Dasgupta, S.; Hu, B.; Sarkar, D.; Fisher, P.B. Chapter Three AEG-1/MTDH/LYRIC Signaling Pathways, Downstream Genes, Interacting Proteins, and Regulation of Tumor Angiogenesis. Adv. Cancer Res. 2013, 120, 75–111. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zhu, D.; Yang, S.; Wang, X.; Xiong, Z.; Zhang, Y.; Brachova, P.; Leslie, K.K. Cytoplasmic Metadherin (MTDH) Provides Survival Advantage under Conditions of Stress by Acting as RNA-binding Protein*. J. Biol. Chem. 2012, 287, 4485–4491. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Bio 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.B.; Jones, L.; Brighton, B.; Ehre, C.; Abdulah, L.; Davis, C.W.; Ron, D.; O’Neal, W.K.; Ribeiro, C.M.P. The ER stress transducer IRE1β is required for airway epithelial mucin production. Mucosal Immunol. 2013, 6, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Grey, M.J.; Cloots, E.; Simpson, M.S.; LeDuc, N.; Serebrenik, Y.V.; Luca, H.D.; Sutter, D.D.; Luong, P.; Thiagarajah, J.R.; Paton, A.W.; et al. IRE1β negatively regulates IRE1α signaling in response to endoplasmic reticulum stress. J Cell Biol. 2020, 219, e201904048. [Google Scholar] [CrossRef]
- Lhomond, S.; Avril, T.; Dejeans, N.; Voutetakis, K.; Doultsinos, D.; McMahon, M.; Pineau, R.; Obacz, J.; Papadodima, O.; Jouan, F.; et al. Dual IRE 1 RN ase functions dictate glioblastoma development. EMBO Mol. Med. 2018, 10, e7929. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Lin, D.-C.; Guo, X.; Masouleh, B.K.; Gery, S.; Cao, Q.; Alkan, S.; Ikezoe, T.; Akiba, C.; Paquette, R.; et al. Inhibition of IRE1α-driven pro-survival pathways is a promising therapeutic application in acute myeloid leukemia. Oncotarget 2016, 7, 18736–18749. [Google Scholar] [CrossRef]
- Zhao, N.; Cao, J.; Xu, L.; Tang, Q.; Dobrolecki, L.E.; Lv, X.; Talukdar, M.; Lu, Y.; Wang, X.; Hu, D.Z.; et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J. Clin. Investig. 2018, 128, 1283–1299. [Google Scholar] [CrossRef]
- Raymundo, D.P.; Doultsinos, D.; Guillory, X.; Carlesso, A.; Eriksson, L.A.; Chevet, E. Pharmacological Targeting of IRE1 in Cancer. Trends Cancer 2020, 6, 1018–1030. [Google Scholar] [CrossRef]
- Butterfield, J.H.; Weiler, D.; Dewald, G.; Gleich, G.J. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leukemia Res. 1988, 12, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Volmer, R.; van der Ploeg, K.; Ron, D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. USA 2013, 110, 4628–4633. [Google Scholar] [CrossRef]
- Lipson, K.L.; Ghosh, R.; Urano, F. The Role of IRE1α in the Degradation of Insulin mRNA in Pancreatic β-Cells. PLoS ONE 2008, 3, e1648. [Google Scholar] [CrossRef]
- Morgenstern, J.P.; Land, H. Advanced mammalian gene transfer: High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990, 18, 3587–3596. [Google Scholar] [CrossRef] [PubMed]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.A.; Dykxhoorn, D.M.; Palliser, D.; Mizuno, H.; Yu, E.Y.; An, D.S.; Sabatini, D.M.; Chen, I.S.Y.; Hahn, W.C.; Sharp, P.A.; et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003, 9, 493–501. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Tresse, E.; Salomons, F.A.; Vesa, J.; Bott, L.C.; Kimonis, V.; Yao, T.-P.; Dantuma, N.P.; Taylor, J.P. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 2010, 6, 217–227. [Google Scholar] [CrossRef]
- Iwawaki, T.; Hosoda, A.; Okuda, T.; Kamigori, Y.; Nomura-Furuwatari, C.; Kimata, Y.; Tsuru, A.; Kohno, K. Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nat. Cell Biol. 2001, 3, 158–164. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Holzer, G.; Magistris, P.D.; Gramminger, C.; Sachdev, R.; Magalska, A.; Schooley, A.; Scheufen, A.; Lennartz, B.; Tatarek-Nossol, M.; Lue, H.; et al. The nucleoporin Nup50 activates the Ran guanine nucleotide exchange factor RCC1 to promote NPC assembly at the end of mitosis. EMBO J. 2021, 40, e108788. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Nolte, H.; MacVicar, T.D.; Tellkamp, F.; Krüger, M. Instant Clue: A Software Suite for Interactive Data Visualization and Analysis. Sci. Rep. 2018, 8, 12648. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.J.; Le Moigne, R.; Djakovic, S.; Kumar, B.; Rice, J.; Wong, S.; Wang, J.; Yao, B.; Valle, E.; Kiss von Soly, S.; et al. Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis. Cancer Cell 2015, 28, 653–665. [Google Scholar] [CrossRef]
- Meyer, H.; Bug, M.; Bremer, S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 2012, 14, 117–123. [Google Scholar] [CrossRef]
- Brown, D.M.; Ruoslahti, E. Metadherin, a cell surface protein in breast tumors that mediates lung metastasis. Cancer Cell 2004, 5, 365–374. [Google Scholar] [CrossRef]
- Sun, S.; Shi, G.; Sha, H.; Ji, Y.; Han, X.; Shu, X.; Ma, H.; Inoue, T.; Gao, B.; Kim, H.; et al. IRE1α is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 2015, 17, 1546–1555. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Axten, J.M.; Patterson, J.B. Pharmacological targeting of the unfolded protein response for disease intervention. Nat. Chem. Biol. 2019, 15, 764–775. [Google Scholar] [CrossRef] [PubMed]
- May, D.G.; Scott, K.L.; Campos, A.R.; Roux, K.J. Comparative Application of BioID and TurboID for Protein-Proximity Biotinylation. Cells 2020, 9, 1070. [Google Scholar] [CrossRef] [PubMed]
- Sears, R.M.; May, D.G.; Roux, K.J. Enzyme-Mediated Ligation Methods. Methods Mol. Biol. 2019, 2012, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Kume, H.; Muraoka, S.; Kuga, T.; Adachi, J.; Narumi, R.; Watanabe, S.; Kuwano, M.; Kodera, Y.; Matsushita, K.; Fukuoka, J.; et al. Discovery of Colorectal Cancer Biomarker Candidates by Membrane Proteomic Analysis and Subsequent Verification using Selected Reaction Monitoring (SRM) and Tissue Microarray (TMA) Analysis*. Mol. Cell. Proteom. 2014, 13, 1471–1484. [Google Scholar] [CrossRef] [PubMed]
- Gholami, A.M.; Hahne, H.; Wu, Z.; Auer, F.J.; Meng, C.; Wilhelm, M.; Kuster, B. Global Proteome Analysis of the NCI-60 Cell Line Panel. Cell Rep. 2013, 4, 609–620. [Google Scholar] [CrossRef]
- Sundaram, A.; Plumb, R.; Appathurai, S.; Mariappan, M. The Sec61 translocon limits IRE1α signaling during the unfolded protein response. eLife 2017, 6, e27187. [Google Scholar] [CrossRef]
- Hetz, C.; Glimcher, L.H. Fine-Tuning of the Unfolded Protein Response: Assembling the IRE1α Interactome. Mol. Cell 2009, 35, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Lisbona, F.; Rojas-Rivera, D.; Thielen, P.; Zamorano, S.; Todd, D.; Martinon, F.; Glavic, A.; Kress, C.; Lin, J.H.; Walter, P.; et al. BAX Inhibitor-1 Is a Negative Regulator of the ER Stress Sensor IRE1α. Mol. Cell 2009, 33, 679–691. [Google Scholar] [CrossRef]
- Pinkaew, D.; Chattopadhyay, A.; King, M.D.; Chunhacha, P.; Liu, Z.; Stevenson, H.L.; Chen, Y.; Sinthujaroen, P.; McDougal, O.M.; Fujise, K. Fortilin binds IRE1α and prevents ER stress from signaling apoptotic cell death. Nat. Commun. 2017, 8, 18. [Google Scholar] [CrossRef]
- Bashir, S.; Banday, M.; Qadri, O.; Pal, D.; Bashir, A.; Hilal, N.; Altaf, M.; Fazili, K.M. The Bcl-2 family protein bid interacts with the ER stress sensor IRE1 to differentially modulate its RNase activity. FEBS Lett. 2023, 597, 962–974. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Bernasconi, P.; Fisher, J.; Lee, A.-H.; Bassik, M.C.; Antonsson, B.; Brandt, G.S.; Iwakoshi, N.N.; Schinzel, A.; Glimcher, L.H.; et al. Proapoptotic BAX and BAK Modulate the Unfolded Protein Response by a Direct Interaction with IRE1α. Science 2006, 312, 572–576. [Google Scholar] [CrossRef]
- Mao, T.; Shao, M.; Qiu, Y.; Huang, J.; Zhang, Y.; Song, B.; Wang, Q.; Jiang, L.; Liu, Y.; Han, J.-D.; et al. PKA phosphorylation couples hepatic inositol-requiring enzyme 1α to glucagon signaling in glucose metabolism. Proc. Natl. Acad. Sci. USA 2011, 108, 15852–15857. [Google Scholar] [CrossRef] [PubMed]
- Jwa, M.; Chang, P. PARP16 is a tail-anchored endoplasmic reticulum protein required for the PERK- and IRE1α-mediated unfolded protein response. Nat. Cell Biol. 2012, 14, 1223–1230. [Google Scholar] [CrossRef]
- Marcu, M.G.; Doyle, M.; Bertolotti, A.; Ron, D.; Hendershot, L.; Neckers, L. Heat Shock Protein 90 Modulates the Unfolded Protein Response by Stabilizing IRE1α. Mol. Cell Biol. 2002, 22, 8506–8513. [Google Scholar] [CrossRef]
- Plumb, R.; Zhang, Z.-R.; Appathurai, S.; Mariappan, M. A functional link between the co-translational protein translocation pathway and the UPR. eLife 2015, 4, e07426. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Pihán, P.; Hetz, C. The UPRosome—Decoding novel biological outputs of IRE1α function. J. Cell Sci. 2020, 133, jcs218107. [Google Scholar] [CrossRef]
- Peter, J.J.; Magnussen, H.M.; DaRosa, P.A.; Millrine, D.; Matthews, S.P.; Lamoliatte, F.; Sundaramoorthy, R.; Kopito, R.R.; Kulathu, Y. A non-canonical scaffold-type E3 ligase complex mediates protein UFMylation. EMBO J. 2022, 41, e111015. [Google Scholar] [CrossRef]
- Liang, J.; Yin, C.; Doong, H.; Fang, S.; Peterhoff, C.; Nixon, R.A.; Monteiro, M.J. Characterization of erasin (UBXD2): A new ER protein that promotes ER-associated protein degradation. J. Cell Sci. 2006, 119, 4011–4024. [Google Scholar] [CrossRef]
- Meyer, H.; Weihl, C.C. The VCP/p97 system at a glance: Connecting cellular function to disease pathogenesis. J. Cell Sci. 2014, 127, 3877–3883. [Google Scholar] [CrossRef]
- Zhou, H.-L.; Geng, C.; Luo, G.; Lou, H. The p97–UBXD8 complex destabilizes mRNA by promoting release of ubiquitinated HuR from mRNP. Genes Dev. 2013, 27, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Kilgas, S.; Ramadan, K. Inhibitors of the ATPase p97/VCP: From basic research to clinical applications. Cell Chem. Biol. 2023, 30, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Gareau, A.; Rico, C.; Boerboom, D.; Nadeau, M.-E. In vitro efficacy of a first-generation valosin-containing protein inhibitor (CB-5083) against canine lymphoma. Vet. Comp. Oncol. 2018, 16, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Willmann, M.; Yuzbasiyan-Gurkan, V.; Marconato, L.; Dacasto, M.; Hadzijusufovic, E.; Hermine, O.; Sadovnik, I.; Gamperl, S.; Schneeweiss-Gleixner, M.; Gleixner, K.V.; et al. Proposed Diagnostic Criteria and Classification of Canine Mast Cell Neoplasms: A Consensus Proposal. Front. Vet. Sci. 2021, 8, 755258. [Google Scholar] [CrossRef] [PubMed]
- Mattia, T.D.; Martinet, A.; Ikhlef, S.; McEwen, A.G.; Nominé, Y.; Wendling, C.; Poussin-Courmontagne, P.; Voilquin, L.; Eberling, P.; Ruffenach, F.; et al. FFAT motif phosphorylation controls formation and lipid transfer function of inter-organelle contacts. EMBO J. 2020, 39, e104369. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.G.; Liu, N.; Miao, G.; Chen, Y.; Zhao, H.; Zhang, H. The ER Contact Proteins VAPA/B Interact with Multiple Autophagy Proteins to Modulate Autophagosome Biogenesis. Curr. Biol. 2018, 28, 1234–1245.e4. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Zhao, D.; Zou, C.-X.; Liu, X.-M.; Jiang, Z.-D.; Yu, Z.-Q.; Suo, F.; Du, T.-Y.; Dong, M.-Q.; He, W.; Du, L.-L. A UPR-Induced Soluble ER-Phagy Receptor Acts with VAPs to Confer ER Stress Resistance. Mol. Cell 2020, 79, 963–977.e3. [Google Scholar] [CrossRef]
- Tschurtschenthaler, M.; Adolph, T.E.; Ashcroft, J.W.; Niederreiter, L.; Bharti, R.; Saveljeva, S.; Bhattacharyya, J.; Flak, M.B.; Shih, D.Q.; Fuhler, G.M.; et al. Defective ATG16L1-mediated removal of IRE1α drives Crohn’s disease–like ileitis. J. Exp. Med. 2017, 214, 401–422. [Google Scholar] [CrossRef]
- Buchwalter, A.; Schulte, R.; Tsai, H.; Capitanio, J.; Hetzer, M. Selective clearance of the inner nuclear membrane protein emerin by vesicular transport during ER stress. eLife 2019, 8, e49796. [Google Scholar] [CrossRef]
- Shen, M.; Smith, H.A.; Wei, Y.; Jiang, Y.-Z.; Zhao, S.; Wang, N.; Rowicki, M.; Tang, Y.; Hang, X.; Wu, S.; et al. Pharmacological disruption of the MTDH–SND1 complex enhances tumor antigen presentation and synergizes with anti-PD-1 therapy in metastatic breast cancer. Nat. Cancer 2022, 3, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.K.; Santhekadur, P.K.; Gredler, R.; Chen, D.; Emdad, L.; Bhutia, S.; Pannell, L.; Fisher, P.B.; Sarkar, D. Increased RNA-induced silencing complex (RISC) activity contributes to hepatocellular carcinoma. Hepatology 2011, 53, 1538–1548. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2021, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, N.; Preisinger, C.; Wilhelm, T.; Huber, M. TurboID-Based IRE1 Interactome Reveals Participants of the Endoplasmic Reticulum-Associated Protein Degradation Machinery in the Human Mast Cell Leukemia Cell Line HMC-1.2. Cells 2024, 13, 747. https://doi.org/10.3390/cells13090747
Ahmed N, Preisinger C, Wilhelm T, Huber M. TurboID-Based IRE1 Interactome Reveals Participants of the Endoplasmic Reticulum-Associated Protein Degradation Machinery in the Human Mast Cell Leukemia Cell Line HMC-1.2. Cells. 2024; 13(9):747. https://doi.org/10.3390/cells13090747
Chicago/Turabian StyleAhmed, Nabil, Christian Preisinger, Thomas Wilhelm, and Michael Huber. 2024. "TurboID-Based IRE1 Interactome Reveals Participants of the Endoplasmic Reticulum-Associated Protein Degradation Machinery in the Human Mast Cell Leukemia Cell Line HMC-1.2" Cells 13, no. 9: 747. https://doi.org/10.3390/cells13090747
APA StyleAhmed, N., Preisinger, C., Wilhelm, T., & Huber, M. (2024). TurboID-Based IRE1 Interactome Reveals Participants of the Endoplasmic Reticulum-Associated Protein Degradation Machinery in the Human Mast Cell Leukemia Cell Line HMC-1.2. Cells, 13(9), 747. https://doi.org/10.3390/cells13090747