
GDF-15 Suppresses Puromycin Aminonucleoside-Induced Podocyte Injury by Reducing Endoplasmic Reticulum Stress and Glomerular Inflammation

 ,
,  , and
, and 
Abstract
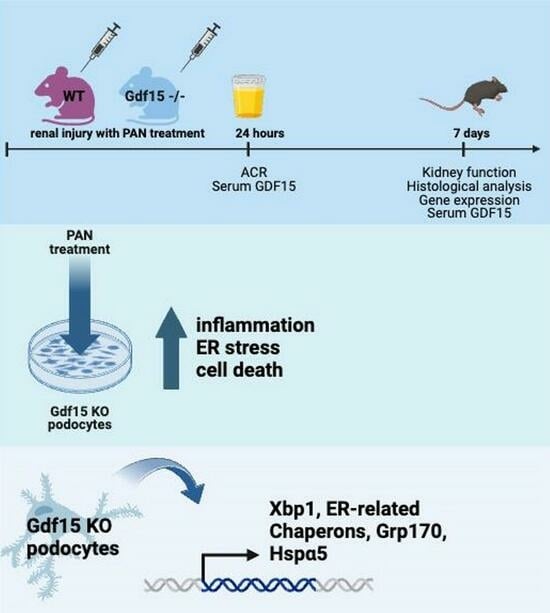
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
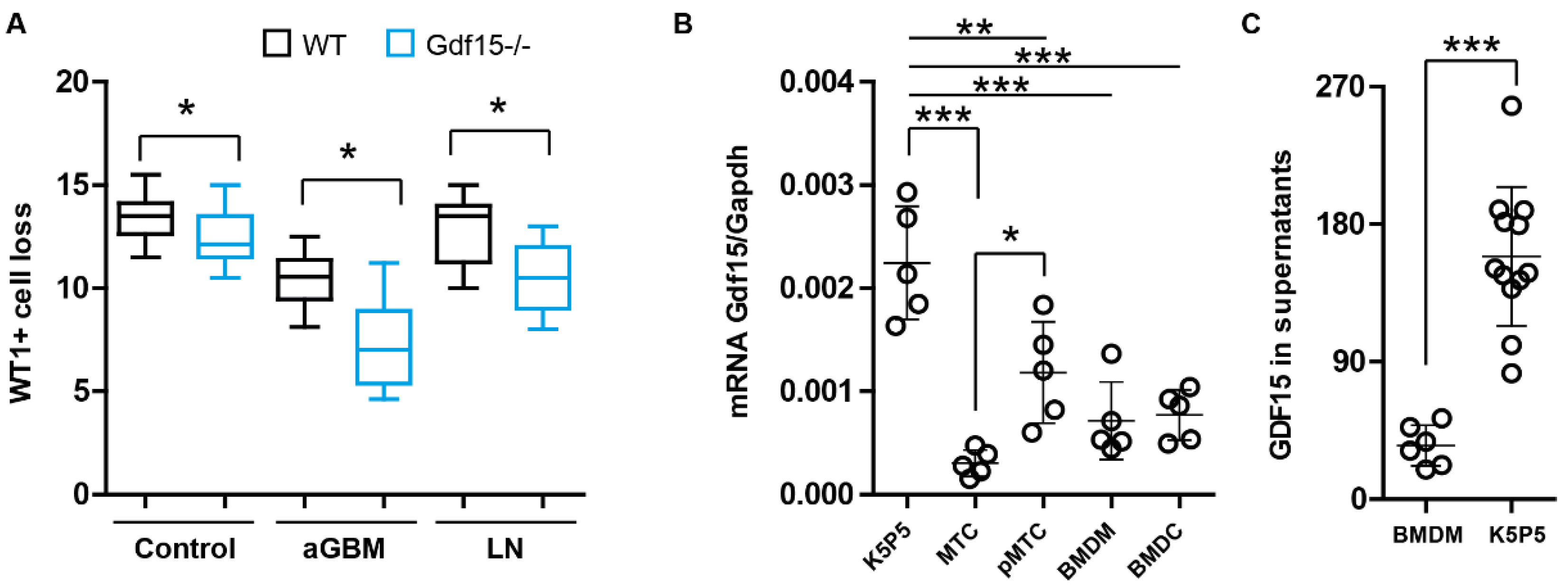
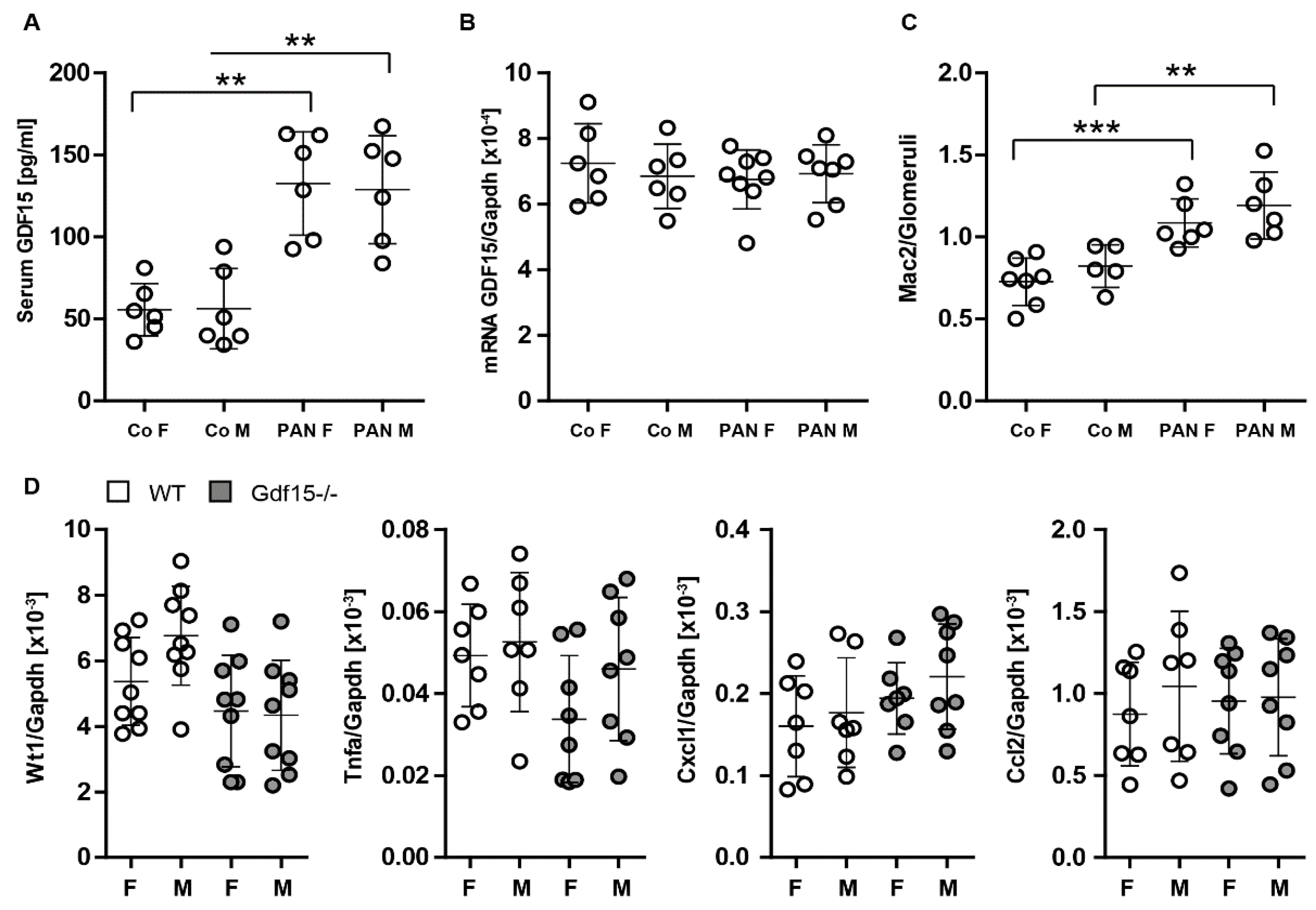
3.1. Podocytes Express and Secrete GDF15 and Its Deficiency Correlates with Podocyte Loss
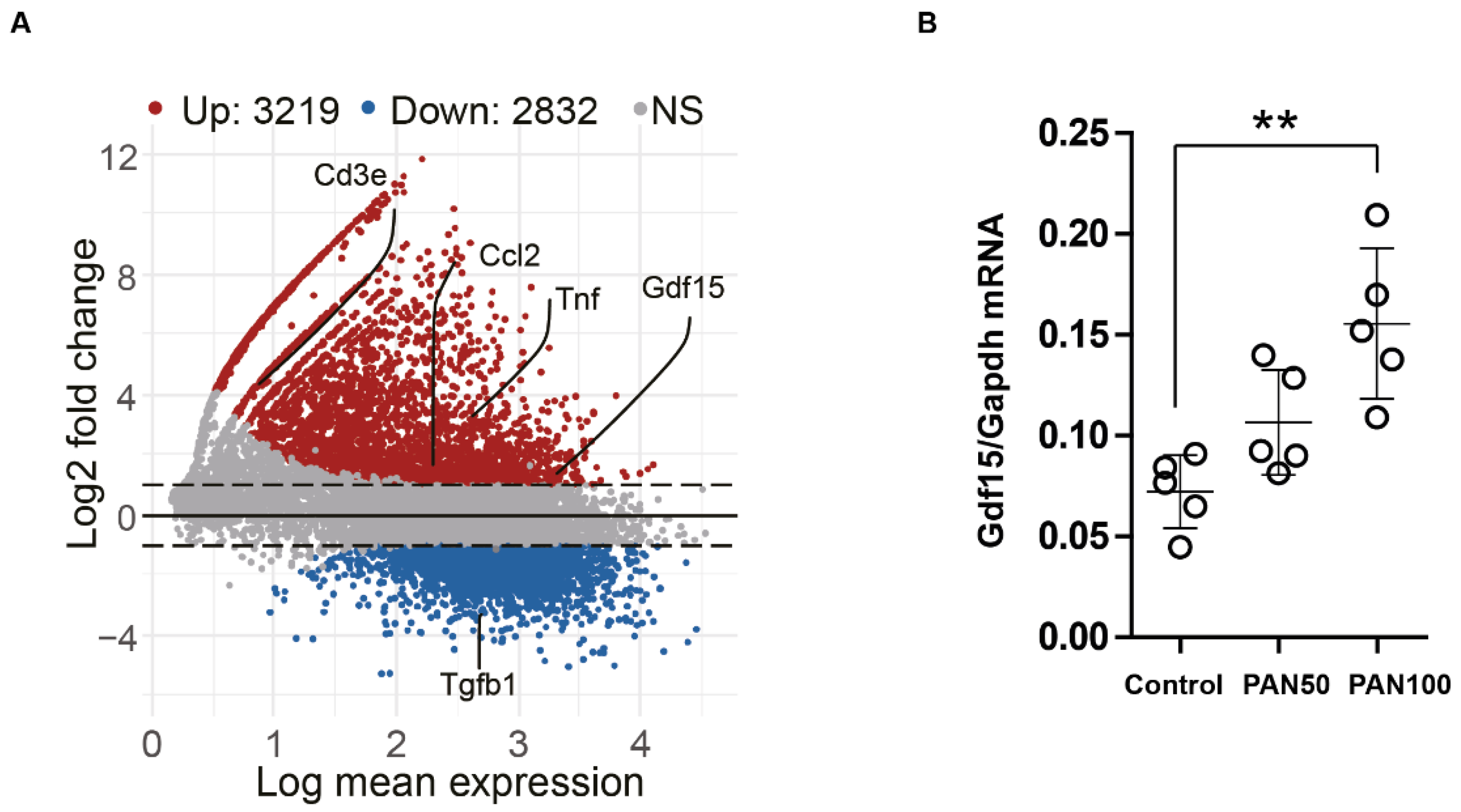
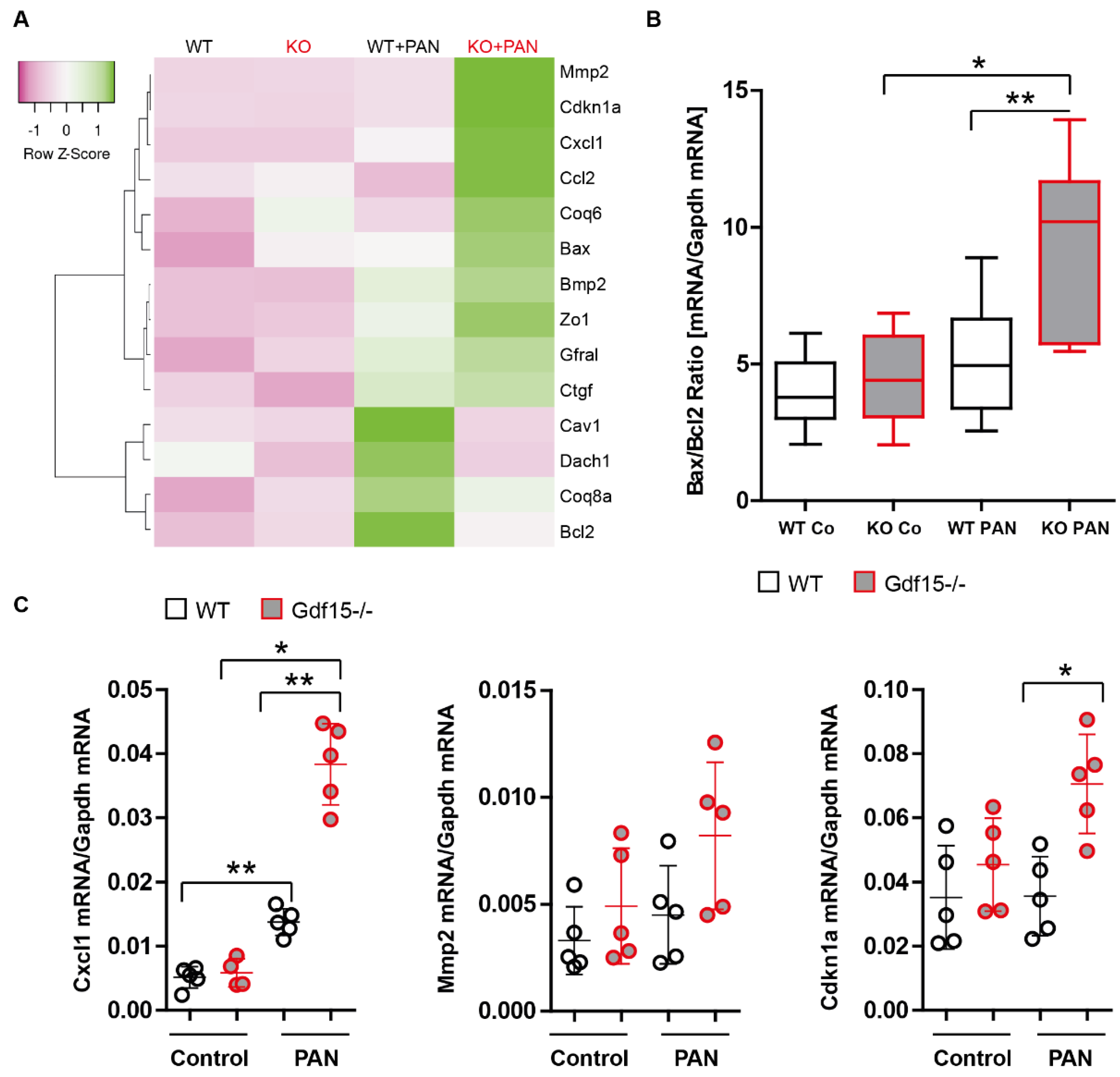
3.2. PAN Induces Gdf15 In Vitro and Gdf15 Knockout Alters Podocyte Expression Pattern
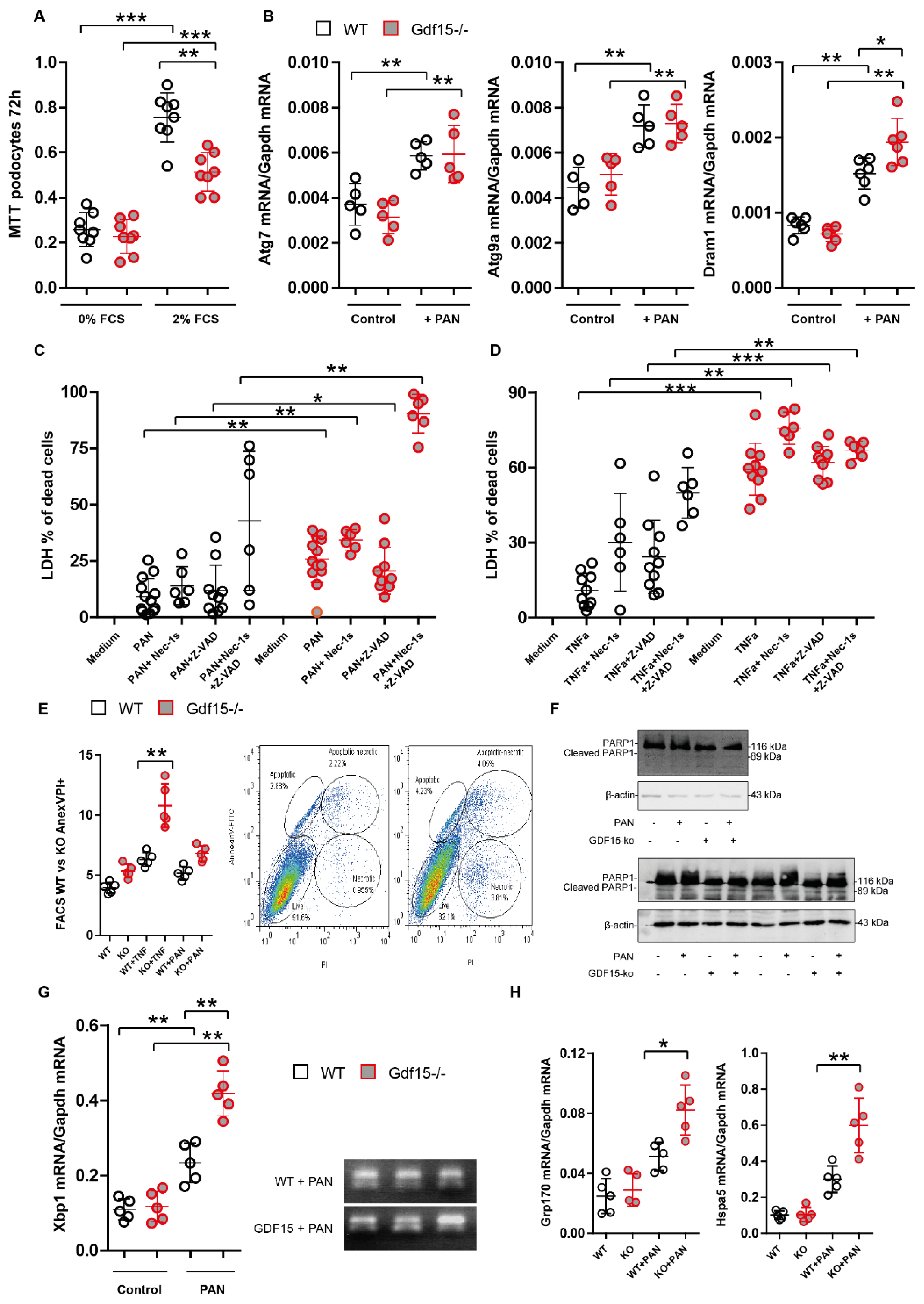
3.3. Gdf15-Deficiency Induces Endoplasmic Reticulum Stress and Podocyte Death In Vitro
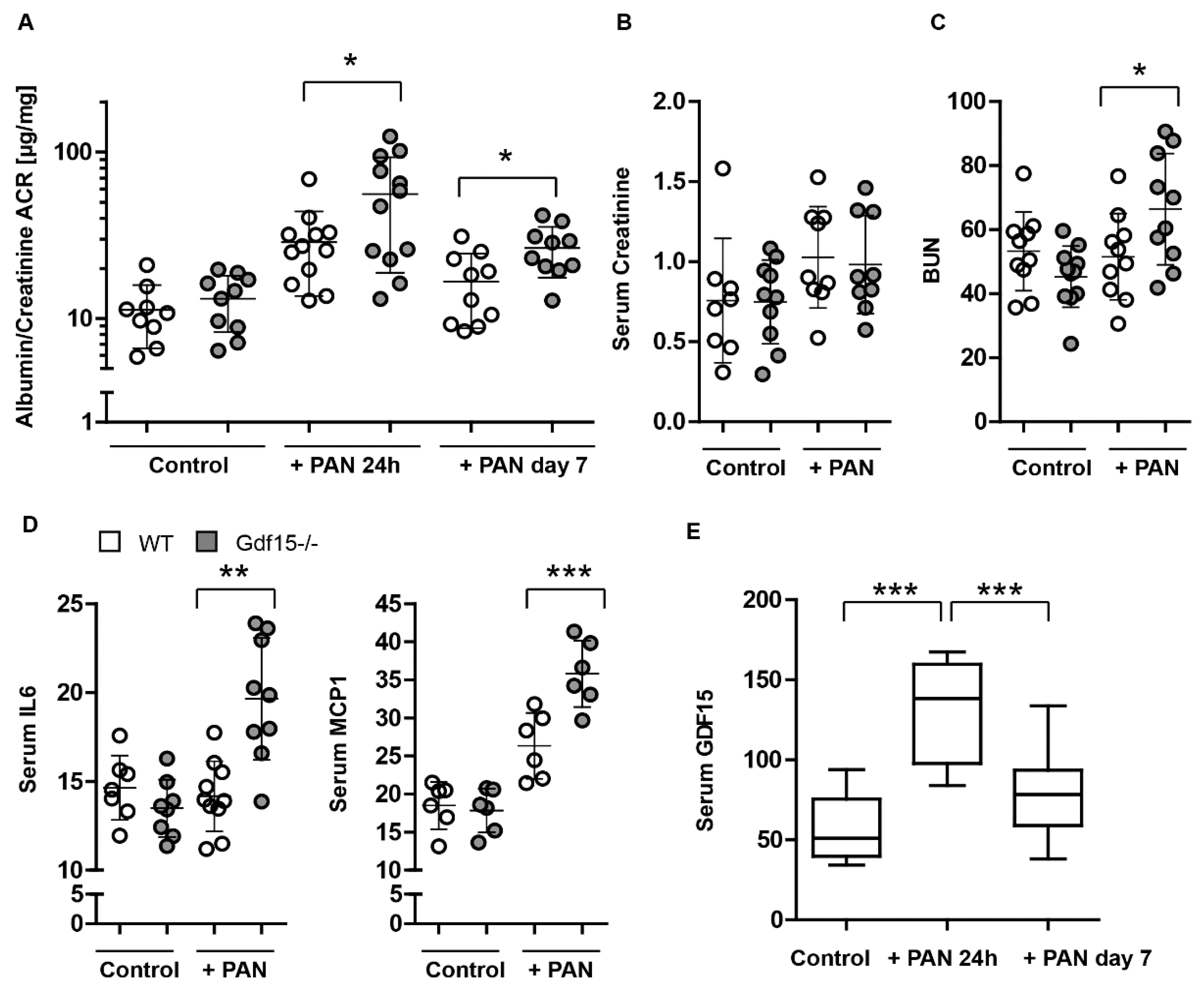
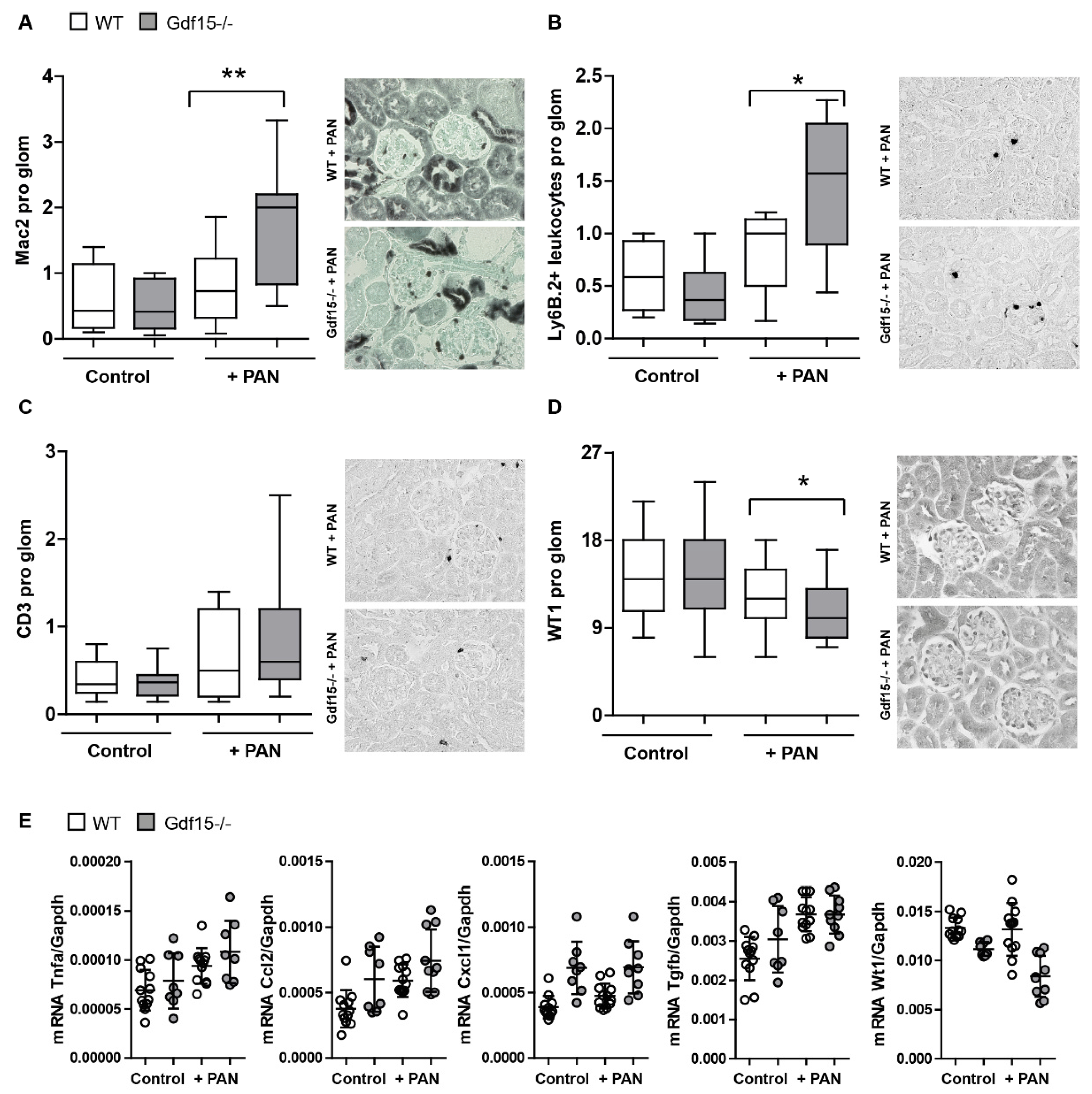
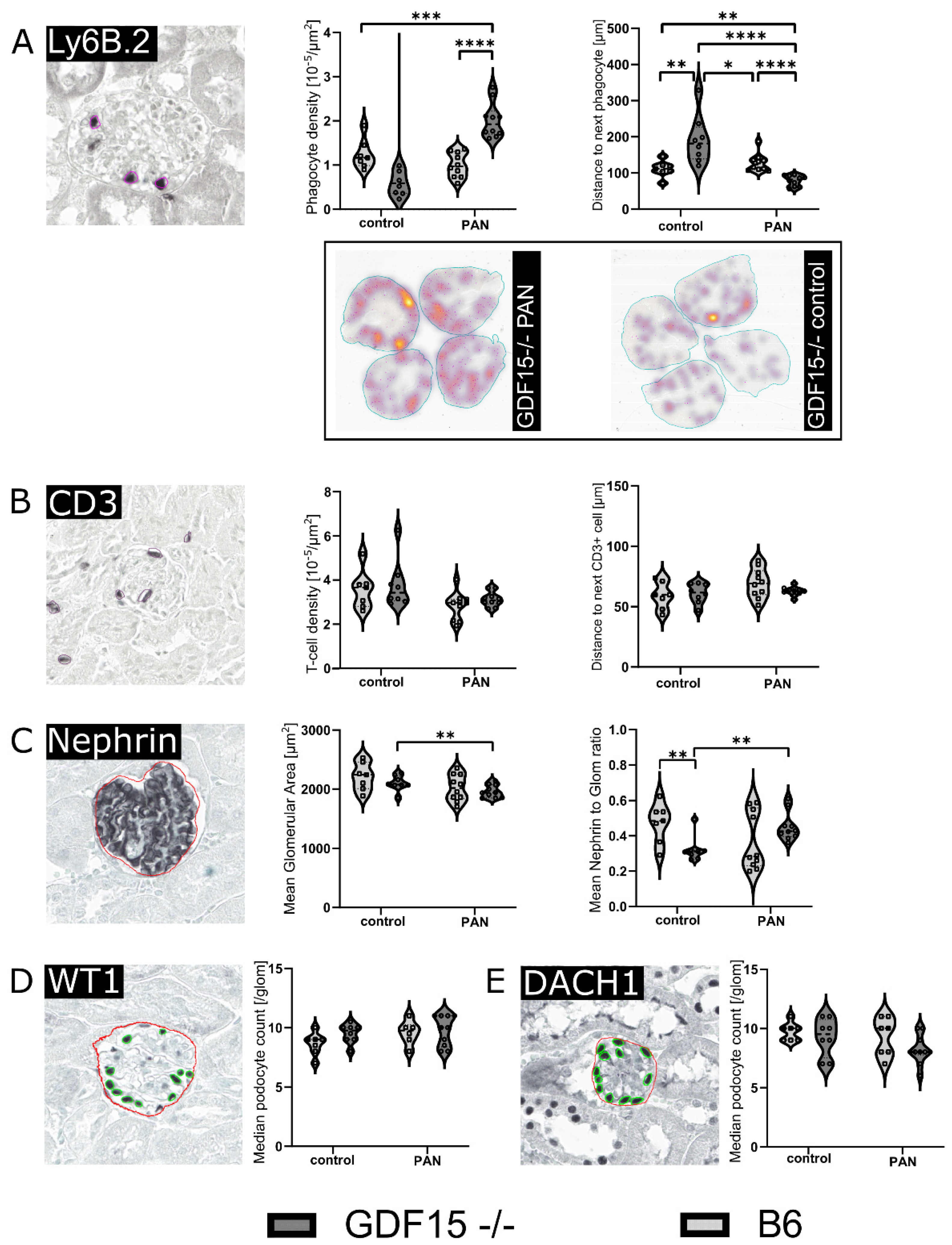
3.4. GDF15-/- Mice Exhibit Mildly Enhanced Glomerular Immune Cell Infiltration after PAN Treatment
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vivarelli, M.; Massella, L.; Ruggiero, B.; Emma, F. Minimal Change Disease. Clin. J. Am. Soc. Nephrol. 2017, 12, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.B.; Anders, H.-J.; Susztak, K.; Podestà, M.A.; Remuzzi, G.; Hildebrandt, F.; Romagnani, P. Podocytopathies. Nat. Rev. Dis. Primers 2020, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.; Singh, G.; Dinda, A.K. Understanding Podocytopathy and Its Relevance to Clinical Nephrology. Indian J. Nephrol. 2015, 25, 1–7. [Google Scholar] [CrossRef]
- Sieber, J.; Lindenmeyer, M.T.; Kampe, K.; Campbell, K.N.; Cohen, C.D.; Hopfer, H.; Mundel, P.; Jehle, A.W. Regulation of Podocyte Survival and Endoplasmic Reticulum Stress by Fatty Acids. Am. J. Physiol. Ren. Physiol. 2010, 299, F821–F829. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R.; Nangaku, M.; Onogi, H.; Ueyama, H.; Kitao, Y.; Nakazato, K.; Ogawa, S.; Kurokawa, K.; Couser, W.G.; Miyata, T. Involvement of Endoplasmic Reticulum (ER) Stress in Podocyte Injury Induced by Excessive Protein Accumulation. Kidney Int. 2005, 68, 2639–2650. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zhang, J.; Xiao, W.; Lee, K.; Li, Z.; Wen, J.; He, L.; Gui, D.; Xue, R.; Jian, G.; et al. Rtn1a-Mediated Endoplasmic Reticulum Stress in Podocyte Injury and Diabetic Nephropathy. Sci. Rep. 2017, 7, 323. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Kumar, V.; Lan, X.; Shoshtari, S.S.M.; Eng, J.M.; Zhou, X.; Wang, F.; Wang, H.; Skorecki, K.; Xing, G.; et al. APOL1 Risk Variants Cause Podocytes Injury through Enhancing Endoplasmic Reticulum Stress. Biosci. Rep. 2018, 38, BSR20171713. [Google Scholar] [CrossRef] [PubMed]
- Unsicker, K.; Spittau, B.; Krieglstein, K. The Multiple Facets of the TGF-β Family Cytokine Growth/Differentiation Factor-15/Macrophage Inhibitory Cytokine-1. Cytokine Growth Factor Rev. 2013, 24, 373–384. [Google Scholar] [CrossRef]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural Biology and Evolution of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2016, 8, a022103. [Google Scholar] [CrossRef]
- Bootcov, M.R.; Bauskin, A.R.; Valenzuela, S.M.; Moore, A.G.; Bansal, M.; He, X.Y.; Zhang, H.P.; Donnellan, M.; Mahler, S.; Pryor, K.; et al. MIC-1, a Novel Macrophage Inhibitory Cytokine, Is a Divergent Member of the TGF-b Superfamily. Proc. Natl. Acad. Sci. USA 1997, 94, 11514–11519. [Google Scholar] [CrossRef]
- Böttner, M.; Laaff, M.; Schechinger, B.; Rappold, G.; Unsicker, K.; Suter-Crazzolara, C. Characterization of the Rat, Mouse, and Human Genes of Growth/Differentiation Factor-15/Macrophage Inhibiting Cytokine-1 (GDF-15/MIC-1). Gene 1999, 237, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.B.; Sapinoso, L.M.; Kern, S.G.; Brown, D.A.; Liu, T.; Bauskin, A.R.; Ward, R.L.; Hawkins, N.J.; Quinn, D.I.; Russell, P.J.; et al. Large-Scale Delineation of Secreted Protein Biomarkers Overexpressed in Cancer Tissue and Serum. Proc. Natl. Acad. Sci. USA 2003, 100, 3410–3415. [Google Scholar] [CrossRef] [PubMed]
- Mullican, S.E.; Lin-Schmidt, X.; Chin, C.-N.; Chavez, J.A.; Furman, J.L.; Armstrong, A.A.; Beck, S.C.; South, V.J.; Dinh, T.Q.; Cash-Mason, T.D.; et al. GFRAL Is the Receptor for GDF15 and the Ligand Promotes Weight Loss in Mice and Nonhuman Primates. Nat. Med. 2017, 23, 1150–1157. [Google Scholar] [CrossRef]
- Patel, S.; Alvarez-Guaita, A.; Melvin, A.; Rimmington, D.; Dattilo, A.; Miedzybrodzka, E.L.; Cimino, I.; Maurin, A.-C.; Roberts, G.P.; Meek, C.L.; et al. GDF15 Provides an Endocrine Signal of Nutritional Stress in Mice and Humans. Cell Metab. 2019, 29, 707–718.e8. [Google Scholar] [CrossRef]
- Kempf, T.; Zarbock, A.; Widera, C.; Butz, S.; Stadtmann, A.; Rossaint, J.; Bolomini-Vittori, M.; Korf-Klingebiel, M.; Napp, L.C.; Hansen, B.; et al. GDF-15 Is an Inhibitor of Leukocyte Integrin Activation Required for Survival after Myocardial Infarction in Mice. Nat. Med. 2011, 17, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Wischhusen, J.; Melero, I.; Fridman, W.H. Growth/Differentiation Factor-15 (GDF-15): From Biomarker to Novel Targetable Immune Checkpoint. Front. Immunol. 2020, 11, 951. [Google Scholar] [CrossRef]
- Lorenz, G.; Ribeiro, A.; Von Rauchhaupt, E.; Würf, V.; Schmaderer, C.; Cohen, C.D.; Vohra, T.; Anders, H.-J.; Lindenmeyer, M.; Lech, M. GDF15 Suppresses Lymphoproliferation and Humoral Autoimmunity in a Murine Model of Systemic Lupus Erythematosus. J. Innate Immun. 2022, 14, 673–689. [Google Scholar] [CrossRef]
- Luan, H.H.; Wang, A.; Hilliard, B.K.; Carvalho, F.; Rosen, C.E.; Ahasic, A.M.; Herzog, E.L.; Kang, I.; Pisani, M.A.; Yu, S.; et al. GDF15 Is an Inflammation-Induced Central Mediator of Tissue Tolerance. Cell 2019, 178, 1231–1244.e11. [Google Scholar] [CrossRef] [PubMed]
- Moschovaki-Filippidou, F.; Steiger, S.; Lorenz, G.; Schmaderer, C.; Ribeiro, A.; von Rauchhaupt, E.; Cohen, C.D.; Anders, H.-J.; Lindenmeyer, M.; Lech, M. Growth Differentiation Factor 15 Ameliorates Anti-Glomerular Basement Membrane Glomerulonephritis in Mice. Int. J. Mol. Sci. 2020, 21, 6978. [Google Scholar] [CrossRef]
- Arif, E.; Solanki, A.K.; Srivastava, P.; Rahman, B.; Tash, B.R.; Holzman, L.B.; Janech, M.G.; Martin, R.; Knölker, H.-J.; Fitzgibbon, W.R.; et al. The Motor Protein Myo1c Regulates Transforming Growth Factor-β–Signaling and Fibrosis in Podocytes. Kidney Int. 2019, 96, 139–158. [Google Scholar] [CrossRef]
- Hsiao, E.C.; Koniaris, L.G.; Zimmers-Koniaris, T.; Sebald, S.M.; Huynh, T.V.; Lee, S.-J. Characterization of Growth-Differentiation Factor 15, a Transforming Growth Factor β Superfamily Member Induced Following Liver Injury. Mol. Cell. Biol. 2000, 20, 3742–3751. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open Source Software for Digital Pathology Image Analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed]
- Leaños-Miranda, A.; Márquez-Acosta, J.; Romero-Arauz, F.; Cárdenas-Mondragón, G.M.; Rivera-Leaños, R.; Isordia-Salas, I.; Ulloa-Aguirre, A. Protein:Creatinine Ratio in Random Urine Samples Is a Reliable Marker of Increased 24-Hour Protein Excretion in Hospitalized Women with Hypertensive Disorders of Pregnancy. Clin. Chem. 2007, 53, 1623–1628. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saleem, M.A.; O’Hare, M.J.; Reiser, J.; Coward, R.J.; Inward, C.D.; Farren, T.; Xing, C.Y.; Ni, L.; Mathieson, P.W.; Mundel, P. A Conditionally Immortalized Human Podocyte Cell Line Demonstrating Nephrin and Podocin Expression. J. Am. Soc. Nephrol. 2002, 13, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Uniyal, A.P.; Mansotra, K.; Yadav, S.K.; Kumar, V. An Overview of Designing and Selection of sgRNAs for Precise Genome Editing by the CRISPR-Cas9 System in Plants. 3 Biotech 2019, 9, 223. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Solanki, A.K.; Srivastava, P.; Rahman, B.; Lipschutz, J.H.; Nihalani, D.; Arif, E. The Use of High-Throughput Transcriptomics to Identify Pathways with Therapeutic Significance in Podocytes. Int. J. Mol. Sci. 2019, 21, 274. [Google Scholar] [CrossRef]
- Solanki, A.K.; Arif, E.; Srivastava, P.; Furcht, C.M.; Rahman, B.; Wen, P.; Singh, A.; Holzman, L.B.; Fitzgibbon, W.R.; Budisavljevic, M.N.; et al. Phosphorylation of Slit Diaphragm Proteins NEPHRIN and NEPH1 upon Binding of HGF Promotes Podocyte Repair. J. Biol. Chem. 2021, 297, 101079. [Google Scholar] [CrossRef]
- Akhiani, A.A.; Werlenius, O.; Aurelius, J.; Movitz, C.; Martner, A.; Hellstrand, K.; Thorén, F.B. Role of the ERK Pathway for Oxidant-Induced Parthanatos in Human Lymphocytes. PLoS ONE 2014, 9, e89646. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER Stress Sensor XBP1 Controls Anti-Tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef]
- So, J.-S. Roles of Endoplasmic Reticulum Stress in Immune Responses. Mol. Cells 2018, 41, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-M.; Kang, T.-I.; So, J.-S. Roles of XBP1s in Transcriptional Regulation of Target Genes. Biomedicines 2021, 9, 791. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.W.; Nguyen, D.N.; Clayton, D.R.; Ruiz, W.G.; Mutchler, S.M.; Ray, E.C.; Marciszyn, A.L.; Nkashama, L.J.; Subramanya, A.R.; Gingras, S.; et al. The Molecular Chaperone GRP170 Protects against ER Stress and Acute Kidney Injury in Mice. JCI Insight 2022, 7, e151869. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, J.; Chen, X.; Kar, G.; Henriksson, J.; Gomes, T.; Park, J.-E.; Natarajan, K.; Meyer, K.B.; Miao, Z.; McKenzie, A.N.J.; et al. Genome-Wide Analyses Reveal the IRE1a-XBP1 Pathway Promotes T Helper Cell Differentiation by Resolving Secretory Stress and Accelerating Proliferation. Genome Med. 2018, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Klaus, M.; Wong, M.N.; Thebille, A.-K.; Gernhold, L.; Kuppe, C.; Halder, M.; Kranz, J.; Wanner, N.; Braun, F.; et al. Deep Learning-Based Molecular Morphometrics for Kidney Biopsies. JCI Insight 2021, 6, e144779. [Google Scholar] [CrossRef] [PubMed]
- Govind, D.; Becker, J.U.; Miecznikowski, J.; Rosenberg, A.Z.; Dang, J.; Tharaux, P.L.; Yacoub, R.; Thaiss, F.; Hoyer, P.F.; Manthey, D.; et al. PodoSighter: A Cloud-Based Tool for Label-Free Podocyte Detection in Kidney Whole-Slide Images. J. Am. Soc. Nephrol. 2021, 32, 2795–2813. [Google Scholar] [CrossRef]
- Hölscher, D.L.; Bouteldja, N.; Joodaki, M.; Russo, M.L.; Lan, Y.-C.; Sadr, A.V.; Cheng, M.; Tesar, V.; Stillfried, S.V.; Klinkhammer, B.M.; et al. Next-Generation Morphometry for Pathomics-Data Mining in Histopathology. Nat. Commun. 2023, 14, 470. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, T.; Sharma, M.; Yew, K.-H.; Sharma, R.; Duncan, R.S.; Saleem, M.A.; McCarthy, E.T.; Kats, A.; Cudmore, P.A.; Alon, U.S.; et al. LPS and PAN-Induced Podocyte Injury in an in Vitro Model of Minimal Change Disease: Changes in TLR Profile. J. Cell Commun. Signal. 2013, 7, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Lin, Q.; Mo, J.; Xiao, Y.; Xie, Y. Hirudin Attenuates Puromycin Aminonucleoside-Induced Glomerular Podocyte Injury by Inhibiting MAPK-Mediated Endoplasmic Reticulum Stress. Drug Dev. Res. 2022, 83, 1047–1056. [Google Scholar] [CrossRef]
- Jo, Y.-I.; Cheng, H.; Wang, S.; Moeckel, G.W.; Harris, R.C. Puromycin Induces Reversible Proteinuric Injury in Transgenic Mice Expressing Cyclooxygenase-2 in Podocytes. Nephron Exp. Nephrol. 2007, 107, e87–e94. [Google Scholar] [CrossRef]
- Cheng, Z.-Z.; Pätäri, A.; Aalto-Setälä, K.; Novikov, D.; Schlöndorff, D.; Holthöfer, H. Hypercholesterolemia Is a Prerequisite for Puromycin Inducible Damage in Mouse Kidney. Kidney Int. 2003, 63, 107–112. [Google Scholar] [CrossRef]
- Ha, T.-S. Roles of Adaptor Proteins in Podocyte Biology. World J. Nephrol. 2013, 2, 1–10. [Google Scholar] [CrossRef]
- Toska, E.; Roberts, S.G.E. Mechanisms of Transcriptional Regulation by WT1 (Wilms’ Tumour 1). Biochem. J. 2014, 461, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Konieczkowski, M.; Mukherjee, A.; Schechtman, S.; Khan, S.; Schelling, J.R.; Ross, M.D.; Bruggeman, L.A.; Sedor, J.R. Podocyte Injury Induces Nuclear Translocation of WTIP via Microtubule-Dependent Transport. J. Biol. Chem. 2010, 285, 9995–10004. [Google Scholar] [CrossRef] [PubMed]
- Schell, C.; Huber, T.B. The Evolving Complexity of the Podocyte Cytoskeleton. J. Am. Soc. Nephrol. 2017, 28, 3166–3174. [Google Scholar] [CrossRef] [PubMed]
- Long, D.A.; Kolatsi-Joannou, M.; Price, K.L.; Dessapt-Baradez, C.; Huang, J.L.; Papakrivopoulou, E.; Hubank, M.; Korstanje, R.; Gnudi, L.; Woolf, A.S. Albuminuria Is Associated with Too Few Glomeruli and Too Much Testosterone. Kidney Int. 2013, 83, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Doublier, S.; Lupia, E.; Catanuto, P.; Periera-Simon, S.; Xia, X.; Korach, K.; Berho, M.; Elliot, S.J.; Karl, M. Testosterone and 17β-Estradiol Have Opposite Effects on Podocyte Apoptosis That Precedes Glomerulosclerosis in Female Estrogen Receptor Knockout Mice. Kidney Int. 2011, 79, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Kummer, S.; Jeruschke, S.; Wegerich, L.V.; Peters, A.; Lehmann, P.; Seibt, A.; Mueller, F.; Koleganova, N.; Halbenz, E.; Schmitt, C.P.; et al. Estrogen Receptor Alpha Expression in Podocytes Mediates Protection against Apoptosis In-Vitro and in-Vivo. PLoS ONE 2011, 6, e27457. [Google Scholar] [CrossRef]
- Gujarati, N.A.; Chow, A.K.; Mallipattu, S.K. Central Role of Podocytes in Mediating Cellular Cross Talk in Glomerular Health and Disease. Am. J. Physiol. Ren. Physiol. 2024, 326, F313–F325. [Google Scholar] [CrossRef]
- Anders, H.-J.; Kitching, A.R.; Leung, N.; Romagnani, P. Glomerulonephritis: Immunopathogenesis and Immunotherapy. Nat. Rev. Immunol. 2023, 23, 453–471. [Google Scholar] [CrossRef]
- Chen, Z.; Gao, L.; Li, C.; Sun, W. GDF15 Interference Regulates Proliferation, Inflammation, and Autophagy of Lipopolysaccharide-Induced Mesangial Cells by Inhibiting PI3K/ AKT/mTOR Signaling. Endocr. Metab. Immune Disord. Drug Targets 2023, 24, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, W.; Qian, J.; Tang, Y. Fasting Exacerbates Hepatic Growth Differentiation Factor 15 to Promote Fatty Acid β-Oxidation and Ketogenesis via Activating XBP1 Signaling in Liver. Redox Biol. 2018, 16, 87–96. [Google Scholar] [CrossRef]
- Hassan, H.; Tian, X.; Inoue, K.; Chai, N.; Liu, C.; Soda, K.; Moeckel, G.; Tufro, A.; Lee, A.-H.; Somlo, S.; et al. Essential Role of X-Box Binding Protein-1 during Endoplasmic Reticulum Stress in Podocytes. J. Am. Soc. Nephrol. 2016, 27, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Betancourt, J.R.; Papillon, J.; Guillemette, J.; Iwawaki, T.; Chung, C.-F.; Cybulsky, A.V. Role of IRE1α in Podocyte Proteostasis and Mitochondrial Health. Cell Death Discov. 2020, 6, 128. [Google Scholar] [CrossRef]
- Kaufman, D.R.; Papillon, J.; Larose, L.; Iwawaki, T.; Cybulsky, A.V. Deletion of Inositol-Requiring Enzyme-1α in Podocytes Disrupts Glomerular Capillary Integrity and Autophagy. Mol. Biol. Cell 2017, 28, 1636–1651. [Google Scholar] [CrossRef] [PubMed]
- Min, S.-Y.; Ha, D.-S.; Ha, T.-S. Puromycin Aminonucleoside Triggers Apoptosis in Podocytes by Inducing Endoplasmic Reticulum Stress. Kidney Res. Clin. Pract. 2018, 37, 210–221. [Google Scholar] [CrossRef]
- Wang, J.; Lee, J.; Liem, D.; Ping, P. HSPA5 Gene Encoding Hsp70 Chaperone BiP in the Endoplasmic Reticulum. Gene 2017, 618, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Spaan, C.N.; Smit, W.L.; van Lidth de Jeude, J.F.; Meijer, B.J.; Muncan, V.; van den Brink, G.R.; Heijmans, J. Expression of UPR Effector Proteins ATF6 and XBP1 Reduce Colorectal Cancer Cell Proliferation and Stemness by Activating PERK Signaling. Cell Death Dis. 2019, 10, 490. [Google Scholar] [CrossRef] [PubMed]
- Hartleben, B.; Gödel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Köbler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy Influences Glomerular Disease Susceptibility and Maintains Podocyte Homeostasis in Aging Mice. J. Clin. Investig. 2010, 120, 1084–1096. [Google Scholar] [CrossRef]
- Inoki, K.; Mori, H.; Wang, J.; Suzuki, T.; Hong, S.; Yoshida, S.; Blattner, S.M.; Ikenoue, T.; Rüegg, M.A.; Hall, M.N.; et al. mTORC1 Activation in Podocytes Is a Critical Step in the Development of Diabetic Nephropathy in Mice. J. Clin. Investig. 2011, 121, 2181–2196. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Tojo, A.; Satonaka, H.; Yamada, N.O.; Senda, T.; Ishimitsu, T. Decreased Podocyte Vesicle Transcytosis and Albuminuria in APC C-Terminal Deficiency Mice with Puromycin-Induced Nephrotic Syndrome. Int. J. Mol. Sci. 2021, 22, 13412. [Google Scholar] [CrossRef]
- Cybulsky, A.V. Endoplasmic Reticulum Stress, the Unfolded Protein Response and Autophagy in Kidney Diseases. Nat. Rev. Nephrol. 2017, 13, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Bek, M.F.; Bayer, M.; Müller, B.; Greiber, S.; Lang, D.; Schwab, A.; August, C.; Springer, E.; Rohrbach, R.; Huber, T.B.; et al. Expression and Function of C/EBP Homology Protein (GADD153) in Podocytes. Am. J. Pathol. 2006, 168, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Markan, S.; Kohli, H.S.; Joshi, K.; Minz, R.W.; Sud, K.; Ahuja, M.; Anand, S.; Khullar, M. Up Regulation of the GRP-78 and GADD-153 and down Regulation of Bcl-2 Proteins in Primary Glomerular Diseases: A Possible Involvement of the ER Stress Pathway in Glomerulonephritis. Mol. Cell. Biochem. 2009, 324, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a P53-Induced Modulator of Autophagy, Is Critical for Apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Valbuena, A.; Castro-Obregón, S.; Lazo, P.A. Downregulation of VRK1 by P53 in Response to DNA Damage Is Mediated by the Autophagic Pathway. PLoS ONE 2011, 6, e17320. [Google Scholar] [CrossRef] [PubMed]
- Nagata, M.; Arakawa, S.; Yamaguchi, H.; Torii, S.; Endo, H.; Tsujioka, M.; Honda, S.; Nishida, Y.; Konishi, A.; Shimizu, S. Dram1 Regulates DNA Damage-Induced Alternative Autophagy. Cell Stress 2018, 2, 55–65. [Google Scholar] [CrossRef]
- Wang, Y.; Kim, N.S.; Haince, J.-F.; Kang, H.C.; David, K.K.; Andrabi, S.A.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-Ribose) (PAR) Binding to Apoptosis-Inducing Factor Is Critical for PAR Polymerase-1-Dependent Cell Death (Parthanatos). Sci. Signal. 2011, 4, ra20. [Google Scholar] [CrossRef]
- Dasari, S.K.; Bialik, S.; Levin-Zaidman, S.; Levin-Salomon, V.; Merrill, A.H.; Futerman, A.H.; Kimchi, A. Signalome-Wide RNAi Screen Identifies GBA1 as a Positive Mediator of Autophagic Cell Death. Cell Death Differ. 2017, 24, 1288–1302. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Zeng, Y.-T.; Liu, W.-F.; Zheng, P.-S.; Li, S. GDF15 Deficiency Hinders Human Trophoblast Invasion to Mediate Pregnancy Loss through Downregulating Smad1/5 Phosphorylation. iScience 2023, 26, 107902. [Google Scholar] [CrossRef] [PubMed]
- Roth, P.; Junker, M.; Tritschler, I.; Mittelbronn, M.; Dombrowski, Y.; Breit, S.N.; Tabatabai, G.; Wick, W.; Weller, M.; Wischhusen, J. GDF-15 Contributes to Proliferation and Immune Escape of Malignant Gliomas. Clin. Cancer Res. 2010, 16, 3851–3859. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, G.; Liu, Y.; Chen, R.; Zhao, D.; McAlister, V.; Mele, T.; Liu, K.; Zheng, X. GDF15 Regulates Malat-1 Circular RNA and Inactivates NFκB Signaling Leading to Immune Tolerogenic DCs for Preventing Alloimmune Rejection in Heart Transplantation. Front. Immunol. 2018, 9, 2407. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-B.; Choi, M.J.; Ryu, D.; Yi, H.-S.; Lee, S.E.; Chang, J.Y.; Chung, H.K.; Kim, Y.K.; Kang, S.G.; Lee, J.H.; et al. Reduced Oxidative Capacity in Macrophages Results in Systemic Insulin Resistance. Nat. Commun. 2018, 9, 1551. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
von Rauchhaupt, E.; Klaus, M.; Ribeiro, A.; Honarpisheh, M.; Li, C.; Liu, M.; Köhler, P.; Adamowicz, K.; Schmaderer, C.; Lindenmeyer, M.; et al. GDF-15 Suppresses Puromycin Aminonucleoside-Induced Podocyte Injury by Reducing Endoplasmic Reticulum Stress and Glomerular Inflammation. Cells 2024, 13, 637. https://doi.org/10.3390/cells13070637
von Rauchhaupt E, Klaus M, Ribeiro A, Honarpisheh M, Li C, Liu M, Köhler P, Adamowicz K, Schmaderer C, Lindenmeyer M, et al. GDF-15 Suppresses Puromycin Aminonucleoside-Induced Podocyte Injury by Reducing Endoplasmic Reticulum Stress and Glomerular Inflammation. Cells. 2024; 13(7):637. https://doi.org/10.3390/cells13070637
Chicago/Turabian Stylevon Rauchhaupt, Ekaterina, Martin Klaus, Andrea Ribeiro, Mohsen Honarpisheh, Chenyu Li, Min Liu, Paulina Köhler, Karina Adamowicz, Christoph Schmaderer, Maja Lindenmeyer, and et al. 2024. "GDF-15 Suppresses Puromycin Aminonucleoside-Induced Podocyte Injury by Reducing Endoplasmic Reticulum Stress and Glomerular Inflammation" Cells 13, no. 7: 637. https://doi.org/10.3390/cells13070637
APA Stylevon Rauchhaupt, E., Klaus, M., Ribeiro, A., Honarpisheh, M., Li, C., Liu, M., Köhler, P., Adamowicz, K., Schmaderer, C., Lindenmeyer, M., Steiger, S., Anders, H.-J., & Lech, M. (2024). GDF-15 Suppresses Puromycin Aminonucleoside-Induced Podocyte Injury by Reducing Endoplasmic Reticulum Stress and Glomerular Inflammation. Cells, 13(7), 637. https://doi.org/10.3390/cells13070637