
Role of the Skin Immune System in Wound Healing
Abstract
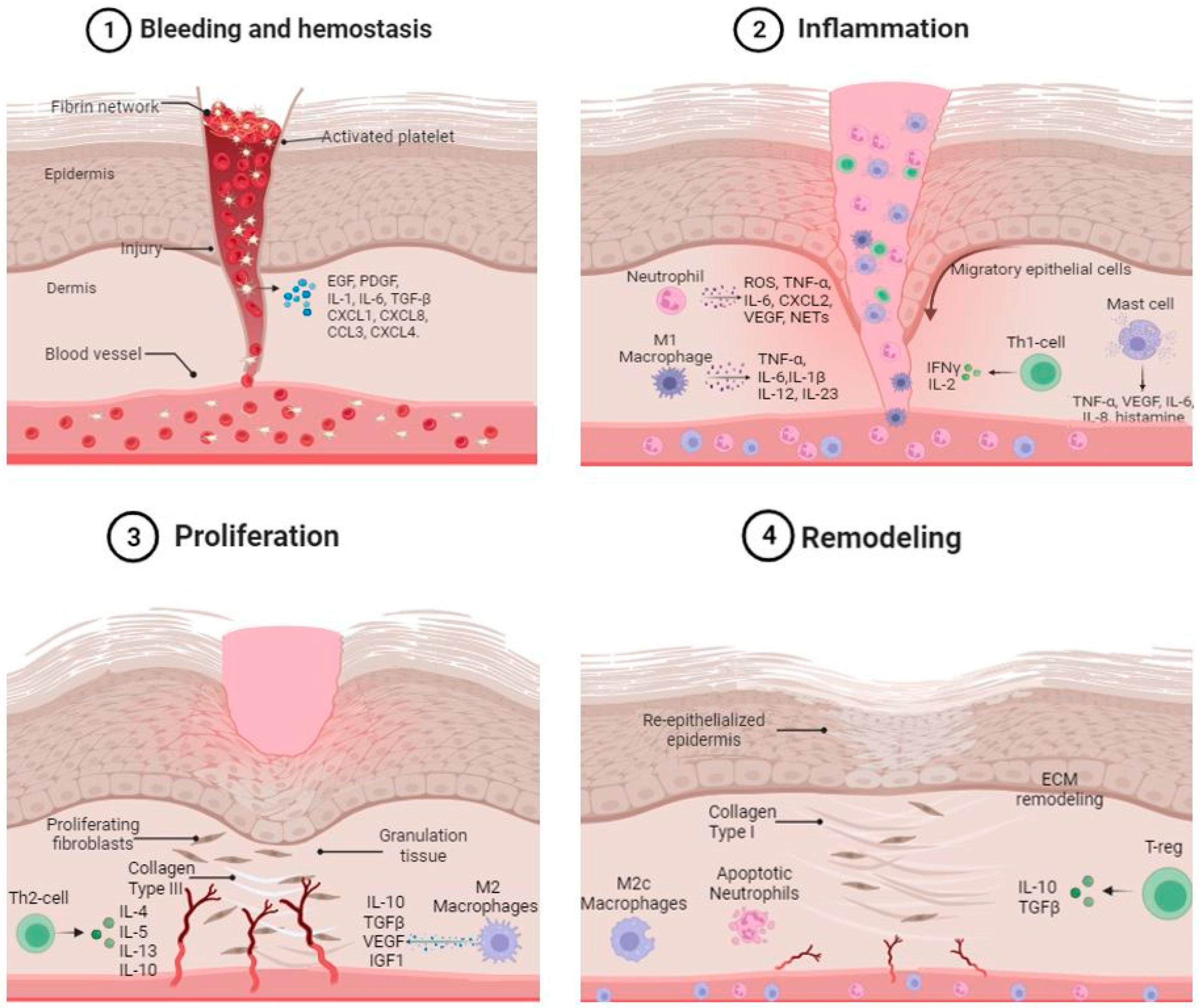
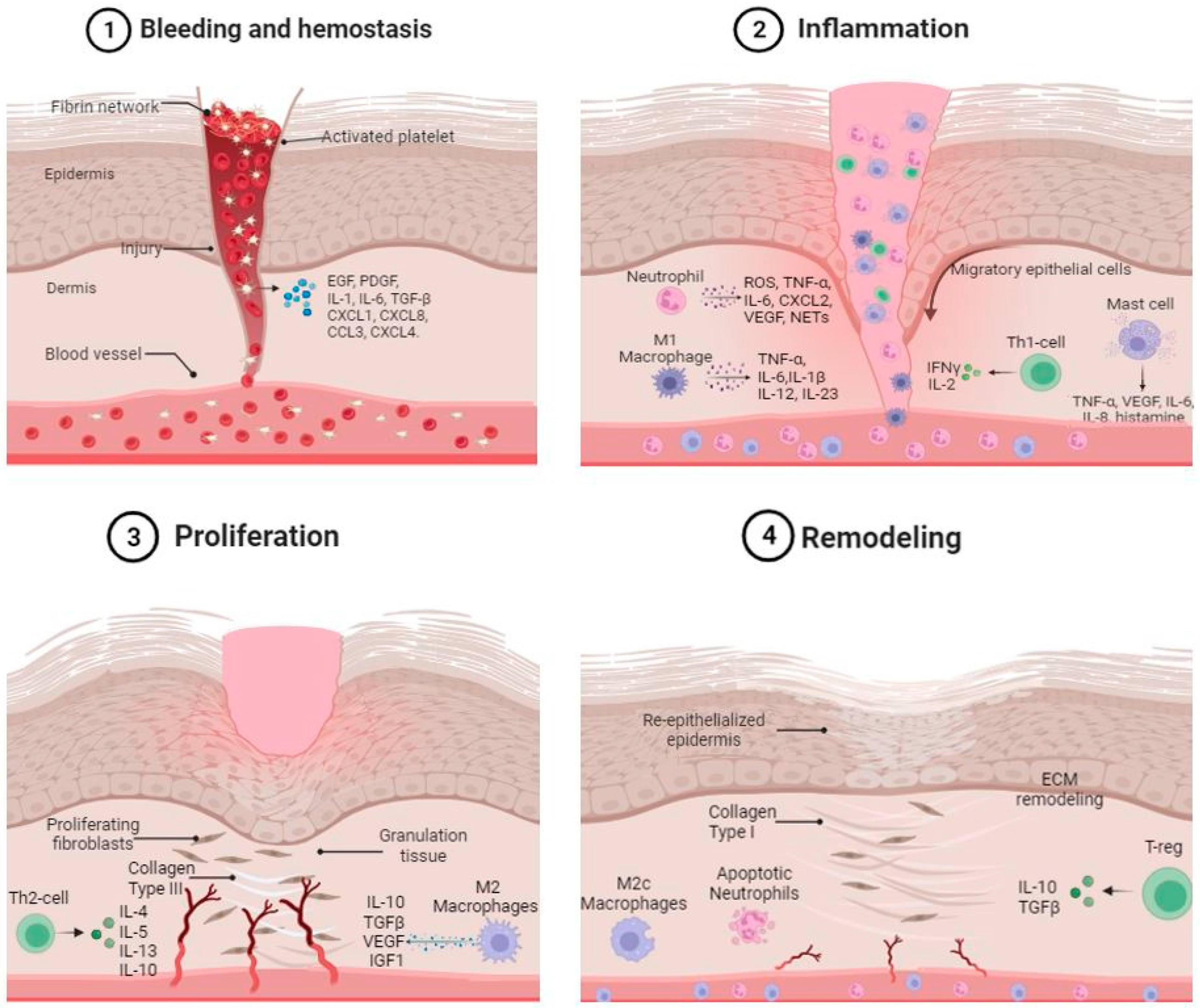
1. Wound Healing Overview
2. Skin Immune System Overview
3. Role of Skin Immune System in Hemostasis
3.1. Platelets
3.2. Platelets and Inflammation
4. Role of the Innate and Adaptive Immune Cells in the Inflammatory and Proliferative Phases of Wound Healing
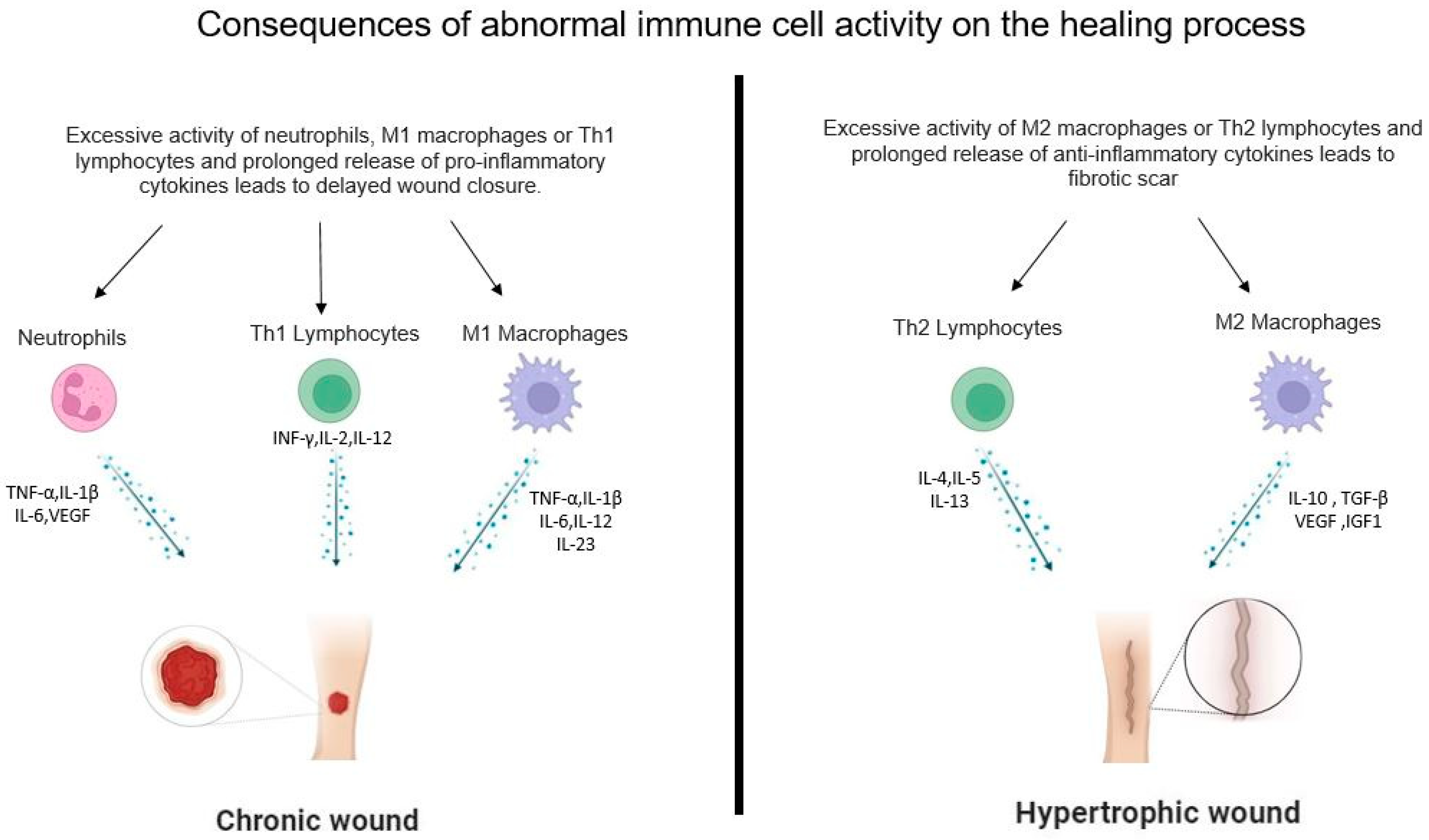
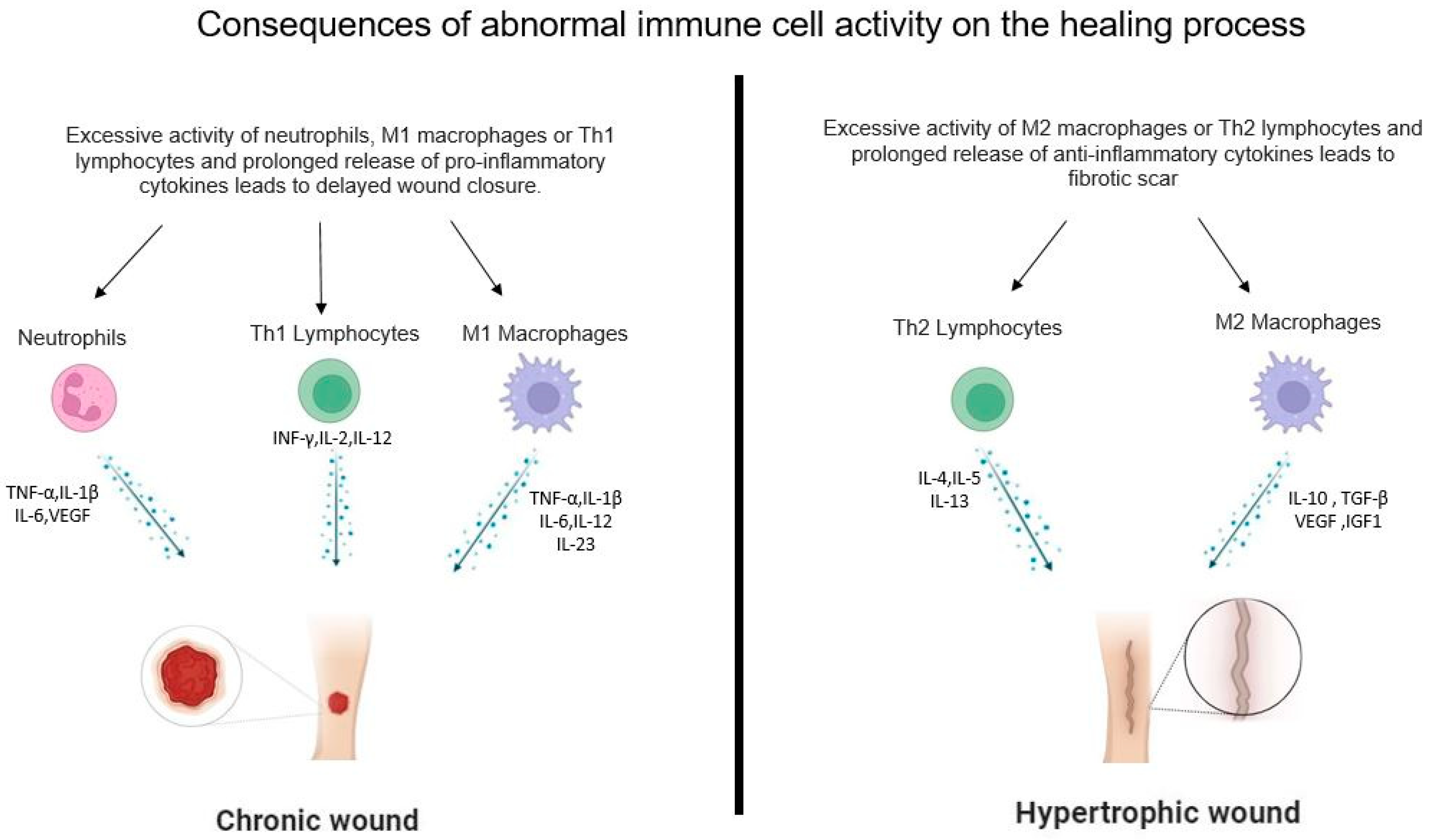
4.1. Neutrophils
4.2. Macrophages
4.2.1. M1 Macrophages
4.2.2. M2 Macrophages
4.3. Mast Cells
4.4. T-Cell Populations
4.4.1. Th1 Lymphocytes
4.4.2. Th2 Lymphocytes
4.4.3. Th17 and Th22 Lymphocytes
4.4.4. T-Reg Lymphocytes
4.4.5. γδ T Lymphocytes
4.5. Langerhans Cells and Dermal Dendritic Cells
5. Role of Immune Cells in the Remodeling Phases of Wound Healing
6. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wilkinson, H.N.; Hardman, M.J. Wound Healing: Cellular Mechanisms and Pathological Outcomes. Open Biol. 2020, 10, 200223. [Google Scholar] [CrossRef] [PubMed]
- Singer, A.J.; Clark, R.A.F. Cutaneous Wound Healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Kosaric, N.; Bonham, C.A.; Gurtner, G.C. Wound Healing: A Cellular Perspective. Physiol. Rev. 2019, 99, 665–706. [Google Scholar] [CrossRef]
- Martins-Green, M.; Petreaca, M.; Wang, L. Chemokines and Their Receptors Are Key Players in the Orchestra That Regulates Wound Healing. Adv. Wound Care 2013, 2, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s That Spur Autophagy and Immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Piipponen, M.; Li, D.; Landén, N.X. The Immune Functions of Keratinocytes in Skin Wound Healing. Int. J. Mol. Sci. 2020, 21, 8790. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Aiba, S.; Yoshino, Y.; Tagami, H. Acute Cutaneous Barrier Disruption Activates Epidermal P44/42 and P38 Mitogen-Activated Protein Kinases in Human and Hairless Guinea Pig Skin. Exp. Dermatol. 2003, 12, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.J.; Martin, P. Wound Repair at a Glance. J. Cell Sci. 2009, 122, 3209–3213. [Google Scholar] [CrossRef] [PubMed]
- Larouche, J.; Sheoran, S.; Maruyama, K.; Martino, M.M. Immune Regulation of Skin Wound Healing: Mechanisms and Novel Therapeutic Targets. Adv. Wound Care 2018, 7, 209–231. [Google Scholar] [CrossRef]
- Schultz, G.S.; Chin, G.A.; Moldawer, L.; Diegelmann, R.F. Principles of Wound Healing. In Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists; Fitridge, R., Thompson, M., Eds.; University of Adelaide Press: Adelaide, Australia, 2011; ISBN 978-0-9871718-2-5. [Google Scholar]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. PERSPECTIVE ARTICLE: Growth Factors and Cytokines in Wound Healing. Wound Repair. Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef]
- Pastar, I.; Stojadinovic, O.; Yin, N.C.; Ramirez, H.; Nusbaum, A.G.; Sawaya, A.; Patel, S.B.; Khalid, L.; Isseroff, R.R.; Tomic-Canic, M. Epithelialization in Wound Healing: A Comprehensive Review. Adv. Wound Care 2014, 3, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Ucuzian, A.A.; Gassman, A.A.; East, A.T.; Greisler, H.P. Molecular Mediators of Angiogenesis. J. Burn Care Res. 2010, 31, 158. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, T.; Kaburagi, Y.; Hamaguchi, Y.; Hasegawa, M.; Takehara, K.; Steeber, D.A.; Tedder, T.F.; Sato, S. Delayed Wound Healing in the Absence of Intercellular Adhesion Molecule-1 or L-Selectin Expression. Am. J. Pathol. 2000, 157, 237–247. [Google Scholar] [CrossRef] [PubMed]
- DiPietro, L.A. Angiogenesis and Wound Repair: When Enough Is Enough. J. Leukoc. Biol. 2016, 100, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Caley, M.P.; Martins, V.L.C.; O’Toole, E.A. Metalloproteinases and Wound Healing. Adv. Wound Care 2015, 4, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, F.; Mijouin, L.; Pichon, C. Skin Immune Landscape: Inside and Outside the Organism. Mediat. Inflamm. 2017, 2017, 5095293. [Google Scholar] [CrossRef] [PubMed]
- Sorg, H.; Tilkorn, D.J.; Hager, S.; Hauser, J.; Mirastschijski, U. Skin Wound Healing: An Update on the Current Knowledge and Concepts. Eur. Surg. Res. 2017, 58, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Del Rosso, J.Q.; Levin, J. The Clinical Relevance of Maintaining the Functional Integrity of the Stratum Corneum in Both Healthy and Disease-Affected Skin. J. Clin. Aesthet. Dermatol. 2011, 4, 22–42. [Google Scholar] [PubMed]
- Kabashima, K.; Honda, T.; Ginhoux, F.; Egawa, G. The Immunological Anatomy of the Skin. Nat. Rev. Immunol. 2019, 19, 19–30. [Google Scholar] [CrossRef]
- Kenshi, Y.; Richard, L.G. Antimicrobial Peptides in Human Skin Disease. Eur. J. Dermatol. 2008, 18, 11–21. [Google Scholar] [CrossRef]
- Nguyen, A.V.; Soulika, A.M. The Dynamics of the Skin’s Immune System. Int. J. Mol. Sci. 2019, 20, 1811. [Google Scholar] [CrossRef]
- Cavallo, I.; Sivori, F.; Mastrofrancesco, A.; Abril, E.; Pontone, M.; Di Domenico, E.G.; Pimpinelli, F. Bacterial Biofilm in Chronic Wounds and Possible Therapeutic Approaches. Biology 2024, 13, 109. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Tsoi, L.C.; Billi, A.C.; Ward, N.L.; Harms, P.W.; Zeng, C.; Maverakis, E.; Kahlenberg, J.M.; Gudjonsson, J.E. Cytokinocytes: The Diverse Contribution of Keratinocytes to Immune Responses in Skin. JCI Insight 2020, 5, e142067. [Google Scholar] [CrossRef] [PubMed]
- Koike, S.; Yamasaki, K. Melanogenesis Connection with Innate Immunity and Toll-Like Receptors. Int. J. Mol. Sci. 2020, 21, 9769. [Google Scholar] [CrossRef] [PubMed]
- Rumbaut, R.E.; Thiagarajan, P. Platelet Adhesion to Vascular Walls. In Platelet-Vessel Wall Interactions in Hemostasis and Thrombosis; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Sang, Y.; Roest, M.; de Laat, B.; de Groot, P.G.; Huskens, D. Interplay between Platelets and Coagulation. Blood Rev. 2021, 46, 100733. [Google Scholar] [CrossRef]
- Scopelliti, F.; Cattani, C.; Dimartino, V.; Mirisola, C.; Cavani, A. Platelet Derivatives and the Immunomodulation of Wound Healing. Int. J. Mol. Sci. 2022, 23, 8370. [Google Scholar] [CrossRef] [PubMed]
- Sinno, H.; Prakash, S. Complements and the Wound Healing Cascade: An Updated Review. Plast. Surg. Int. 2013, 2013, 146764. [Google Scholar] [CrossRef] [PubMed]
- Deuel, T.F.; Senior, R.M.; Chang, D.; Griffin, G.L.; Heinrikson, R.L.; Kaiser, E.T. Platelet Factor 4 Is Chemotactic for Neutrophils and Monocytes. Proc. Natl. Acad. Sci. USA 1981, 78, 4584–4587. [Google Scholar] [CrossRef] [PubMed]
- Chicharro-Alcántara, D.; Rubio-Zaragoza, M.; Damiá-Giménez, E.; Carrillo-Poveda, J.M.; Cuervo-Serrato, B.; Peláez-Gorrea, P.; Sopena-Juncosa, J.J. Platelet Rich Plasma: New Insights for Cutaneous Wound Healing Management. J. Funct. Biomater. 2018, 9, 10. [Google Scholar] [CrossRef]
- Chong, D.L.W.; Trinder, S.; Labelle, M.; Rodriguez-Justo, M.; Hughes, S.; Holmes, A.M.; Scotton, C.J.; Porter, J.C. Platelet-Derived Transforming Growth Factor-Β1 Promotes Keratinocyte Proliferation in Cutaneous Wound Healing. J. Tissue Eng. Regen. Med. 2020, 14, 645–649. [Google Scholar] [CrossRef]
- Raziyeva, K.; Kim, Y.; Zharkinbekov, Z.; Kassymbek, K.; Jimi, S.; Saparov, A. Immunology of Acute and Chronic Wound Healing. Biomolecules 2021, 11, 700. [Google Scholar] [CrossRef] [PubMed]
- Garraud, O.; Hamzeh-Cognasse, H.; Pozzetto, B.; Cavaillon, J.-M.; Cognasse, F. Bench-to-Bedside Review: Platelets and Active Immune Functions—New Clues for Immunopathology? Crit. Care 2013, 17, 236. [Google Scholar] [CrossRef] [PubMed]
- Balaji, S.; Watson, C.L.; Ranjan, R.; King, A.; Bollyky, P.L.; Keswani, S.G. Chemokine Involvement in Fetal and Adult Wound Healing. Adv. Wound Care 2015, 4, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Dickhout, A.; Kaczor, D.M.; Heinzmann, A.C.A.; Brouns, S.L.N.; Heemskerk, J.W.M.; van Zandvoort, M.A.M.J.; Koenen, R.R. Rapid Internalization and Nuclear Translocation of CCL5 and CXCL4 in Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 7332. [Google Scholar] [CrossRef] [PubMed]
- Blair, P.; Flaumenhaft, R. Platelet α–Granules: Basic Biology and Clinical Correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef]
- Scherlinger, M.; Richez, C.; Tsokos, G.C.; Boilard, E.; Blanco, P. The Role of Platelets in Immune-Mediated Inflammatory Diseases. Nat. Rev. Immunol. 2023, 23, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Elzey, B.D.; Tian, J.; Jensen, R.J.; Swanson, A.K.; Lees, J.R.; Lentz, S.R.; Stein, C.S.; Nieswandt, B.; Wang, Y.; Davidson, B.L.; et al. Platelet-Mediated Modulation of Adaptive Immunity: A Communication Link between Innate and Adaptive Immune Compartments. Immunity 2003, 19, 9–19. [Google Scholar] [CrossRef]
- Scopelliti, F.; Caterina, C.; Valentina, D.; Gianfranco, C.; Concetta, M.; Andrea, C. Platelet Lysate Converts M (IFNγ+LPS) Macrophages in CD206+ TGF-Β+ Arginase+ M2-like Macrophages That Affect Fibroblast Activity and T Lymphocyte Migration. J. Tissue Eng. Regen. Med. 2021, 15, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Scopelliti, F.; Cattani, C.; Dimartino, V.; Scarponi, C.; Madonna, S.; Albanesi, C.; Costanzo, G.; Mirisola, C.; Cavani, A. Platelet Lysate Promotes the Expansion of T Regulatory Cells That Favours In Vitro Wound Healing by Increasing Keratinocyte Migration and Fibroblast Production of Extracellular Matrix Components. Eur. J. Dermatol. 2020, 30, 3–11. [Google Scholar] [CrossRef]
- Faurschou, M.; Borregaard, N. Neutrophil Granules and Secretory Vesicles in Inflammation. Microbes Infect. 2003, 5, 1317–1327. [Google Scholar] [CrossRef]
- Wilgus, T.A.; Roy, S.; McDaniel, J.C. Neutrophils and Wound Repair: Positive Actions and Negative Reactions. Adv. Wound Care 2013, 2, 379. [Google Scholar] [CrossRef]
- Scozzi, D.; Liao, F.; Krupnick, A.S.; Kreisel, D.; Gelman, A.E. The Role of Neutrophil Extracellular Traps in Acute Lung Injury. Front. Immunol. 2022, 13, 953195. [Google Scholar] [CrossRef]
- Mutua, V.; Gershwin, L.J. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin. Rev. Allergy Immunol. 2021, 61, 194–211. [Google Scholar] [CrossRef] [PubMed]
- Theilgaard-Mönch, K.; Knudsen, S.; Follin, P.; Borregaard, N. The Transcriptional Activation Program of Human Neutrophils in Skin Lesions Supports Their Important Role in Wound Healing. J. Immunol. 2004, 172, 7684–7693. [Google Scholar] [CrossRef] [PubMed]
- Boniakowski, A.E.; Kimball, A.S.; Jacobs, B.N.; Kunkel, S.L.; Gallagher, K.A. Macrophage-Mediated Inflammation in Normal and Diabetic Wound Healing. J. Immunol. 2017, 199, 17–24. [Google Scholar] [CrossRef]
- Snyder, R.J.; Lantis, J.; Kirsner, R.S.; Shah, V.; Molyneaux, M.; Carter, M.J. Macrophages: A Review of Their Role in Wound Healing and Their Therapeutic Use. Wound Repair. Regen. 2016, 24, 613–629. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage Plasticity and Polarization: In Vivo Veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Slauch, J.M. How Does the Oxidative Burst of Macrophages Kill Bacteria? Still an Open Question. Mol. Microbiol. 2011, 80, 580–583. [Google Scholar] [CrossRef]
- Aitcheson, S.M.; Frentiu, F.D.; Hurn, S.E.; Edwards, K.; Murray, R.Z. Skin Wound Healing: Normal Macrophage Function and Macrophage Dysfunction in Diabetic Wounds. Molecules 2021, 26, 4917. [Google Scholar] [CrossRef]
- Aristorena, M.; Gallardo-Vara, E.; Vicen, M.; de Las Casas-Engel, M.; Ojeda-Fernandez, L.; Nieto, C.; Blanco, F.J.; Valbuena-Diez, A.C.; Botella, L.M.; Nachtigal, P.; et al. MMP-12, Secreted by Pro-Inflammatory Macrophages, Targets Endoglin in Human Macrophages and Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 3107. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Landén, N.X.; Li, D.; Ståhle, M. Transition from Inflammation to Proliferation: A Critical Step during Wound Healing. Cell. Mol. Life Sci. 2016, 73, 3861–3885. [Google Scholar] [CrossRef]
- Leask, A. Potential Therapeutic Targets for Cardiac Fibrosis. Circ. Res. 2010, 106, 1675–1680. [Google Scholar] [CrossRef]
- Darby, I.A.; Laverdet, B.; Bonté, F.; Desmoulière, A. Fibroblasts and Myofibroblasts in Wound Healing. Clin. Cosmet. Investig. Dermatol. 2014, 7, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound Repair and Regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Veith, A.P.; Henderson, K.; Spencer, A.; Sligar, A.D.; Baker, A.B. Therapeutic Strategies for Enhancing Angiogenesis in Wound Healing. Adv. Drug Deliv. Rev. 2019, 146, 97–125. [Google Scholar] [CrossRef]
- Komi, D.E.A.; Rambasek, T.; Wöhrl, S. Mastocytosis: From a Molecular Point of View. Clin. Rev. Allergy Immunol. 2018, 54, 397–411. [Google Scholar] [CrossRef]
- Trautmann, A.; Toksoy, A.; Engelhardt, E.; Bröcker, E.-B.; Gillitzer, R. Mast Cell Involvement in Normal Human Skin Wound Healing: Expression of Monocyte Chemoattractant Protein-1 Is Correlated with Recruitment of Mast Cells Which Synthesize Interleukin-4 In Vivo. J. Pathol. 2000, 190, 100–106. [Google Scholar] [CrossRef]
- Ng, M.F. The Role of Mast Cells in Wound Healing. Int. Wound J. 2010, 7, 55–61. [Google Scholar] [CrossRef]
- Komi, D.E.A.; Khomtchouk, K.; Santa Maria, P.L. A Review of the Contribution of Mast Cells in Wound Healing: Involved Molecular and Cellular Mechanisms. Clin. Rev. Allergy Immunol. 2020, 58, 298–312. [Google Scholar] [CrossRef]
- Metz, M.; Siebenhaar, F.; Maurer, M. Mast Cell Functions in the Innate Skin Immune System. Immunobiology 2008, 213, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Sabat, R.; Wolk, K.; Loyal, L.; Döcke, W.-D.; Ghoreschi, K. T Cell Pathology in Skin Inflammation. Semin. Immunopathol. 2019, 41, 359–377. [Google Scholar] [CrossRef]
- Ishida, Y.; Kondo, T.; Takayasu, T.; Iwakura, Y.; Mukaida, N. The Essential Involvement of Cross-Talk between IFN-γ and TGF-β in the Skin Wound-Healing Process. J. Immunol. 2004, 172, 1848–1855. [Google Scholar] [CrossRef] [PubMed]
- Short, W.D.; Wang, X.; Keswani, S.G. The Role of T Lymphocytes in Cutaneous Scarring. Adv. Wound Care 2022, 11, 121–131. [Google Scholar] [CrossRef]
- Fujita, H. The Role of IL-22 and Th22 Cells in Human Skin Diseases. J. Dermatol. Sci. 2013, 72, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Avitabile, S.; Odorisio, T.; Madonna, S.; Eyerich, S.; Guerra, L.; Eyerich, K.; Zambruno, G.; Cavani, A.; Cianfarani, F. Interleukin-22 Promotes Wound Repair in Diabetes by Improving Keratinocyte Pro-Healing Functions. J. Investig. Dermatol. 2015, 135, 2862–2870. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, D.M.; Minutti, C.M.; Knipper, J.A. Immune- and Non-immune-mediated Roles of Regulatory T-cells during Wound Healing. Immunology 2019, 157, 190–197. [Google Scholar] [CrossRef]
- Li, J.; Tan, J.; Martino, M.M.; Lui, K.O. Regulatory T-Cells: Potential Regulator of Tissue Repair and Regeneration. Front. Immunol. 2018, 9, 585. [Google Scholar] [CrossRef] [PubMed]
- Munoz, L.D.; Sweeney, M.J.; Jameson, J.M. Skin Resident Γδ T Cell Function and Regulation in Wound Repair. Int. J. Mol. Sci. 2020, 21, 9286. [Google Scholar] [CrossRef]
- Jameson, J.M.; Cauvi, G.; Witherden, D.A.; Havran, W.L. A Keratinocyte-Responsive Γδ TCR Is Necessary for Dendritic Epidermal T Cell Activation by Damaged Keratinocytes and Maintenance in the Epidermis. J. Immunol. 2004, 172, 3573–3579. [Google Scholar] [CrossRef]
- Jameson, J.M.; Cauvi, G.; Sharp, L.L.; Witherden, D.A.; Havran, W.L. Γδ T Cell–Induced Hyaluronan Production by Epithelial Cells Regulates Inflammation. J. Exp. Med. 2005, 201, 1269–1279. [Google Scholar] [CrossRef]
- Cruz, M.S.; Diamond, A.; Russell, A.; Jameson, J.M. Human Aβ and Γδ T Cells in Skin Immunity and Disease. Front. Immunol. 2018, 9, 1304. [Google Scholar] [CrossRef] [PubMed]
- Romani, N.; Clausen, B.E.; Stoitzner, P. Langerhans Cells and More: Langerin-Expressing Dendritic Cell Subsets in the Skin. Immunol. Rev. 2010, 234, 120–141. [Google Scholar] [CrossRef] [PubMed]
- Kabashima, K.; Shiraishi, N.; Sugita, K.; Mori, T.; Onoue, A.; Kobayashi, M.; Sakabe, J.; Yoshiki, R.; Tamamura, H.; Fujii, N.; et al. CXCL12-CXCR4 Engagement Is Required for Migration of Cutaneous Dendritic Cells. Am. J. Pathol. 2007, 171, 1249–1257. [Google Scholar] [CrossRef]
- Adib, Y.; Bensussan, A.; Michel, L. Cutaneous Wound Healing: A Review about Innate Immune Response and Current Therapeutic Applications. Mediat. Inflamm. 2022, 2022, 5344085. [Google Scholar] [CrossRef] [PubMed]
- Galkowska, H.; Olszewski, W.L.; Wojewodzka, U. Expression of Natural Antimicrobial Peptide Beta-Defensin-2 and Langerhans Cell Accumulation in Epidermis from Human Non-Healing Leg Ulcers. Folia Histochem. Cytobiol. 2005, 43, 133–136. [Google Scholar] [PubMed]
- Niessen, F.B.; Schalkwijk, J.; Vos, H.; Timens, W. Hypertrophic Scar Formation Is Associated with an Increased Number of Epidermal Langerhans Cells. J. Pathol. 2004, 202, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, A.; Stuart, G.; Real, N.; Ahn, J.; Tschirley, A.; Wise, L.; Hibma, M. Depletion of Langerin+ Cells Enhances Cutaneous Wound Healing. Immunology 2020, 160, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Di Meglio, P.; Qin, J.-Z.; Nickoloff, B.J. Skin Immune Sentinels in Health and Disease. Nat. Rev. Immunol. 2009, 9, 679–691. [Google Scholar] [CrossRef]
- Gregorio, J.; Meller, S.; Conrad, C.; Di Nardo, A.; Homey, B.; Lauerma, A.; Arai, N.; Gallo, R.L.; DiGiovanni, J.; Gilliet, M. Plasmacytoid Dendritic Cells Sense Skin Injury and Promote Wound Healing through Type I Interferons. J. Exp. Med. 2010, 207, 2921–2930. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Wound Healing Phase | Cell Type | Cytokines | Signaling Pathways |
|---|---|---|---|
| INFLAMMATION | NEUTROPHILES | TNF-α IL-1β IL-6 VEGF | NF-kB NF-kB STAT3 ERK |
| M1 MACROPHAGES | TNF-α IL-1β IL-6 IL-12 IL-23 | NF-kB NF-kB STAT3 STAT4 STAT4 | |
| MAST CELL | TNF-α IL-6 VEGF IL-8 | NF-kB STAT3 ERK STAT3/ERK | |
| TH1 LYMPHOCYTES | INF-γ IL-2 IL-12 | STAT1 STAT3/ERK STAT4 | |
| PROLIFERATION | M2 MACROPHAGES | IL-10 TGF-β VEGF IGF1 | STAT3 SMAD ERK ERK |
| TH2 LYMPHOCYTES | IL-4 IL-5 IL-13 | STAT6 ERK/NF-kB STAT6 | |
| REMODELING | T-REG | IL-10 TGF-β | STAT3 SMAD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cioce, A.; Cavani, A.; Cattani, C.; Scopelliti, F. Role of the Skin Immune System in Wound Healing. Cells 2024, 13, 624. https://doi.org/10.3390/cells13070624
Cioce A, Cavani A, Cattani C, Scopelliti F. Role of the Skin Immune System in Wound Healing. Cells. 2024; 13(7):624. https://doi.org/10.3390/cells13070624
Chicago/Turabian StyleCioce, Angela, Andrea Cavani, Caterina Cattani, and Fernanda Scopelliti. 2024. "Role of the Skin Immune System in Wound Healing" Cells 13, no. 7: 624. https://doi.org/10.3390/cells13070624
APA StyleCioce, A., Cavani, A., Cattani, C., & Scopelliti, F. (2024). Role of the Skin Immune System in Wound Healing. Cells, 13(7), 624. https://doi.org/10.3390/cells13070624