
Hematopoietic Stem Cells as an Integrative Hub Linking Lifestyle to Cardiovascular Health


Abstract
1. Introduction
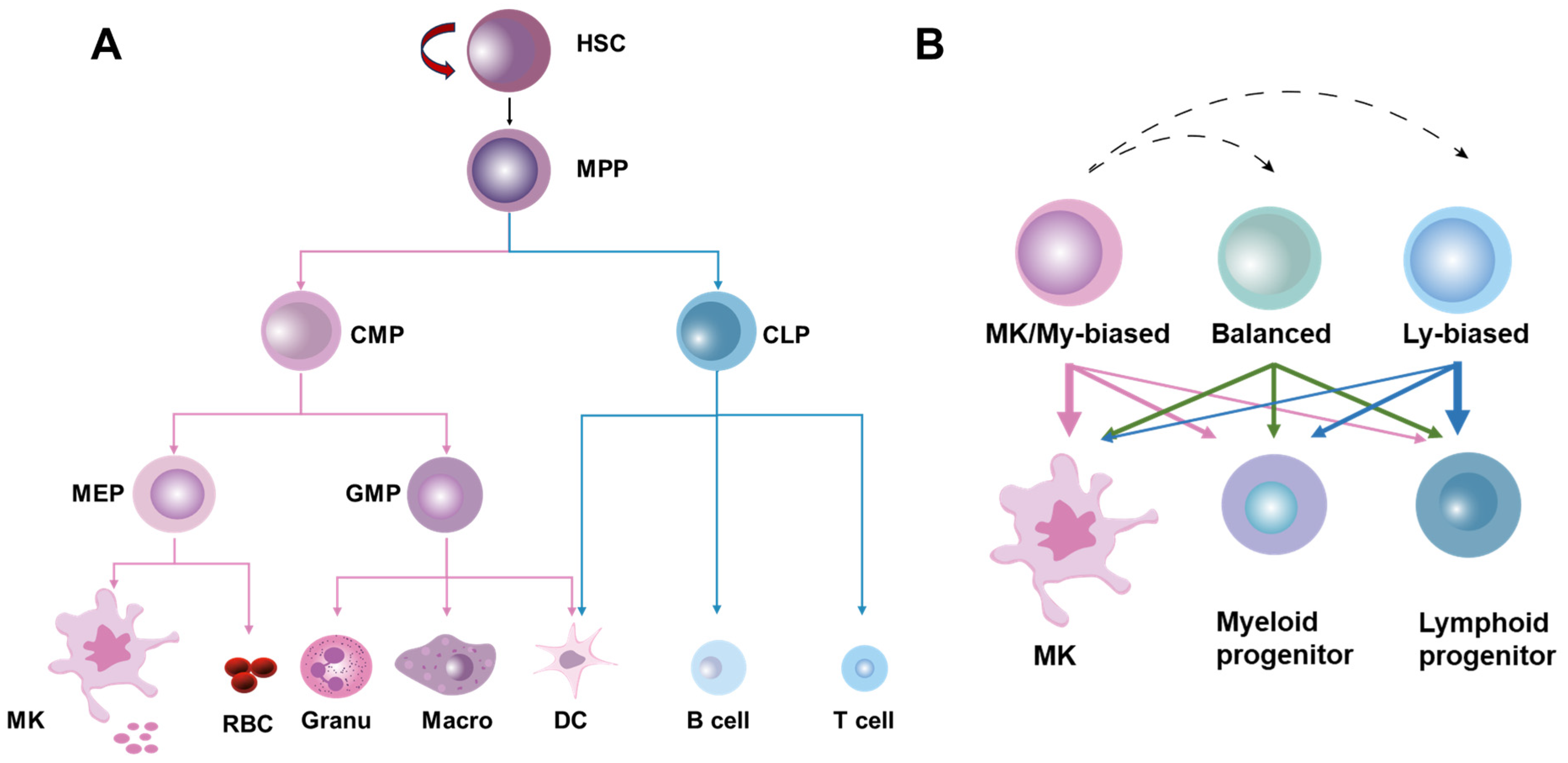
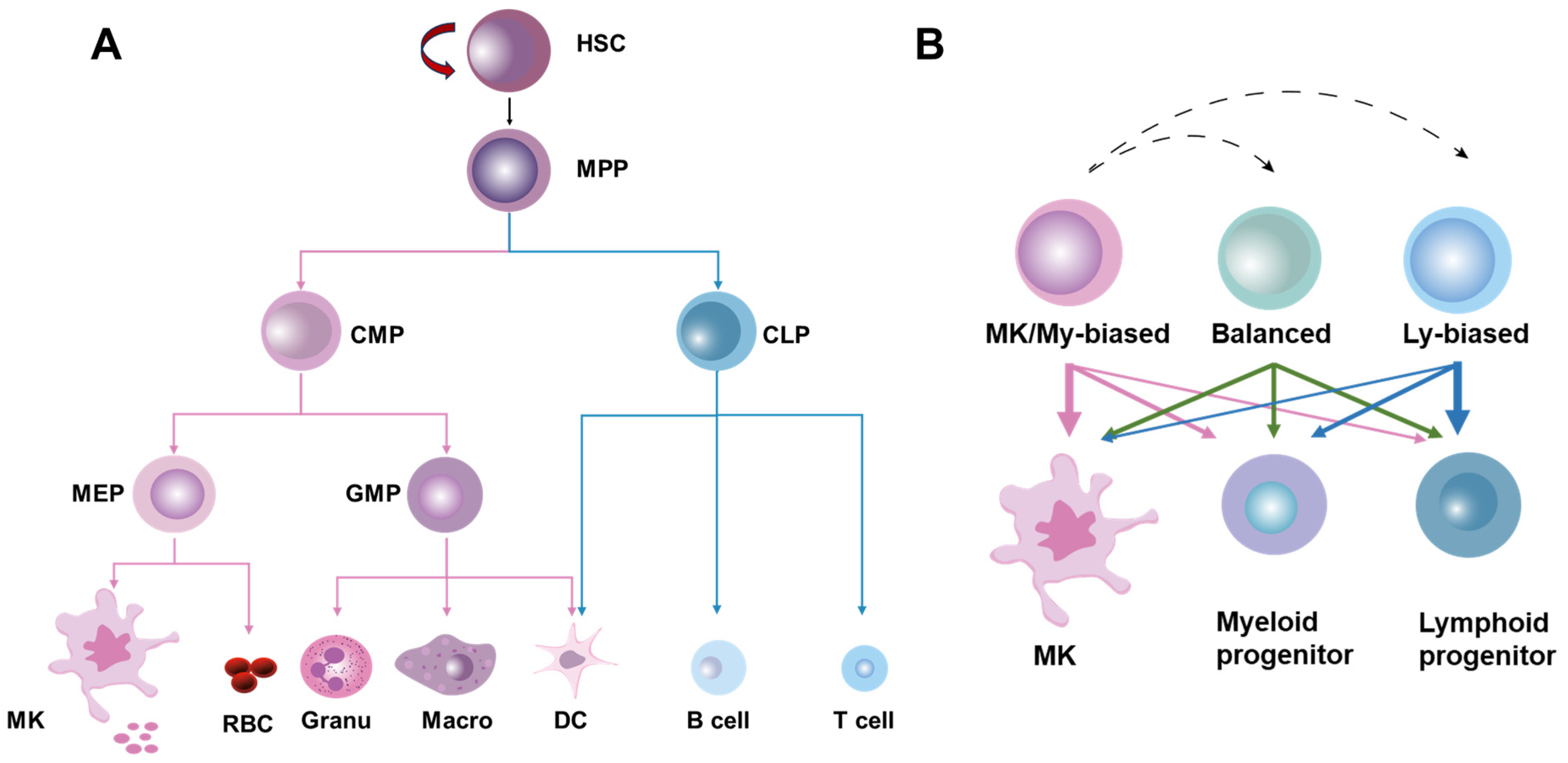
2. Overview of HSC Homeostasis
3. Hematopoietic Underpinnings of CVD
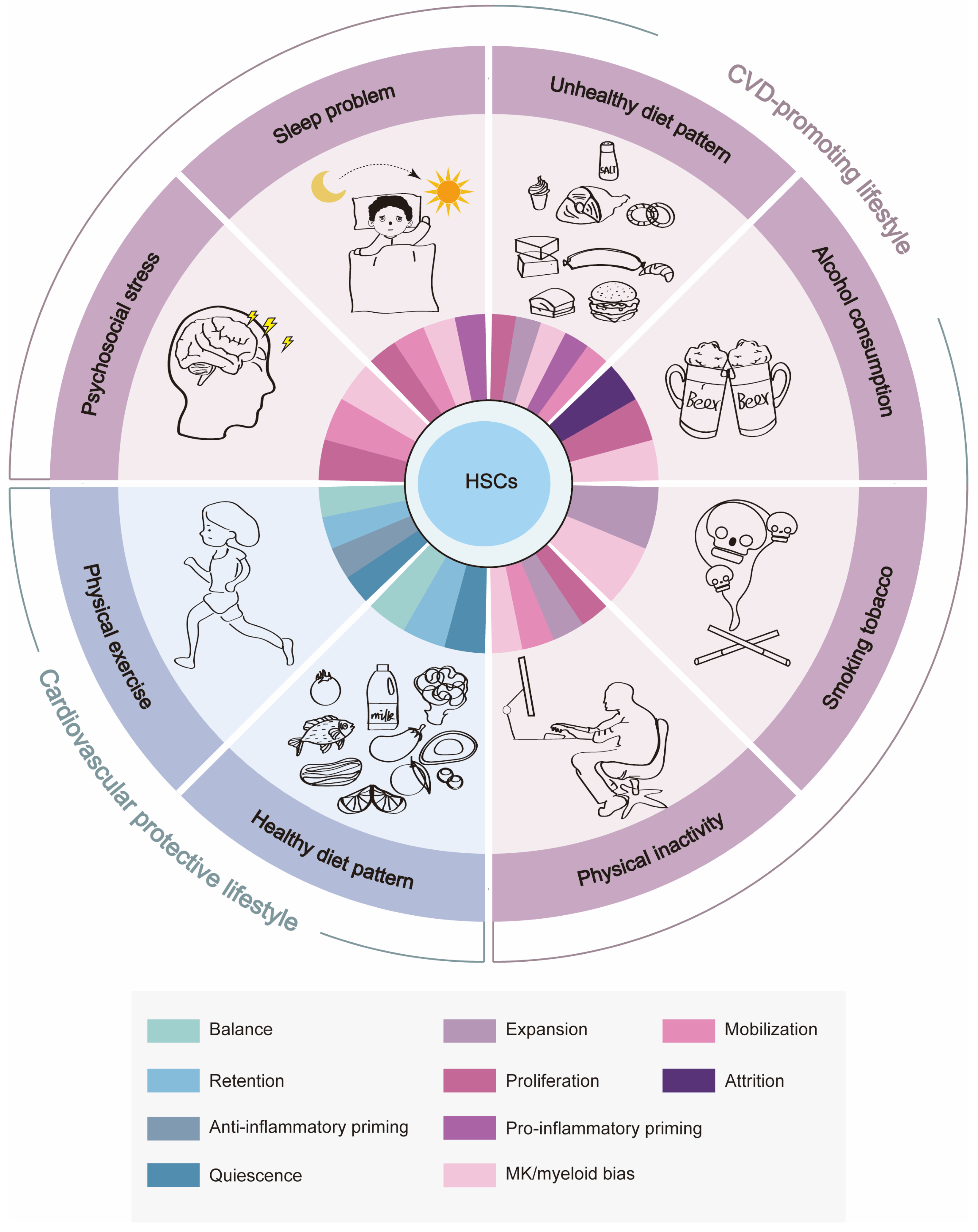
4. CVD-Promoting Lifestyle Remodels BM HSC Homeostasis
4.1. Psychosocial Stress
4.2. Sleep Problems
4.3. Unhealthy Diet Pattern
4.4. Alcohol Consumption and Tobacco Smoking
4.5. Physical Inactivity/Sedentary Behavior
5. Cardiovascular Protective Lifestyle Benefits BM HSC Homeostasis
5.1. Healthy Diet Pattern

{kind=link}
{kind=link}
| Bioactive Compound | Influences on BM HSCs | Potential Mechanisms | Species |
|---|---|---|---|
| Vitamin A | Quiescence Balanced differentiation | Restriction of protein translation and levels of ROS and Myc in HSCs through retinoic acid signaling [129,130]. | Mouse |
| Ascorbate | Balanced differentiation | Stimulation of Tet2 activity [131,132]. | Mouse Human |
| Nicotinamide riboside | Balanced differentiation | Restoration of mitochondrial metabolic activity and promotion of mitochondrial recycling [133,134]. | Mouse Human |
| Vitamin E | Maintenance | Promotion of lipid redox balance in HSCs [135]. | Mouse |
| Urolithin A | Balanced differentiation | Induction of mitochondrial recycling [136]. | Mouse |
| Coffee and tea | Maintenance | Promotion of lipid redox balance in HSCs [139,140,141,142,143]. | Mouse Human |
| Fasting | Balanced differentiation Retention | Reduction of circulating levels of IGF-1 and PKA activity in HSCs [145]; AMPK activation in hepatocytes suppresses systemic CCL2 production via PPARα [146]? Remodeling of CXCL12–CXCR4 and S1P–S1P1R interactions in the BM niche [147,148]? | Mouse Human |
5.2. Physical Exercise
6. HSC Plasticity and Its Association with CVD
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Björkegren, J.L.M.; Lusis, A.J. Atherosclerosis: Recent developments. Cell 2022, 185, 1630–1645. [Google Scholar] [PubMed]
- Vaduganathan, M.; Mensah George, A.; Turco Justine, V.; Fuster, V.; Roth Gregory, A. The Global Burden of Cardiovascular Diseases and Risk. J. Am. Coll. Cardiol. 2022, 80, 2361–2371. [Google Scholar] [PubMed]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [PubMed]
- Magnussen, C.; Ojeda, F.M.; Leong, D.P.; Alegre-Diaz, J.; Amouyel, P.; Aviles-Santa, L.; De Bacquer, D.; Ballantyne, C.M.; Bernabé-Ortiz, A.; Bobak, M.; et al. Global Effect of Modifiable Risk Factors on Cardiovascular Disease and Mortality. N. Engl. J. Med. 2023, 389, 1273–1285. [Google Scholar] [PubMed]
- Schloss, M.J.; Swirski, F.K.; Nahrendorf, M. Modifiable Cardiovascular Risk, Hematopoiesis, and Innate Immunity. Circ. Res. 2020, 126, 1242–1259. [Google Scholar] [PubMed]
- Janssen, H.; Koekkoek, L.L.; Swirski, F.K. Effects of lifestyle factors on leukocytes in cardiovascular health and disease. Nat. Rev. Cardiol. 2024, 21, 157–169. [Google Scholar] [PubMed]
- Nahrendorf, M. Myeloid cell contributions to cardiovascular health and disease. Nat. Med. 2018, 24, 711–720. [Google Scholar] [PubMed]
- Poller, W.C.; Nahrendorf, M.; Swirski, F.K. Hematopoiesis and Cardiovascular Disease. Circ. Res. 2020, 126, 1061–1085. [Google Scholar] [PubMed]
- Vanhie, J.J.; De Lisio, M. How Does Lifestyle Affect Hematopoiesis and the Bone Marrow Microenvironment? Toxicol. Pathol. 2022, 50, 858–866. [Google Scholar]
- Liu, C.; Liao, W.; Chen, J.; Yu, K.; Wu, Y.; Zhang, S.; Chen, M.; Chen, F.; Wang, S.; Cheng, T.; et al. Cholesterol confers ferroptosis resistance onto myeloid-biased hematopoietic stem cells and prevents irradiation-induced myelosuppression. Redox Biol. 2023, 62, 102661. [Google Scholar]
- Du, C.; Wang, X.; Wu, Y.; Liao, W.; Xiong, J.; Zhu, Y.; Liu, C.; Han, W.; Wang, Y.; Han, S.; et al. Renal Klotho and inorganic phosphate are extrinsic factors that antagonistically regulate hematopoietic stem cell maintenance. Cell Rep. 2022, 38, 110392. [Google Scholar]
- Kasbekar, M.; Mitchell, C.A.; Proven, M.A.; Passegué, E. Hematopoietic stem cells through the ages: A lifetime of adaptation to organismal demands. Cell Stem Cell 2023, 30, 1403–1420. [Google Scholar]
- Cheng, H.; Zheng, Z.; Cheng, T. New paradigms on hematopoietic stem cell differentiation. Protein Cell 2020, 11, 34–44. [Google Scholar]
- Haas, S.; Trumpp, A.; Milsom, M.D. Causes and Consequences of Hematopoietic Stem Cell Heterogeneity. Cell Stem Cell 2018, 22, 627–638. [Google Scholar] [PubMed]
- Zeng, Y.; He, J.; Bai, Z.; Li, Z.; Gong, Y.; Liu, C.; Ni, Y.; Du, J.; Ma, C.; Bian, L.; et al. Tracing the first hematopoietic stem cell generation in human embryo by single-cell RNA sequencing. Cell Res. 2019, 29, 881–894. [Google Scholar]
- Li, J.-J.; Liu, J.; Li, Y.E.; Chen, L.V.; Cheng, H.; Li, Y.; Cheng, T.; Wang, Q.-F.; Zhou, B.O. Differentiation route determines the functional outputs of adult megakaryopoiesis. Immunity 2024, 57, 478–494.e476. [Google Scholar] [PubMed]
- Nakamura-Ishizu, A.; Ito, K.; Suda, T. Hematopoietic Stem Cell Metabolism during Development and Aging. Dev. Cell 2020, 54, 239–255. [Google Scholar]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [PubMed]
- Lee-Thedieck, C.; Schertl, P.; Klein, G. The extracellular matrix of hematopoietic stem cell niches. Adv. Drug Deliv. Rev. 2022, 181, 114069. [Google Scholar]
- Zhang, Y.W.; Mess, J.; Aizarani, N.; Mishra, P.; Johnson, C.; Romero-Mulero, M.C.; Rettkowski, J.; Schönberger, K.; Obier, N.; Jäcklein, K.; et al. Hyaluronic acid-GPRC5C signalling promotes dormancy in haematopoietic stem cells. Nat. Cell Biol. 2022, 24, 1038–1048. [Google Scholar]
- Pierce, H.; Zhang, D.; Magnon, C.; Lucas, D.; Christin, J.R.; Huggins, M.; Schwartz, G.J.; Frenette, P.S. Cholinergic Signals from the CNS Regulate G-CSF-Mediated HSC Mobilization from Bone Marrow via a Glucocorticoid Signaling Relay. Cell Stem Cell 2017, 20, 648–658.e644. [Google Scholar] [PubMed]
- Decker, M.; Leslie, J.; Liu, Q.; Ding, L. Hepatic thrombopoietin is required for bone marrow hematopoietic stem cell maintenance. Science 2018, 360, 106–110. [Google Scholar] [PubMed]
- Nakada, D.; Oguro, H.; Levi, B.P.; Ryan, N.; Kitano, A.; Saitoh, Y.; Takeichi, M.; Wendt, G.R.; Morrison, S.J. Oestrogen increases haematopoietic stem-cell self-renewal in females and during pregnancy. Nature 2014, 505, 555–558. [Google Scholar] [PubMed]
- Speer, T.; Dimmeler, S.; Schunk, S.J.; Fliser, D.; Ridker, P.M. Targeting innate immunity-driven inflammation in CKD and cardiovascular disease. Nat. Rev. Nephrol. 2022, 18, 762–778. [Google Scholar] [PubMed]
- Tall, A.R.; Fuster, J.J. Clonal hematopoiesis in cardiovascular disease and therapeutic implications. Nat. Cardiovasc. Res. 2022, 1, 116–124. [Google Scholar] [PubMed]
- Rohde, D.; Vandoorne, K.; Lee, I.H.; Grune, J.; Zhang, S.; McAlpine, C.S.; Schloss, M.J.; Nayar, R.; Courties, G.; Frodermann, V.; et al. Bone marrow endothelial dysfunction promotes myeloid cell expansion in cardiovascular disease. Nat. Cardiovasc. Res. 2022, 1, 28–44. [Google Scholar] [PubMed]
- Chavakis, T.; Mitroulis, I.; Hajishengallis, G. Hematopoietic progenitor cells as integrative hubs for adaptation to and fine-tuning of inflammation. Nat. Immunol. 2019, 20, 802–811. [Google Scholar] [PubMed]
- Kovtonyuk, L.V.; Caiado, F.; Garcia-Martin, S.; Manz, E.-M.; Helbling, P.; Takizawa, H.; Boettcher, S.; Al-Shahrour, F.; Nombela-Arrieta, C.; Slack, E.; et al. IL-1 mediates microbiome-induced inflammaging of hematopoietic stem cells in mice. Blood 2022, 139, 44–58. [Google Scholar]
- Pietras, E.M.; Mirantes-Barbeito, C.; Fong, S.; Loeffler, D.; Kovtonyuk, L.V.; Zhang, S.; Lakshminarasimhan, R.; Chin, C.P.; Techner, J.-M.; Will, B.; et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016, 18, 607–618. [Google Scholar]
- Mitchell, C.A.; Verovskaya, E.V.; Calero-Nieto, F.J.; Olson, O.C.; Swann, J.W.; Wang, X.; Hérault, A.; Dellorusso, P.V.; Zhang, S.Y.; Svendsen, A.F.; et al. Stromal niche inflammation mediated by IL-1 signalling is a targetable driver of haematopoietic ageing. Nat. Cell Biol. 2023, 25, 30–41. [Google Scholar]
- Grusanovic, S.; Danek, P.; Kuzmina, M.; Adamcova, M.K.; Burocziova, M.; Mikyskova, R.; Vanickova, K.; Kosanovic, S.; Pokorna, J.; Reinis, M.; et al. Chronic inflammation decreases HSC fitness by activating the druggable Jak/Stat3 signaling pathway. EMBO Rep. 2023, 24, e54729. [Google Scholar]
- Cheong, J.-G.; Ravishankar, A.; Sharma, S.; Parkhurst, C.N.; Grassmann, S.A.; Wingert, C.K.; Laurent, P.; Ma, S.; Paddock, L.; Miranda, I.C.; et al. Epigenetic memory of coronavirus infection in innate immune cells and their progenitors. Cell 2023, 186, 3882–3902.e3824. [Google Scholar] [PubMed]
- Yamashita, M.; Passegué, E. TNF-α Coordinates Hematopoietic Stem Cell Survival and Myeloid Regeneration. Cell Stem Cell 2019, 25, 357–372.e357. [Google Scholar] [PubMed]
- He, H.; Xu, P.; Zhang, X.; Liao, M.; Dong, Q.; Cong, T.; Tang, B.; Yang, X.; Ye, M.; Chang, Y.; et al. Aging-induced IL27Ra signaling impairs hematopoietic stem cells. Blood 2020, 136, 183–198. [Google Scholar]
- Scicchitano, P.; Marzullo, A.; Santoro, A.; Zito, A.; Cortese, F.; Galeandro, C.; Ciccone, A.S.; Angiletta, D.; Manca, F.; Pulli, R.; et al. The Prognostic Role of ST2L and sST2 in Patients Who Underwent Carotid Plaque Endarterectomy: A Five-Year Follow-Up Study. J. Clin. Med. 2022, 11, 3142. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, M.M.; Cortese, F.; Gesualdo, M.; Riccardi, R.; Di Nunzio, D.; Moncelli, M.; Iacoviello, M.; Scicchitano, P. A Novel Cardiac Bio-Marker: ST2: A Review. Molecules 2013, 18, 15314–15328. [Google Scholar] [CrossRef] [PubMed]
- Le, H.; Kim, W.; Kim, J.; Cho, H.R.; Kwon, B. Interleukin-33: A Mediator of Inflammation Targeting Hematopoietic Stem and Progenitor Cells and Their Progenies. Front. Immunol. 2013, 4, 104. [Google Scholar] [PubMed]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar]
- Liao, W.; Du, C.; Wang, J. The cGAS-STING Pathway in Hematopoiesis and Its Physiopathological Significance. Front. Immunol. 2020, 11, 573915. [Google Scholar]
- Yang, K.; Wang, C.; Nie, L.; Zhao, X.; Gu, J.; Guan, X.; Wang, S.; Xiao, T.; Xu, X.; He, T.; et al. Klotho Protects Against Indoxyl Sulphate-Induced Myocardial Hypertrophy. J. Am. Soc. Nephrol. 2015, 26, 2434–2446. [Google Scholar] [PubMed]
- Han, W.; Du, C.; Zhu, Y.; Ran, L.; Wang, Y.; Xiong, J.; Wu, Y.; Lan, Q.; Wang, Y.; Wang, L.; et al. Targeting Myocardial Mitochondria-STING-Polyamine Axis Prevents Cardiac Hypertrophy in Chronic Kidney Disease. JACC Basic Transl. Sci. 2022, 7, 820–840. [Google Scholar] [PubMed]
- Bi, X.; Du, C.; Wang, X.; Wang, X.; Han, W.; Wang, Y.; Qiao, Y.; Zhu, Y.; Ran, L.; Liu, Y.; et al. Mitochondrial Damage-Induced Innate Immune Activation in Vascular Smooth Muscle Cells Promotes Chronic Kidney Disease-Associated Plaque Vulnerability. Adv. Sci. 2021, 8, 2002738. [Google Scholar]
- Yang, K.; Du, C.; Wang, X.; Li, F.; Xu, Y.; Wang, S.; Chen, S.; Chen, F.; Shen, M.; Chen, M.; et al. Indoxyl sulfate induces platelet hyperactivity and contributes to chronic kidney disease—Associated thrombosis in mice. Blood 2017, 129, 2667–2679. [Google Scholar] [PubMed]
- Lan, Q.; Du, C.; Xiong, J.; Wu, Y.; Liao, W.; Liu, C.; Chen, J.; Ran, L.; Wang, Y.; Wang, Y.; et al. Renal Klotho safeguards platelet lifespan in advanced chronic kidney disease through restraining Bcl-xL ubiquitination and degradation. J. Thromb. Haemost. 2022, 20, 2972–2987. [Google Scholar] [PubMed]
- Du, C.-H.; Wu, Y.-D.; Yang, K.; Liao, W.-N.; Ran, L.; Liu, C.-N.; Zhang, S.-Z.; Yu, K.; Chen, J.; Quan, Y.; et al. Apoptosis-resistant megakaryocytes produce large and hyperreactive platelets in response to radiation injury. Mil. Med. Res. 2023, 10, 66. [Google Scholar]
- Heyde, A.; Rohde, D.; McAlpine, C.S.; Zhang, S.; Hoyer, F.F.; Gerold, J.M.; Cheek, D.; Iwamoto, Y.; Schloss, M.J.; Vandoorne, K.; et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell 2021, 184, 1348–1361.e22. [Google Scholar] [PubMed]
- McKim, D.B.; Yin, W.; Wang, Y.; Cole, S.W.; Godbout, J.P.; Sheridan, J.F. Social Stress Mobilizes Hematopoietic Stem Cells to Establish Persistent Splenic Myelopoiesis. Cell Rep. 2018, 25, 2552–2562.e2553. [Google Scholar] [PubMed]
- Heidt, T.; Sager, H.B.; Courties, G.; Dutta, P.; Iwamoto, Y.; Zaltsman, A.; von zur Muhlen, C.; Bode, C.; Fricchione, G.L.; Denninger, J.; et al. Chronic variable stress activates hematopoietic stem cells. Nat. Med. 2014, 20, 754–758. [Google Scholar]
- Annas, A.-S.; Man, K.S.L.; Alexandra, W.; Danielle, L.M.; Waled, A.S.; Alyce, J.N.; Olivia, D.C.; Michael, J.K.; Camilla Bertuzzo, V.; Ann-Maree, J.; et al. Chronic sympathetic driven hypertension promotes atherosclerosis by enhancing hematopoiesis. Haematologica 2019, 104, 456–467. [Google Scholar]
- Liu, Y.; Chen, Q.; Jeong, H.-W.; Han, D.; Fabian, J.; Drexler, H.C.A.; Stehling, M.; Schöler, H.R.; Adams, R.H. Dopamine signaling regulates hematopoietic stem and progenitor cell function. Blood 2021, 138, 2051–2065. [Google Scholar] [PubMed]
- Niraula, A.; Wang, Y.; Godbout, J.P.; Sheridan, J.F. Corticosterone Production during Repeated Social Defeat Causes Monocyte Mobilization from the Bone Marrow, Glucocorticoid Resistance, and Neurovascular Adhesion Molecule Expression. J. Neurosci. 2018, 38, 2328–2340. [Google Scholar] [PubMed]
- McAlpine, C.S.; Kiss, M.G.; Rattik, S.; He, S.; Vassalli, A.; Valet, C.; Anzai, A.; Chan, C.T.; Mindur, J.E.; Kahles, F.; et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature 2019, 566, 383–387. [Google Scholar] [PubMed]
- Sang, D.; Lin, K.; Yang, Y.; Ran, G.; Li, B.; Chen, C.; Li, Q.; Ma, Y.; Lu, L.; Cui, X.-Y.; et al. Prolonged sleep deprivation induces a cytokine-storm-like syndrome in mammals. Cell 2023, 186, 5500–5516.e5521. [Google Scholar] [PubMed]
- McAlpine, C.S.; Kiss, M.G.; Zuraikat, F.M.; Cheek, D.; Schiroli, G.; Amatullah, H.; Huynh, P.; Bhatti, M.Z.; Wong, L.P.; Yates, A.G.; et al. Sleep exerts lasting effects on hematopoietic stem cell function and diversity. J. Exp. Med. 2022, 219, e20220081. [Google Scholar] [PubMed]
- Méndez-Ferrer, S.; Lucas, D.; Battista, M.; Frenette, P.S. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 2008, 452, 442–447. [Google Scholar] [PubMed]
- Golan, K.; Kumari, A.; Kollet, O.; Khatib-Massalha, E.; Subramaniam, M.D.; Ferreira, Z.S.; Avemaria, F.; Rzeszotek, S.; García-García, A.; Xie, S.; et al. Daily Onset of Light and Darkness Differentially Controls Hematopoietic Stem Cell Differentiation and Maintenance. Cell Stem Cell 2018, 23, 572–585.e577. [Google Scholar]
- García-García, A.; Korn, C.; García-Fernández, M.; Domingues, O.; Villadiego, J.; Martín-Pérez, D.; Isern, J.; Bejarano-García, J.A.; Zimmer, J.; Pérez-Simón, J.A.; et al. Dual cholinergic signals regulate daily migration of hematopoietic stem cells and leukocytes. Blood 2019, 133, 224–236. [Google Scholar]
- Budkowska, M.; Ostrycharz, E.; Wojtowicz, A.; Marcinowska, Z.; Woźniak, J.; Ratajczak, M.Z.; Dołęgowska, B. A Circadian Rhythm in both Complement Cascade (ComC) Activation and Sphingosine-1-Phosphate (S1P) Levels in Human Peripheral Blood Supports a Role for the ComC–S1P Axis in Circadian Changes in the Number of Stem Cells Circulating in Peripheral Blood. Stem Cell Rev. Rep. 2018, 14, 677–685. [Google Scholar]
- Ratajczak, M.Z.; Kim, C.H.; Wojakowski, W.; Janowska-Wieczorek, A.; Kucia, M.; Ratajczak, J. Innate immunity as orchestrator of stem cell mobilization. Leukemia 2010, 24, 1667–1675. [Google Scholar]
- Borkowska, S.; Suszynska, M.; Mierzejewska, K.; Ismail, A.; Budkowska, M.; Salata, D.; Dolegowska, B.; Kucia, M.; Ratajczak, J.; Ratajczak, M.Z. Novel evidence that crosstalk between the complement, coagulation and fibrinolysis proteolytic cascades is involved in mobilization of hematopoietic stem/progenitor cells (HSPCs). Leukemia 2014, 28, 2148–2154. [Google Scholar]
- Adamiak, M.; Ciechanowicz, A.; Skoda, M.; Cymer, M.; Tracz, M.; Xu, B.; Ratajczak, M.Z. Novel Evidence that Purinergic Signaling—Nlrp3 Inflammasome Axis Regulates Circadian Rhythm of Hematopoietic Stem/Progenitor Cells Circulation in Peripheral Blood. Stem Cell Rev. Rep. 2020, 16, 335–343. [Google Scholar]
- Adamiak, M.; Abdel-Latif, A.; Bujko, K.; Thapa, A.; Anusz, K.; Tracz, M.; Brzezniakiewicz-Janus, K.; Ratajczak, J.; Kucia, M.; Ratajczak, M.Z. Nlrp3 Inflammasome Signaling Regulates the Homing and Engraftment of Hematopoietic Stem Cells (HSPCs) by Enhancing Incorporation of CXCR4 Receptor into Membrane Lipid Rafts. Stem Cell Rev. Rep. 2020, 16, 954–967. [Google Scholar] [PubMed]
- Stiekema, L.C.A.; Willemsen, L.; Kaiser, Y.; Prange, K.H.M.; Wareham, N.J.; Boekholdt, S.M.; Kuijk, C.; de Winther, M.P.J.; Voermans, C.; Nahrendorf, M.; et al. Impact of cholesterol on proinflammatory monocyte production by the bone marrow. Eur. Heart J. 2021, 42, 4309–4320. [Google Scholar] [PubMed]
- Gu, Q.; Yang, X.; Lv, J.; Zhang, J.; Xia, B.; Kim, J.-d.; Wang, R.; Xiong, F.; Meng, S.; Clements, T.P.; et al. AIBP-mediated cholesterol efflux instructs hematopoietic stem and progenitor cell fate. Science 2019, 363, 1085–1088. [Google Scholar]
- Westerterp, M.; Gourion-Arsiquaud, S.; Murphy, A.J.; Shih, A.; Cremers, S.; Levine, R.L.; Tall, A.R.; Yvan-Charvet, L. Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell Stem Cell 2012, 11, 195–206. [Google Scholar] [PubMed]
- Oguro, H.; McDonald, J.G.; Zhao, Z.; Umetani, M.; Shaul, P.W.; Morrison, S.J. 27-Hydroxycholesterol induces hematopoietic stem cell mobilization and extramedullary hematopoiesis during pregnancy. J. Clin. Investig. 2017, 127, 3392–3401. [Google Scholar]
- Singer, K.; DelProposto, J.; Lee Morris, D.; Zamarron, B.; Mergian, T.; Maley, N.; Cho, K.W.; Geletka, L.; Subbaiah, P.; Muir, L.; et al. Diet-induced obesity promotes myelopoiesis in hematopoietic stem cells. Mol. Metab. 2014, 3, 664–675. [Google Scholar]
- Lee, J.-M.; Govindarajah, V.; Goddard, B.; Hinge, A.; Muench, D.E.; Filippi, M.-D.; Aronow, B.; Cancelas, J.A.; Salomonis, N.; Grimes, H.L.; et al. Obesity alters the long-term fitness of the hematopoietic stem cell compartment through modulation of Gfi1 expression. J. Exp. Med. 2017, 215, 627–644. [Google Scholar]
- Hermetet, F.; Buffière, A.; Aznague, A.; Pais de Barros, J.-P.; Bastie, J.-N.; Delva, L.; Quéré, R. High-fat diet disturbs lipid raft/TGF-β signaling-mediated maintenance of hematopoietic stem cells in mouse bone marrow. Nat. Commun. 2019, 10, 523. [Google Scholar]
- Ambrosi, T.H.; Scialdone, A.; Graja, A.; Gohlke, S.; Jank, A.M.; Bocian, C.; Woelk, L.; Fan, H.; Logan, D.W.; Schürmann, A.; et al. Adipocyte Accumulation in the Bone Marrow during Obesity and Aging Impairs Stem Cell-Based Hematopoietic and Bone Regeneration. Cell Stem Cell 2017, 20, 771–784.e776. [Google Scholar] [PubMed]
- Pasupuleti, S.K.; Ramdas, B.; Burns, S.S.; Palam, L.R.; Kanumuri, R.; Kumar, R.; Pandhiri, T.R.; Dave, U.P.; Yellapu, N.K.; Zhou, X.; et al. Obesity-induced inflammation exacerbates clonal hematopoiesis. J. Clin. Investig. 2023, 133, e163968. [Google Scholar] [PubMed]
- Luo, Y.; Chen, G.-L.; Hannemann, N.; Ipseiz, N.; Krönke, G.; Bäuerle, T.; Munos, L.; Wirtz, S.; Schett, G.; Bozec, A. Microbiota from Obese Mice Regulate Hematopoietic Stem Cell Differentiation by Altering the Bone Niche. Cell Metab. 2015, 22, 886–894. [Google Scholar] [PubMed]
- Lee, M.K.S.; Murphy, A.J. A high-salt diet promotes atherosclerosis by altering haematopoiesis. Nat. Rev. Cardiol. 2023, 20, 435–436. [Google Scholar] [PubMed]
- Côrte-Real, B.F.; Hamad, I.; Arroyo Hornero, R.; Geisberger, S.; Roels, J.; Van Zeebroeck, L.; Dyczko, A.; van Gisbergen, M.W.; Kurniawan, H.; Wagner, A.; et al. Sodium perturbs mitochondrial respiration and induces dysfunctional Tregs. Cell Metab. 2023, 35, 299–315.e298. [Google Scholar] [PubMed]
- Wu, Y.; Liao, W.; Chen, J.; Liu, C.; Zhang, S.; Yu, K.; Wang, X.; Chen, M.; Wang, S.; Ran, X.; et al. Phosphate Metabolic Inhibition Contributes to Irradiation-Induced Myelosuppression through Dampening Hematopoietic Stem Cell Survival. Nutrients 2022, 14, 3395. [Google Scholar] [CrossRef] [PubMed]
- Seufert, A.L.; Hickman, J.W.; Traxler, S.K.; Peterson, R.M.; Waugh, T.A.; Lashley, S.J.; Shulzhenko, N.; Napier, R.J.; Napier, B.A. Enriched dietary saturated fatty acids induce trained immunity via ceramide production that enhances severity of endotoxemia and clearance of infection. Elife 2022, 11, e76744. [Google Scholar] [PubMed]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Daly, M.; Arends, M.J.; Patel, K.J. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 2012, 489, 571–575. [Google Scholar] [PubMed]
- Pontel, L.B.; Rosado, I.V.; Burgos-Barragan, G.; Garaycoechea, J.I.; Yu, R.; Arends, M.J.; Chandrasekaran, G.; Broecker, V.; Wei, W.; Liu, L.; et al. Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Mol. Cell 2015, 60, 177–188. [Google Scholar]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018, 553, 171–177. [Google Scholar]
- Kaastrup, K.; Grønbæk, K. The Impact of Sedentary Lifestyle, High-fat Diet, Tobacco Smoke, and Alcohol Intake on the Hematopoietic Stem Cell Niches. Hemasphere 2021, 5, e615. [Google Scholar] [PubMed]
- Chang, E.; Forsberg, E.C.; Wu, J.; Bingyin, W.; Prohaska, S.S.; Allsopp, R.; Weissman, I.L.; Cooke, J.P. Cholinergic activation of hematopoietic stem cells: Role in tobacco-related disease? Vasc. Med. 2010, 15, 375–385. [Google Scholar]
- Cool, T.; Baena, A.R.; Forsberg, E.C. Clearing the Haze: How Does Nicotine Affect Hematopoiesis before and after Birth? Cancers 2022, 14, 184. [Google Scholar]
- Frodermann, V.; Rohde, D.; Courties, G.; Severe, N.; Schloss, M.J.; Amatullah, H.; McAlpine, C.S.; Cremer, S.; Hoyer, F.F.; Ji, F.; et al. Exercise reduces inflammatory cell production and cardiovascular inflammation via instruction of hematopoietic progenitor cells. Nat. Med. 2019, 25, 1761–1771. [Google Scholar] [PubMed]
- Osborne, M.T.; Shin, L.M.; Mehta, N.N.; Pitman, R.K.; Fayad, Z.A.; Tawakol, A. Disentangling the Links Between Psychosocial Stress and Cardiovascular Disease. Circ. Cardiovasc. Imaging 2020, 13, e010931. [Google Scholar] [PubMed]
- Barry, M.J.; Nicholson, W.K.; Silverstein, M.; Coker, T.R.; Davidson, K.W.; Davis, E.M.; Donahue, K.E.; Jaén, C.R.; Li, L.; Ogedegbe, G.; et al. Screening for Anxiety Disorders in Adults: US Preventive Services Task Force Recommendation Statement. JAMA 2023, 329, 2163–2170. [Google Scholar] [PubMed]
- Kataoka, N.; Shima, Y.; Nakajima, K.; Nakamura, K. A central master driver of psychosocial stress responses in the rat. Science 2020, 367, 1105–1112. [Google Scholar] [PubMed]
- Hanoun, M.; Maryanovich, M.; Arnal-Estapé, A.; Frenette, P.S. Neural regulation of hematopoiesis, inflammation, and cancer. Neuron 2015, 86, 360–373. [Google Scholar] [PubMed]
- Fu, W.; Meng, G.; Yang, X.; Yu, L.; Jiang, H. Bone marrow sympathetic activation regulates post-myocardial infarction megakaryocyte expansion but not platelet production. Biochem. Biophys. Res. Commun. 2019, 513, 99–104. [Google Scholar]
- Liu, W.; Chen, W.; Xie, M.; Chen, C.; Shao, Z.; Zhang, Y.; Zhao, H.; Song, Q.; Hu, H.; Xing, X.; et al. Traumatic brain injury stimulates sympathetic tone-mediated bone marrow myelopoiesis to favor fracture healing. Signal Transduct. Target. Ther. 2023, 8, 260. [Google Scholar]
- Chen, S.; Du, C.; Shen, M.; Zhao, G.; Xu, Y.; Yang, K.; Wang, X.; Li, F.; Zeng, D.; Chen, F.; et al. Sympathetic stimulation facilitates thrombopoiesis by promoting megakaryocyte adhesion, migration, and proplatelet formation. Blood 2016, 127, 1024–1035. [Google Scholar]
- Vasamsetti, S.B.; Florentin, J.; Coppin, E.; Stiekema, L.C.A.; Zheng, K.H.; Nisar, M.U.; Sembrat, J.; Levinthal, D.J.; Rojas, M.; Stroes, E.S.G.; et al. Sympathetic Neuronal Activation Triggers Myeloid Progenitor Proliferation and Differentiation. Immunity 2018, 49, 93–106.e107. [Google Scholar]
- Gao, X.; Zhang, D.; Xu, C.; Li, H.; Caron, K.M.; Frenette, P.S. Nociceptive nerves regulate haematopoietic stem cell mobilization. Nature 2021, 589, 591–596. [Google Scholar]
- Dong, Z.; Hou, L.; Luo, W.; Pan, L.-H.; Li, X.; Tan, H.-P.; Wu, R.-D.; Lu, H.; Yao, K.; Mu, M.-D.; et al. Myocardial infarction drives trained immunity of monocytes, accelerating atherosclerosis. Eur. Heart J. 2023, 45, 669–684. [Google Scholar]
- Guo, B.; Huang, X.; Cooper, S.; Broxmeyer, H.E. Glucocorticoid hormone-induced chromatin remodeling enhances human hematopoietic stem cell homing and engraftment. Nat. Med. 2017, 23, 424–428. [Google Scholar]
- Meyer, N.; Harvey, A.G.; Lockley, S.W.; Dijk, D.-J. Circadian rhythms and disorders of the timing of sleep. Lancet 2022, 400, 1061–1078. [Google Scholar]
- Makarem, N.; Castro-Diehl, C.; St-Onge, M.P.; Redline, S.; Shea, S.; Lloyd-Jones, D.; Ning, H.; Aggarwal, B. Redefining Cardiovascular Health to Include Sleep: Prospective Associations with Cardiovascular Disease in the MESA Sleep Study. J. Am. Heart Assoc. 2022, 11, e025252. [Google Scholar]
- Ziegler, K.A.; Ahles, A.; Dueck, A.; Esfandyari, D.; Pichler, P.; Weber, K.; Kotschi, S.; Bartelt, A.; Sinicina, I.; Graw, M.; et al. Immune-mediated denervation of the pineal gland underlies sleep disturbance in cardiac disease. Science 2023, 381, 285–290. [Google Scholar]
- Lane, J.M.; Qian, J.; Mignot, E.; Redline, S.; Scheer, F.A.J.L.; Saxena, R. Genetics of circadian rhythms and sleep in human health and disease. Nat. Rev. Genet. 2023, 24, 4–20. [Google Scholar] [PubMed]
- Carson, J.A.S.; Lichtenstein, A.H.; Anderson, C.A.M.; Appel, L.J.; Kris-Etherton, P.M.; Meyer, K.A.; Petersen, K.; Polonsky, T.; Van Horn, L.; American Heart Association Nutrition Committee of the Council on Lifestyle and Cardiometabolic Health. Dietary Cholesterol and Cardiovascular Risk: A Science Advisory From the American Heart Association. Circulation 2020, 141, e39–e53. [Google Scholar] [PubMed]
- Hunter, R.W.; Dhaun, N.; Bailey, M.A. The impact of excessive salt intake on human health. Nat. Rev. Nephrol. 2022, 18, 321–335. [Google Scholar] [PubMed]
- Martínez-González, M.A.; Gea, A.; Ruiz-Canela, M. The Mediterranean Diet and Cardiovascular Health. Circ. Res. 2019, 124, 779–798. [Google Scholar] [PubMed]
- O’Neill, B.; Raggi, P. The ketogenic diet: Pros and cons. Atherosclerosis 2020, 292, 119–126. [Google Scholar] [PubMed]
- Vasim, I.; Majeed, C.N.; DeBoer, M.D. Intermittent Fasting and Metabolic Health. Nutrients 2022, 14, 631. [Google Scholar] [CrossRef] [PubMed]
- Guasch-Ferré, M.; Satija, A.; Blondin, S.A.; Janiszewski, M.; Emlen, E.; O’Connor, L.E.; Campbell, W.W.; Hu, F.B.; Willett, W.C.; Stampfer, M.J. Meta-Analysis of Randomized Controlled Trials of Red Meat Consumption in Comparison with Various Comparison Diets on Cardiovascular Risk Factors. Circulation 2019, 139, 1828–1845. [Google Scholar] [PubMed]
- Bernard, S.; Léopold, K.F.; Emmanuelle, K.-G.; Benjamin, A.; Caroline, M.; Roland, M.A.; Eloi, C.; Mélanie, D.; Serge, H.; Pilar, G.; et al. Ultra-processed food intake and risk of cardiovascular disease: Prospective cohort study (NutriNet-Santé). BMJ 2019, 365, l1451. [Google Scholar]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.N.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9–Niemann-Pick C1 signaling complex. Science 2017, 355, 1306–1311. [Google Scholar] [PubMed]
- Labella, R.; Vujačić, M.; Trivanović, D. Bone Marrow Adipose Tissue: Regulation of Osteoblastic Niche, Hematopoiesis and Hematological Malignancies. Stem Cell Rev. Rep. 2023, 19, 1135–1151. [Google Scholar] [PubMed]
- Tencerova, M.; Figeac, F.; Ditzel, N.; Taipaleenmäki, H.; Nielsen, T.K.; Kassem, M. High-Fat Diet-Induced Obesity Promotes Expansion of Bone Marrow Adipose Tissue and Impairs Skeletal Stem Cell Functions in Mice. J. Bone Miner. Res. 2018, 33, 1154–1165. [Google Scholar]
- Baldo, M.P.; Rodrigues, S.L.; Mill, J.G. High salt intake as a multifaceted cardiovascular disease: New support from cellular and molecular evidence. Heart Fail. Rev. 2015, 20, 461–474. [Google Scholar]
- Müller, D.N.; Wilck, N.; Haase, S.; Kleinewietfeld, M.; Linker, R.A. Sodium in the microenvironment regulates immune responses and tissue homeostasis. Nat. Rev. Immunol. 2019, 19, 243–254. [Google Scholar] [PubMed]
- Erem, S.; Razzaque, M.S. Dietary phosphate toxicity: An emerging global health concern. Histochem. Cell Biol. 2018, 150, 711–719. [Google Scholar] [PubMed]
- Beam, A.; Clinger, E.; Hao, L. Effect of Diet and Dietary Components on the Composition of the Gut Microbiota. Nutrients 2021, 13, 2795. [Google Scholar] [CrossRef]
- Josefsdottir, K.S.; Baldridge, M.T.; Kadmon, C.S.; King, K.Y. Antibiotics impair murine hematopoiesis by depleting the intestinal microbiota. Blood 2017, 129, 729–739. [Google Scholar] [PubMed]
- Iwamura, C.; Bouladoux, N.; Belkaid, Y.; Sher, A.; Jankovic, D. Sensing of the microbiota by NOD1 in mesenchymal stromal cells regulates murine hematopoiesis. Blood 2017, 129, 171–176. [Google Scholar]
- Zhang, D.; Gao, X.; Li, H.; Borger, D.K.; Wei, Q.; Yang, E.; Xu, C.; Pinho, S.; Frenette, P.S. The microbiota regulates hematopoietic stem cell fate decisions by controlling iron availability in bone marrow. Cell Stem Cell 2022, 29, 232–247.e237. [Google Scholar] [PubMed]
- Zeng, X.; Li, X.; Li, X.; Wei, C.; Shi, C.; Hu, K.; Kong, D.; Luo, Q.; Xu, Y.; Shan, W.; et al. Fecal microbiota transplantation from young mice rejuvenates aged hematopoietic stem cells by suppressing inflammation. Blood 2023, 141, 1691–1707. [Google Scholar]
- Charakida, M.; Georgiopoulos, G.; Dangardt, F.; Chiesa, S.T.; Hughes, A.D.; Rapala, A.; Davey Smith, G.; Lawlor, D.; Finer, N.; Deanfield, J.E. Early vascular damage from smoking and alcohol in teenage years: The ALSPAC study. Eur. Heart J. 2019, 40, 345–353. [Google Scholar]
- Varlamov, O.; Bucher, M.; Myatt, L.; Newman, N.; Grant, K.A. Daily Ethanol Drinking Followed by an Abstinence Period Impairs Bone Marrow Niche and Mitochondrial Function of Hematopoietic Stem/Progenitor Cells in Rhesus Macaques. Alcohol. Clin. Exp. Res. 2020, 44, 1088–1098. [Google Scholar]
- Eeden, S.F.v.; Hogg, J.C. The response of human bone marrow to chronic cigarette smoking. Eur. Respir. J. 2000, 15, 915. [Google Scholar]
- Kerr, N.R.; Booth, F.W. Contributions of physical inactivity and sedentary behavior to metabolic and endocrine diseases. Trends Endocrinol. Metab. 2022, 33, 817–827. [Google Scholar] [PubMed]
- Yousefzadeh, M.J.; Flores, R.R.; Zhu, Y.; Schmiechen, Z.C.; Brooks, R.W.; Trussoni, C.E.; Cui, Y.; Angelini, L.; Lee, K.-A.; McGowan, S.J.; et al. An aged immune system drives senescence and ageing of solid organs. Nature 2021, 594, 100–105. [Google Scholar]
- Bogeska, R.; Mikecin, A.-M.; Kaschutnig, P.; Fawaz, M.; Büchler-Schäff, M.; Le, D.; Ganuza, M.; Vollmer, A.; Paffenholz, S.V.; Asada, N.; et al. Inflammatory exposure drives long-lived impairment of hematopoietic stem cell self-renewal activity and accelerated aging. Cell Stem Cell 2022, 29, 1273–1284.e1278. [Google Scholar]
- Jeon, O.H.; Mehdipour, M.; Gil, T.-H.; Kang, M.; Aguirre, N.W.; Robinson, Z.R.; Kato, C.; Etienne, J.; Lee, H.G.; Alimirah, F.; et al. Systemic induction of senescence in young mice after single heterochronic blood exchange. Nat. Metab. 2022, 4, 995–1006. [Google Scholar] [PubMed]
- Raffin, J.; de Souto Barreto, P.; Le Traon, A.P.; Vellas, B.; Aubertin-Leheudre, M.; Rolland, Y. Sedentary behavior and the biological hallmarks of aging. Ageing Res. Rev. 2023, 83, 101807. [Google Scholar]
- Cena, H.; Calder, P.C. Defining a Healthy Diet: Evidence for the Role of Contemporary Dietary Patterns in Health and Disease. Nutrients 2020, 12, 334. [Google Scholar] [CrossRef]
- Wang, P.; Song, M.; Eliassen, A.H.; Wang, M.; Fung, T.T.; Clinton, S.K.; Rimm, E.B.; Hu, F.B.; Willett, W.C.; Tabung, F.K.; et al. Optimal dietary patterns for prevention of chronic disease. Nat. Med. 2023, 29, 719–728. [Google Scholar] [PubMed]
- Bhattacharya, R.; Zekavat, S.M.; Uddin, M.M.; Pirruccello, J.; Niroula, A.; Gibson, C.; Griffin, G.K.; Libby, P.; Ebert, B.L.; Bick, A.; et al. Association of Diet Quality With Prevalence of Clonal Hematopoiesis and Adverse Cardiovascular Events. JAMA Cardiol. 2021, 6, 1069–1077. [Google Scholar]
- Cabezas-Wallscheid, N.; Buettner, F.; Sommerkamp, P.; Klimmeck, D.; Ladel, L.; Thalheimer, F.B.; Pastor-Flores, D.; Roma, L.P.; Renders, S.; Zeisberger, P.; et al. Vitamin A-Retinoic Acid Signaling Regulates Hematopoietic Stem Cell Dormancy. Cell 2017, 169, 807–823.e819. [Google Scholar]
- Schönberger, K.; Obier, N.; Romero-Mulero, M.C.; Cauchy, P.; Mess, J.; Pavlovich, P.V.; Zhang, Y.W.; Mitterer, M.; Rettkowski, J.; Lalioti, M.-E.; et al. Multilayer omics analysis reveals a non-classical retinoic acid signaling axis that regulates hematopoietic stem cell identity. Cell Stem Cell 2022, 29, 131–148.e110. [Google Scholar]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [PubMed]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e1020. [Google Scholar] [PubMed]
- Sun, X.; Cao, B.; Naval-Sanchez, M.; Pham, T.; Sun, Y.B.Y.; Williams, B.; Heazlewood, S.Y.; Deshpande, N.; Li, J.; Kraus, F.; et al. Nicotinamide riboside attenuates age-associated metabolic and functional changes in hematopoietic stem cells. Nat. Commun. 2021, 12, 2665. [Google Scholar] [PubMed]
- Vannini, N.; Campos, V.; Girotra, M.; Trachsel, V.; Rojas-Sutterlin, S.; Tratwal, J.; Ragusa, S.; Stefanidis, E.; Ryu, D.; Rainer, P.Y.; et al. The NAD-Booster Nicotinamide Riboside Potently Stimulates Hematopoiesis through Increased Mitochondrial Clearance. Cell Stem Cell 2019, 24, 405–418.e407. [Google Scholar] [PubMed]
- Hu, Q.; Zhang, Y.; Lou, H.; Ou, Z.; Liu, J.; Duan, W.; Wang, H.; Ge, Y.; Min, J.; Wang, F.; et al. GPX4 and vitamin E cooperatively protect hematopoietic stem and progenitor cells from lipid peroxidation and ferroptosis. Cell Death Dis. 2021, 12, 706. [Google Scholar] [PubMed]
- Girotra, M.; Chiang, Y.-H.; Charmoy, M.; Ginefra, P.; Hope, H.C.; Bataclan, C.; Yu, Y.-R.; Schyrr, F.; Franco, F.; Geiger, H.; et al. Induction of mitochondrial recycling reverts age-associated decline of the hematopoietic and immune systems. Nat. Aging 2023, 3, 1057–1066. [Google Scholar] [PubMed]
- Ye, S.; Shah, B.R.; Li, J.; Liang, H.; Zhan, F.; Geng, F.; Li, B. A critical review on interplay between dietary fibers and gut microbiota. Trends Food Sci. Technol. 2022, 124, 237–249. [Google Scholar]
- Chieng, D.; Kistler, P.M. Coffee and tea on cardiovascular disease (CVD) prevention. Trends Cardiovasc. Med. 2022, 32, 399–405. [Google Scholar] [PubMed]
- Wang, X.; Liao, W.; Chen, J.; Wu, Y.; Liu, C.; Chen, S.; Xu, Y.; Wang, S.; Su, Y.; Du, C.; et al. Caffeic acid attenuates irradiation-induced hematopoietic stem cell apoptosis through inhibiting mitochondrial damage. Exp. Cell Res. 2021, 409, 112934. [Google Scholar]
- Han, X.; Zhang, J.; Xue, X.; Zhao, Y.; Lu, L.; Cui, M.; Miao, W.; Fan, S. Theaflavin ameliorates ionizing radiation-induced hematopoietic injury via the NRF2 pathway. Free Radic. Biol. Med. 2017, 113, 59–70. [Google Scholar]
- Ferdousi, F.; Araki, R.; Hashimoto, K.; Isoda, H. Olive leaf tea may have hematological health benefit over green tea. Clin. Nutr. 2019, 38, 2952–2955. [Google Scholar] [PubMed]
- Tiwari, M.; Dixit, B.; Parvez, S.; Agrawala, P.K. EGCG, a tea polyphenol, as a potential mitigator of hematopoietic radiation injury in mice. Biomed. Pharmacother. 2017, 88, 203–209. [Google Scholar] [PubMed]
- Long, W.; Zhang, G.; Dong, Y.; Li, D. Dark tea extract mitigates hematopoietic radiation injury with antioxidative activity. J. Radiat. Res. 2018, 59, 387–394. [Google Scholar] [PubMed]
- Longo, V.D.; Di Tano, M.; Mattson, M.P.; Guidi, N. Intermittent and periodic fasting, longevity and disease. Nat. Aging 2021, 1, 47–59. [Google Scholar] [PubMed]
- Cheng, C.-W.; Adams, G.B.; Perin, L.; Wei, M.; Zhou, X.; Lam, B.S.; Da Sacco, S.; Mirisola, M.; Quinn, D.I.; Dorff, T.B.; et al. Prolonged Fasting Reduces IGF-1/PKA to Promote Hematopoietic-Stem-Cell-Based Regeneration and Reverse Immunosuppression. Cell Stem Cell 2014, 14, 810–823. [Google Scholar] [PubMed]
- Jordan, S.; Tung, N.; Casanova-Acebes, M.; Chang, C.; Cantoni, C.; Zhang, D.; Wirtz, T.H.; Naik, S.; Rose, S.A.; Brocker, C.N.; et al. Dietary Intake Regulates the Circulating Inflammatory Monocyte Pool. Cell 2019, 178, 1102–1114.e1117. [Google Scholar] [PubMed]
- Janssen, H.; Kahles, F.; Liu, D.; Downey, J.; Koekkoek, L.L.; Roudko, V.; D’Souza, D.; McAlpine, C.S.; Halle, L.; Poller, W.C.; et al. Monocytes re-enter the bone marrow during fasting and alter the host response to infection. Immunity 2023, 56, 783–796.e787. [Google Scholar] [PubMed]
- Collins, N.; Han, S.J.; Enamorado, M.; Link, V.M.; Huang, B.; Moseman, E.A.; Kishton, R.J.; Shannon, J.P.; Dixit, D.; Schwab, S.R.; et al. The Bone Marrow Protects and Optimizes Immunological Memory during Dietary Restriction. Cell 2019, 178, 1088–1101.e1015. [Google Scholar] [PubMed]
- Moreira, J.B.N.; Wohlwend, M.; Wisløff, U. Exercise and cardiac health: Physiological and molecular insights. Nat. Metab. 2020, 2, 829–839. [Google Scholar]
- Peng, H.; Hu, B.; Xie, L.Q.; Su, T.; Li, C.J.; Liu, Y.; Yang, M.; Xiao, Y.; Feng, X.; Zhou, R.; et al. A mechanosensitive lipolytic factor in the bone marrow promotes osteogenesis and lymphopoiesis. Cell Metab. 2022, 34, 1168–1182.e1166. [Google Scholar]
- Liu, L.; Kim, S.; Buckley, M.T.; Reyes, J.M.; Kang, J.; Tian, L.; Wang, M.; Lieu, A.; Mao, M.; Rodriguez-Mateo, C.; et al. Exercise reprograms the inflammatory landscape of multiple stem cell compartments during mammalian aging. Cell Stem Cell 2023, 30, 689–705.e684. [Google Scholar] [PubMed]
- Shen, B.; Tasdogan, A.; Ubellacker, J.M.; Zhang, J.; Nosyreva, E.D.; Du, L.; Murphy, M.M.; Hu, S.; Yi, Y.; Kara, N.; et al. A mechanosensitive peri-arteriolar niche for osteogenesis and lymphopoiesis. Nature 2021, 591, 438–444. [Google Scholar] [PubMed]
- Avots, A.; Harder, F.; Schmittwolf, C.; Petrovic, S.; Mueller, A.M. Plasticity of hematopoietic stem cells and cellular memory. Immunol. Rev. 2002, 187, 9–21. [Google Scholar] [PubMed]
- Sugden, W.W.; North, T.E. Making Blood from the Vessel: Extrinsic and Environmental Cues Guiding the Endothelial-to-Hematopoietic Transition. Life 2021, 11, 1027. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.S.; Jiang, S.; Afentoulis, M.; Baumann, C.I.; Schroeder, D.A.; Olson, S.B.; Wong, M.H.; Fleming, W.H. Transplanted adult hematopoietic stems cells differentiate into functional endothelial cells. Blood 2004, 103, 13–19. [Google Scholar] [PubMed]
- Visconti, R.P.; Ebihara, Y.; LaRue, A.C.; Fleming, P.A.; McQuinn, T.C.; Masuya, M.; Minamiguchi, H.; Markwald, R.R.; Ogawa, M.; Drake, C.J. An In Vivo Analysis of Hematopoietic Stem Cell Potential. Circ. Res. 2006, 98, 690–696. [Google Scholar] [PubMed]
- Burt, R.K.; Pearce, W.H.; Luo, K.; Oyama, Y.; Davidson, C.; Beohar, N.; Gheorghiade, M. Hematopoietic stem cell transplantation for cardiac and peripheral vascular disease. Bone Marrow Transplant. 2003, 32, S29–S31. [Google Scholar]
- Kavanagh, D.P.J.; Durant, L.E.; Crosby, H.A.; Lalor, P.F.; Frampton, J.; Adams, D.H.; Kalia, N. Haematopoietic stem cell recruitment to injured murine liver sinusoids depends on (alpha)4(beta)1 integrin/VCAM-1 interactions. Gut 2010, 59, 79. [Google Scholar]
- Kavanagh, D.P.J.; Lokman, A.B.; Neag, G.; Colley, A.; Kalia, N. Imaging the injured beating heart intravitally and the vasculoprotection afforded by haematopoietic stem cells. Cardiovasc. Res. 2019, 115, 1918–1932. [Google Scholar]
- Jackson, K.; Majka, S.; Wang, H.; Pocius, J.; Hartley, C.; Majesky, M.; Entman, M.; Michael, L.; Hirschi, K.; Goodell, M. Regeneration of ischemic cardiac muscle and vascular endothelium by adult stem cells. J. Clin. Investig. 2001, 107, 1395–1402. [Google Scholar]
- Soonpaa, M.H.; Lafontant, P.J.; Reuter, S.; Scherschel, J.A.; Srour, E.F.; Zaruba, M.-M.; der Lohe, M.R.-V.; Field, L.J. Absence of Cardiomyocyte Differentiation Following Transplantation of Adult Cardiac-Resident Sca-1+ Cells Into Infarcted Mouse Hearts. Circulation 2018, 138, 2963–2966. [Google Scholar] [PubMed]
- Murry, C.E.; Soonpaa, M.H.; Reinecke, H.; Nakajima, H.; Nakajima, H.O.; Rubart, M.; Pasumarthi, K.B.S.; Ismail Virag, J.; Bartelmez, S.H.; Poppa, V.; et al. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature 2004, 428, 664–668. [Google Scholar] [PubMed]
- Ishikawa, F.; Shimazu, H.; Shultz, L.D.; Fukata, M.; Nakamura, R.; Lyons, B.; Shimoda, K.; Shimoda, S.; Kanemaru, T.; Nakamura, K.-I.; et al. Purified human hematopoietic stem cells contribute to the generation of cardiomyocytes through cell fusion. FASEB J. 2006, 20, 950–952. [Google Scholar] [PubMed]
- Chiodi, I.; Mondello, C. Life style factors, tumor cell plasticity and cancer stem cells. Mutat. Res./Rev. Mutat. Res. 2020, 784, 108308. [Google Scholar] [PubMed]
- Lo Martire, V.; Caruso, D.; Palagini, L.; Zoccoli, G.; Bastianini, S. Stress & sleep: A relationship lasting a lifetime. Neurosci. Biobehav. Rev. 2020, 117, 65–77. [Google Scholar]
- Godos, J.; Grosso, G.; Castellano, S.; Galvano, F.; Caraci, F.; Ferri, R. Association between diet and sleep quality: A systematic review. Sleep Med. Rev. 2021, 57, 101430. [Google Scholar]
| Lifestyle | Influences on BM HSCs | Potential Mechanisms | Species |
|---|---|---|---|
| Psychosocial stress | Proliferation; Mobilization; Myeloid-biased differentiation. | Noradrenaline released by SNS signals niche cells via β-AR signaling to disrupt CXCL12-CXCR4 axis [48,49,50]; dopamine released by SNS activates PKA-Lck-ERK axis in HSCs via D2-type receptor [51]; glucocorticoids released by HPA axis upregulate actin-organizing molecules in HSCs via NR3C1 [21]; decrease in CXCL12 expression in niche cells [52]. | Mouse |
| Sleep problems | Proliferation; Myeloid-biased differentiation; Pro-inflammatory priming; Mobilization? | Less production of hypocretin by hypothalamus promotes CSF1 production by BM pre-neutrophils [53]; brain PGD2 elevation and efflux induce systemic inflammation via DP1 [54]; epigenetic reprogramming and promotion of clonal hematopoiesis through accelerated genetic drift in HSCs [55]; disruption of circadian rhythm leading to deregulated SNS activation [56,57,58], serum proteolytic cascades [59,60,61], and HSC inflammasome signaling [62,63]? | Mouse Human |
| High-cholesterol diet | Proliferation; Expansion; Mobilization; Myeloid-biased differentiation; Pro-inflammatory priming. | Epigenetic reprogramming in HSCs [64]? SREBP2 activation-mediated Notch upregulation in HSCs [65]; SLC38A9-mTOR axis activation in HSCs [10]; promotion of clonal hematopoiesis through expediting somatic evolution in HSCs [47]; elevated serum levels of CSF3 due to IL-23 generation by splenic dendritic cells and macrophages and decreased CXCL12 production by MSCs [66]; 27-hydroxycholesterol downregulates CXCR4 on HSCs via ERα [67]. | Mouse Human |
| High-fat diet | Proliferation; Expansion; Myeloid-biased differentiation; Pro-inflammatory priming. | MyD88 activation and epigenetic reprogramming in HSCs [68]? Oxidative stress-induced GFI1 upregulation in HSCs [69]; disruption of TGF-β receptor signaling within lipid rafts of HSCs [70]; expanded BM adipocytes produce excessive DPP4 [71]; inflammation-induced clonal hematopoiesis [72]; structural changes in microbiota alters HSC niche via activation of PPARγ2 [73]. | Mouse Human |
| High sodium intake | Mobilization? Myeloid-biased differentiation? | Increased IL-17 release by Th17 cells [74]? Perturbation of mitochondrial respiration in HSCs [75]? | Mouse |
| High-Pi diet | Expansion; MK/myeloid-biased differentiation. | Activation of PPIP5K2-Akt axis in HSCs [11]; Akt-mediated increase in apoptosis resistance of HSCs [76]. | Mouse |
| Ketogenic diet | Expansion Myeloid-biased differentiation Pro-inflammatory priming | Increased circulating levels of free PA and PA-associated lipids [77]; epigenetic reprogramming in HSCs [77]? | Mouse |
| Alcohol consumption | Attrition Proliferation Myeloid-biased differentiation | Acetaldehyde-toxicity-induced DNA damage activates p53 to induce apoptosis [78,79,80]; remodeling of HSC niche [81]. | Mouse |
| Tobacco smoking | Expansion Myeloid-biased differentiation | Nicotine directly acts on nicotinic acetylcholine receptors on HSCs [82]? Remodeling of HSC niche [81,83]. | Mouse |
|
Physical inactivity/ sedentary behavior | Proliferation Expansion Mobilization Myeloid-biased differentiation | More leptin is produced by increased body fat and interacts with LepR-positive BM stromal cells to decrease expression of quiescence- and retention-promoting hematopoietic niche factors [84]; epigenetic reprogramming in HSCs [84]. | Mouse Human |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Liu, C.; Wang, J.; Du, C. Hematopoietic Stem Cells as an Integrative Hub Linking Lifestyle to Cardiovascular Health. Cells 2024, 13, 712. https://doi.org/10.3390/cells13080712
Chen X, Liu C, Wang J, Du C. Hematopoietic Stem Cells as an Integrative Hub Linking Lifestyle to Cardiovascular Health. Cells. 2024; 13(8):712. https://doi.org/10.3390/cells13080712
Chicago/Turabian StyleChen, Xinliang, Chaonan Liu, Junping Wang, and Changhong Du. 2024. "Hematopoietic Stem Cells as an Integrative Hub Linking Lifestyle to Cardiovascular Health" Cells 13, no. 8: 712. https://doi.org/10.3390/cells13080712
APA StyleChen, X., Liu, C., Wang, J., & Du, C. (2024). Hematopoietic Stem Cells as an Integrative Hub Linking Lifestyle to Cardiovascular Health. Cells, 13(8), 712. https://doi.org/10.3390/cells13080712