
Challenges and Considerations of Preclinical Development for iPSC-Based Myogenic Cell Therapy

 ,
,
Abstract
1. Introduction
2. Differentiation and Maintenance of Myogenic Cells from Pluripotent Stem Cells
2.1. Myogenic Lineage Specification from hiPSCs
2.1.1. Direct Programming
2.1.2. Transgene-Free 2D Myogenic Differentiation
2.1.3. Three-Dimensional Myogenic Differentiation
2.2. Lineage Specification Efficiency, Purification Process, and Tumorigenicity
2.3. Maintaining Muscle Progenitor Cell Characteristics in Culture
3. Genetic Modification
Genetic Modification Strategies
4. Development and Challenges of Disease Models for Preclinical Studies
4.1. Cellular Models
4.2. Animal Models
4.2.1. Murine Models
Duchenne Muscular Dystrophy
Facioscapulohumeral Muscular Dystrophy
Oculopharyngeal Muscular Dystrophy
Limb–Girdle Muscular Dystrophies
Myotonic Dystrophy
4.2.2. Other Animal Models (Zebrafish, Dogs, Pigs)
4.3. Animal Models for Myogenic Cell Transplantation Therapy
5. Delivery Strategies
6. Manufacturing Considerations for Clinically Compatible Cell Product
6.1. Manufacturing Challenges for Myogenic Lineage Specification
6.2. Manufacturing Challenges for Cell Expansion
7. Future Challenges and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Bradley, A.; Evans, M.; Kaufman, M.H.; Robertson, E. Formation of germ-line chimaeras from embryo-derived teratocarcinoma cell lines. Nature 1984, 309, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Markoulaki, S.; Hanna, J.; Saha, K.; Gao, Q.; Mitalipova, M.; Jaenisch, R. Reprogramming of murine and human somatic cells using a single polycistronic vector. Proc. Natl. Acad. Sci. USA 2009, 106, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Lai, Y.S.; Pawlik, K.M.; Liu, K.; Sun, C.W.; Li, C.; Schoeb, T.R.; Townes, T.M. Polycistronic lentiviral vector for “hit and run” reprogramming of adult skin fibroblasts to induced pluripotent stem cells. Stem Cells 2009, 27, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Nefzger, C.M.; Rossello, F.J.; Chen, J.; Knaupp, A.S.; Firas, J.; Ford, E.; Pflueger, J.; Paynter, J.M.; Chy, H.S.; et al. Comprehensive characterization of distinct states of human naive pluripotency generated by reprogramming. Nat. Methods 2017, 14, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Weltner, J.; Anisimov, A.; Alitalo, K.; Otonkoski, T.; Trokovic, R. Induced pluripotent stem cell clones reprogrammed via recombinant adeno-associated virus-mediated transduction contain integrated vector sequences. J. Virol. 2012, 86, 4463–4467. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Xu, Y.; Cao, S.; Shi, K. Inducing somatic cells into pluripotent stem cells is an important platform to study the mechanism of early embryonic development. Mol. Reprod. Dev. 2022, 89, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Nam, Y.; Rim, Y.A.; Ju, J.H. Review of the Current Trends in Clinical Trials Involving Induced Pluripotent Stem Cells. Stem Cell Rev. Rep. 2022, 18, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Relaix, F. PAX3 and PAX7 as upstream regulators of myogenesis. Semin. Cell Dev. Biol. 2015, 44, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, F.; Chen, Q.; Wan, X.; Shi, M.; Chen, A.K.; Ma, Z.; Li, G.; Wang, M.; Ying, Y.; et al. MyoD is a 3D genome structure organizer for muscle cell identity. Nat. Commun. 2022, 13, 205. [Google Scholar] [CrossRef] [PubMed]
- Kablar, B.; Krastel, K.; Ying, C.; Asakura, A.; Tapscott, S.J.; Rudnicki, M.A. MyoD and Myf-5 differentially regulate the development of limb versus trunk skeletal muscle. Development 1997, 124, 4729–4738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Mendieta-Esteban, J.; Magli, A.; Lilja, K.C.; Perlingeiro, R.C.R.; Marti-Renom, M.A.; Tsirigos, A.; Dynlacht, B.D. Muscle progenitor specification and myogenic differentiation are associated with changes in chromatin topology. Nat. Commun. 2020, 11, 6222. [Google Scholar] [CrossRef] [PubMed]
- Magli, A.; Baik, J.; Mills, L.J.; Kwak, I.Y.; Dillon, B.S.; Mondragon Gonzalez, R.; Stafford, D.A.; Swanson, S.A.; Stewart, R.; Thomson, J.A.; et al. Time-dependent Pax3-mediated chromatin remodeling and cooperation with Six4 and Tead2 specify the skeletal myogenic lineage in developing mesoderm. PLoS Biol. 2019, 17, e3000153. [Google Scholar] [CrossRef] [PubMed]
- Ozasa, S.; Kimura, S.; Ito, K.; Ueno, H.; Ikezawa, M.; Matsukura, M.; Yoshioka, K.; Araki, K.; Yamamura, K.I.; Abe, K.; et al. Efficient conversion of ES cells into myogenic lineage using the gene-inducible system. Biochem. Biophys. Res. Commun. 2007, 357, 957–963. [Google Scholar] [CrossRef]
- Abujarour, R.; Bennett, M.; Valamehr, B.; Lee, T.T.; Robinson, M.; Robbins, D.; Le, T.; Lai, K.; Flynn, P. Myogenic differentiation of muscular dystrophy-specific induced pluripotent stem cells for use in drug discovery. Stem Cells Transl. Med. 2014, 3, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Darabi, R.; Arpke, R.W.; Irion, S.; Dimos, J.T.; Grskovic, M.; Kyba, M.; Perlingeiro, R.C. Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell 2012, 10, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Darabi, R.; Santos, F.N.; Filareto, A.; Pan, W.; Koene, R.; Rudnicki, M.A.; Kyba, M.; Perlingeiro, R.C. Assessment of the myogenic stem cell compartment following transplantation of Pax3/Pax7-induced embryonic stem cell-derived progenitors. Stem Cells 2011, 29, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Selvaraj, S.; Kiley, J.; Azzag, K.; Garay, B.I.; Perlingeiro, R.C.R. Genomic Safe Harbor Expression of PAX7 for the Generation of Engraftable Myogenic Progenitors. Stem Cell Rep. 2021, 16, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Oliveira, V.K.P.; Yamamoto, A.; Perlingeiro, R.C.R. Generation of skeletal myogenic progenitors from human pluripotent stem cells using non-viral delivery of minicircle DNA. Stem Cell Res. 2017, 23, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wakamiya, M.; Shea, M.J.; Albrecht, U.; Behringer, R.R.; Bradley, A. Requirement for Wnt3 in vertebrate axis formation. Nat. Genet. 1999, 22, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Shelton, M.; Metz, J.; Liu, J.; Carpenedo, R.L.; Demers, S.P.; Stanford, W.L.; Skerjanc, I.S. Derivation and expansion of PAX7-positive muscle progenitors from human and mouse embryonic stem cells. Stem Cell Rep. 2014, 3, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Al Tanoury, Z.; Oginuma, M.; Moncuquet, P.; Gobert, B.; Miyanari, A.; Tassy, O.; Guevara, G.; Hubaud, A.; Bera, A.; et al. Recapitulating early development of mouse musculoskeletal precursors of the paraxial mesoderm in vitro. Development 2018, 145, dev157339. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.Y.; Lim, H.; Estrellas, K.; Mula, J.; Cohen, T.V.; Zhang, Y.; Donnelly, C.J.; Richard, J.P.; Kim, Y.J.; Kim, H.; et al. Concordant but Varied Phenotypes among Duchenne Muscular Dystrophy Patient-Specific Myoblasts Derived using a Human iPSC-Based Model. Cell Rep. 2016, 15, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Tazumi, A.; Takayama, S.; Takenaka-Ninagawa, N.; Nalbandian, M.; Nagai, M.; Nakamura, Y.; Nakasa, M.; Watanabe, A.; Ikeya, M.; et al. Induced Fetal Human Muscle Stem Cells with High Therapeutic Potential in a Mouse Muscular Dystrophy Model. Stem Cell Rep. 2020, 15, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Hicks, M.R.; Hiserodt, J.; Paras, K.; Fujiwara, W.; Eskin, A.; Jan, M.; Xi, H.; Young, C.S.; Evseenko, D.; Nelson, S.F.; et al. ERBB3 and NGFR mark a distinct skeletal muscle progenitor cell in human development and hPSCs. Nat. Cell Biol. 2018, 20, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Nalbandian, M.; Zhao, M.; Sasaki-Honda, M.; Jonouchi, T.; Lucena-Cacace, A.; Mizusawa, T.; Yasuda, M.; Yoshida, Y.; Hotta, A.; Sakurai, H. Characterization of hiPSC-Derived Muscle Progenitors Reveals Distinctive Markers for Myogenic Cell Purification Toward Cell Therapy. Stem Cell Rep. 2021, 16, 883–898. [Google Scholar] [CrossRef]
- Shin, M.K.; Bang, J.S.; Lee, J.E.; Tran, H.D.; Park, G.; Lee, D.R.; Jo, J. Generation of Skeletal Muscle Organoids from Human Pluripotent Stem Cells to Model Myogenesis and Muscle Regeneration. Int. J. Mol. Sci. 2022, 23, 5108. [Google Scholar] [CrossRef]
- Mavrommatis, L.; Jeong, H.-W.; Gomez-Giro, G.; Stehling, M.; Kienitz, M.-C.; Psathaki, O.E.; Bixel, M.G.; Morosan-Puopolo, G.; Gerovska, D.; Araúzo-Bravo, M.J.; et al. Human skeletal muscle organoids model fetal myogenesis and sustain uncommitted PAX7 myogenic progenitors. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sakai-Takemura, F.; Narita, A.; Masuda, S.; Wakamatsu, T.; Watanabe, N.; Nishiyama, T.; Nogami, K.; Blanc, M.; Takeda, S.; Miyagoe-Suzuki, Y. Premyogenic progenitors derived from human pluripotent stem cells expand in floating culture and differentiate into transplantable myogenic progenitors. Sci. Rep. 2018, 8, 6555. [Google Scholar] [CrossRef] [PubMed]
- Faustino Martins, J.M.; Fischer, C.; Urzi, A.; Vidal, R.; Kunz, S.; Ruffault, P.L.; Kabuss, L.; Hube, I.; Gazzerro, E.; Birchmeier, C.; et al. Self-Organizing 3D Human Trunk Neuromuscular Organoids. Cell Stem Cell 2020, 27, 498. [Google Scholar] [CrossRef] [PubMed]
- Mashinchian, O.; De Franceschi, F.; Nassiri, S.; Michaud, J.; Migliavacca, E.; Aouad, P.; Metairon, S.; Pruvost, S.; Karaz, S.; Fabre, P.; et al. An engineered multicellular stem cell niche for the 3D derivation of human myogenic progenitors from iPSCs. EMBO J. 2022, 41, e110655. [Google Scholar] [CrossRef] [PubMed]
- Tani, S.; Chung, U.I.; Ohba, S.; Hojo, H. Understanding paraxial mesoderm development and sclerotome specification for skeletal repair. Exp. Mol. Med. 2020, 52, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Langerman, J.; Sabri, S.; Chien, P.; Young, C.S.; Younesi, S.; Hicks, M.; Gonzalez, K.; Fujiwara, W.; Marzi, J.; et al. A Human Skeletal Muscle Atlas Identifies the Trajectories of Stem and Progenitor Cells across Development and from Human Pluripotent Stem Cells. Cell Stem Cell 2020, 27, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Magli, A.; Chan, S.S.K.; Oliveira, V.K.P.; Wu, J.; Darabi, R.; Kyba, M.; Perlingeiro, R.C.R. Expansion and Purification Are Critical for the Therapeutic Application of Pluripotent Stem Cell-Derived Myogenic Progenitors. Stem Cell Rep. 2017, 9, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Campbell, H.M.; Dittel, B.N.; Ray, A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J. Vis. Exp. 2010, 10, 1546. [Google Scholar] [CrossRef]
- Magli, A.; Incitti, T.; Kiley, J.; Swanson, S.A.; Darabi, R.; Rinaldi, F.; Selvaraj, S.; Yamamoto, A.; Tolar, J.; Yuan, C.; et al. PAX7 Targets, CD54, Integrin alpha9beta1, and SDC2, Allow Isolation of Human ESC/iPSC-Derived Myogenic Progenitors. Cell Rep. 2017, 19, 2867–2877. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Matthias, N.; Lo, J.; Ortiz-Vitali, J.L.; Shieh, A.W.; Wang, S.H.; Darabi, R. A Myogenic Double-Reporter Human Pluripotent Stem Cell Line Allows Prospective Isolation of Skeletal Muscle Progenitors. Cell Rep. 2018, 25, 1966–1981. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.Y.; Lim, H.; Cho, H.J.; Oh, Y.; Chou, B.K.; Bai, H.; Cheng, L.; Kim, Y.J.; Hyun, S.; Kim, H.; et al. Transcriptional landscape of myogenesis from human pluripotent stem cells reveals a key role of TWIST1 in maintenance of skeletal muscle progenitors. Elife 2020, 9, e46981. [Google Scholar] [CrossRef] [PubMed]
- Valli, H.; Sukhwani, M.; Dovey, S.L.; Peters, K.A.; Donohue, J.; Castro, C.A.; Chu, T.; Marshall, G.R.; Orwig, K.E. Fluorescence- and magnetic-activated cell sorting strategies to isolate and enrich human spermatogonial stem cells. Fertil. Steril. 2014, 102, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Schraivogel, D.; Kuhn, T.M.; Rauscher, B.; Rodriguez-Martinez, M.; Paulsen, M.; Owsley, K.; Middlebrook, A.; Tischer, C.; Ramasz, B.; Ordonez-Rueda, D.; et al. High-speed fluorescence image-enabled cell sorting. Science 2022, 375, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K. Muscle as a secretory organ. Compr. Physiol. 2013, 3, 1337–1362. [Google Scholar] [CrossRef]
- Miura, K.; Okada, Y.; Aoi, T.; Okada, A.; Takahashi, K.; Okita, K.; Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Ohnuki, M.; et al. Variation in the safety of induced pluripotent stem cell lines. Nat. Biotechnol. 2009, 27, 743–745. [Google Scholar] [CrossRef]
- Zhong, C.; Liu, M.; Pan, X.; Zhu, H. Tumorigenicity risk of iPSCs in vivo: Nip it in the bud. Precis. Clin. Med. 2022, 5, pbac004. [Google Scholar] [CrossRef] [PubMed]
- Sekine, K.; Tsuzuki, S.; Yasui, R.; Kobayashi, T.; Ikeda, K.; Hamada, Y.; Kanai, E.; Camp, J.G.; Treutlein, B.; Ueno, Y.; et al. Robust detection of undifferentiated iPSC among differentiated cells. Sci. Rep. 2020, 10, 10293. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, T.; Yasuda, S.; Kusakawa, S.; Hirata, N.; Kanda, Y.; Suzuki, K.; Takahashi, M.; Nishikawa, S.; Kawamata, S.; Sato, Y. Highly sensitive in vitro methods for detection of residual undifferentiated cells in retinal pigment epithelial cells derived from human iPS cells. PLoS ONE 2012, 7, e37342. [Google Scholar] [CrossRef] [PubMed]
- Gussoni, E.; Pavlath, G.K.; Lanctot, A.M.; Sharma, K.R.; Miller, R.G.; Steinman, L.; Blau, H.M. Normal Dys-trophin Transcripts Detected in Duchenne Muscular Dystrophy Patients after Myoblast Transplantation. Nature 1992, 356, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Huard, J.; Bouchard, J.P.; Roy, R.; Malouin, F.; Dansereau, G.; Labrecque, C.; Albert, N.; Richards, C.L.; Lemieux, B.; Tremblay, J.P. Human Myoblast Transplantation: Preliminary Results of 4 Cases. Muscle Nerve 1992, 15, 550–560. [Google Scholar] [CrossRef]
- Karpati, G.; Ajdukovic, D.; Arnold, D.; Gledhill, R.B.; Guttmann, R.; Holland, P.; Koch, P.A.; Shoubridge, E.; Spence, D.; Vanasse, M.; et al. Myoblast Transfer in Duchenne Muscular Dystrophy. Ann. Neurol. 1993, 34, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Kissel, J.T.; Amato, A.A.; King, W.; Signore, L.; Prior, T.W.; Sahenk, Z.; Benson, S.; McAndrew, P.E.; Rice, R.; et al. Myoblast Transfer in the Treatment of Duchenne’s Muscular Dystrophy. N. Engl. J. Med. 1995, 333, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Morandi, L.; Bernasconi, P.; Gebbia, M.; Mora, M.; Crosti, F.; Mantegazza, R.; Cornelio, F. Lack of Mrna and Dystrophin Expression in Dmd Patients Three Months after Myoblast Transfer. Neuromuscul. Disord. 1995, 5, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Skuk, D.; Goulet, M.; Roy, B.; Chapdelaine, P.; Bouchard, J.P.; Roy, R.; Dugré, F.J.; Sylvain, M.; Lachance, J.G.; Deschênes, L.; et al. Dystrophin Expression in Muscles of Duchenne Muscular Dystrophy Patients after High-Density Injections of Normal Myogenic Cells. J. Neuropathol. Exp. Neurol. 2006, 65, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Skuk, D.; Goulet, M.; Roy, B.; Piette, V.; Côté, C.H.; Chapdelaine, P.; Hogrel, J.Y.; Paradis, M.; Bouchard, J.P.; Sylvain, M.; et al. First Test of a "High-Density Injection" Protocol for Myogenic Cell Transplantation Throughout Large Volumes of Muscles in a Duchenne Muscular Dystrophy Patient: Eighteen Months Follow-Up. Neuromuscul. Disord. 2007, 17, 38–46. [Google Scholar] [CrossRef]
- Skuk, D.; Roy, B.; Goulet, M.; Chapdelaine, P.; Bouchard, J.P.; Roy, R.; Dugré, F.J.; Lachance, J.G.; Deschênes, L.; Hélène, S.; et al. Dystrophin Expression in Myofibers of Duchenne Muscular Dystrophy Patients Following Intramuscular Injections of Normal Myogenic Cells. Mol. Ther. 2004, 9, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, J.P.; Malouin, F.; Roy, R.; Huard, J.; Bouchard, J.P.; Satoh, A.; Richards, C.L. Results of a Triple Blind Clinical Study of Myoblast Transplantations without Immunosuppressive Treatment in Young Boys with Duchenne Muscular Dystrophy. Cell Transplant. 1993, 2, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Hauschka, S.D. Clonal analysis of vertebrate myogenesis. II. Environmental influences upon human muscle differentiation. Dev. Biol. 1974, 37, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Ham, R.G.; St Clair, J.A.; Webster, C.; Blau, H.M. Improved media for normal human muscle satellite cells: Serum-free clonal growth and enhanced growth with low serum. In Vitro Cell Dev. Biol. 1988, 24, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Conboy, I.M.; Conboy, M.J.; Smythe, G.M.; Rando, T.A. Notch-mediated restoration of regenerative potential to aged muscle. Science 2003, 302, 1575–1577. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.; Kuroda, K.; Le Grand, F.; Rudnicki, M.A. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 2007, 129, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.M.; Emerson, C.P., Jr.; Owens, J.; Christoforou, N. p38 MAPKs—Roles in skeletal muscle physiology, disease mechanisms, and as potential therapeutic targets. JCI Insight 2021, 6, e149915. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.; Gillespie, M.A.; Rudnicki, M.A. Niche regulation of muscle satellite cell self-renewal and differentiation. Cell Stem Cell 2008, 2, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cheung, D.; Zhou, Y.; Han, C.; Fennelly, C.; Criswell, T.; Soker, S. An in vitro culture system that supports robust expansion and maintenance of in vivo engraftment capabilities for myogenic progenitor cells from adult mice. Biores Open Access 2014, 3, 79–87. [Google Scholar] [CrossRef]
- Quarta, M.; Brett, J.O.; DiMarco, R.; De Morree, A.; Boutet, S.C.; Chacon, R.; Gibbons, M.C.; Garcia, V.A.; Su, J.; Shrager, J.B.; et al. An artificial niche preserves the quiescence of muscle stem cells and enhances their therapeutic efficacy. Nat. Biotechnol. 2016, 34, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; De Mello, V.; Mohamed, A.; Ortuste Quiroga, H.P.; Garcia-Munoz, A.; Al Bloshi, A.; Tremblay, A.M.; von Kriegsheim, A.; Collie-Duguid, E.; Vargesson, N.; et al. Common and Distinctive Functions of the Hippo Effectors Taz and Yap in Skeletal Muscle Stem Cell Function. Stem Cells 2017, 35, 1958–1972. [Google Scholar] [CrossRef] [PubMed]
- Judson, R.N.; Tremblay, A.M.; Knopp, P.; White, R.B.; Urcia, R.; De Bari, C.; Zammit, P.S.; Camargo, F.D.; Wackerhage, H. The Hippo pathway member Yap plays a key role in influencing fate decisions in muscle satellite cells. J. Cell Sci. 2012, 125, 6009–6019. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Rikeit, P.; Knaus, P.; Coirault, C. YAP-Mediated Mechanotransduction in Skeletal Muscle. Front. Physiol. 2016, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, A.; Cera, G.; De Meo, D.; Villani, C.; Bouche, M.; Lozanoska-Ochser, B. A novel approach for the isolation and long-term expansion of pure satellite cells based on ice-cold treatment. Skelet. Muscle 2021, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Hendrie, P.C.; Russell, D.W. Gene targeting with viral vectors. Mol. Ther. 2005, 12, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Walther, W.; Stein, U. Viral vectors for gene transfer: A review of their use in the treatment of human diseases. Drugs 2000, 60, 249–271. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Izsvak, Z. The expanding universe of transposon technologies for gene and cell engineering. Mob. DNA 2010, 1, 25. [Google Scholar] [CrossRef] [PubMed]
- Tipanee, J.; VandenDriessche, T.; Chuah, M.K. Transposons: Moving Forward from Preclinical Studies to Clinical Trials. Hum. Gene Ther. 2017, 28, 1087–1104. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.E.; Chicaybam, L.; Stein, R.T.; Tanuri, A.; Delgado-Canedo, A.; Bonamino, M.H. Retroviral vectors and transposons for stable gene therapy: Advances, current challenges and perspectives. J. Transl. Med. 2016, 14, 288. [Google Scholar] [CrossRef] [PubMed]
- Loperfido, M.; Jarmin, S.; Dastidar, S.; Di Matteo, M.; Perini, I.; Moore, M.; Nair, N.; Samara-Kuko, E.; Athanasopoulos, T.; Tedesco, F.S.; et al. piggyBac transposons expressing full-length human dystrophin enable genetic correction of dystrophic mesoangioblasts. Nucleic Acids Res. 2016, 44, 744–760. [Google Scholar] [CrossRef]
- Iyer, P.S.; Mavoungou, L.O.; Ronzoni, F.; Zemla, J.; Schmid-Siegert, E.; Antonini, S.; Neff, L.A.; Dorchies, O.M.; Jaconi, M.; Lekka, M.; et al. Autologous Cell Therapy Approach for Duchenne Muscular Dystrophy using PiggyBac Transposons and Mesoangioblasts. Mol. Ther. 2018, 26, 1093–1108. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, J.; Xiao, X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc. Natl. Acad. Sci. USA 2000, 97, 13714–13719. [Google Scholar] [CrossRef]
- Maggio, I.; Chen, X.; Goncalves, M.A. The emerging role of viral vectors as vehicles for DMD gene editing. Genome Med. 2016, 8, 59. [Google Scholar] [CrossRef]
- Elangkovan, N.; Dickson, G. Gene Therapy for Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2021, 8, S303–S316. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.H. Genome-Editing Technologies: Concept, Pros, and Cons of Various Genome-Editing Techniques and Bioethical Concerns for Clinical Application. Mol. Ther. Nucleic Acids 2019, 16, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef]
- Hillary, V.E.; Ceasar, S.A. A Review on the Mechanism and Applications of CRISPR/Cas9/Cas12/Cas13/Cas14 Proteins Utilized for Genome Engineering. Mol. Biotechnol. 2023, 65, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Ghosh, A.; Chakravarti, R.; Singh, R.; Ravichandiran, V.; Swarnakar, S.; Ghosh, D. Cas13d: A New Molecular Scissor for Transcriptome Engineering. Front. Cell Dev. Biol. 2022, 10, 866800. [Google Scholar] [CrossRef] [PubMed]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Casas-Mollano, J.A.; Zinselmeier, M.H.; Erickson, S.E.; Smanski, M.J. CRISPR-Cas Activators for Engineering Gene Expression in Higher Eukaryotes. CRISPR J. 2020, 3, 350–364. [Google Scholar] [CrossRef]
- Jensen, T.I.; Mikkelsen, N.S.; Gao, Z.; Fosselteder, J.; Pabst, G.; Axelgaard, E.; Laustsen, A.; Konig, S.; Reinisch, A.; Bak, R.O. Targeted regulation of transcription in primary cells using CRISPRa and CRISPRi. Genome Res. 2021, 31, 2120–2130. [Google Scholar] [CrossRef] [PubMed]
- Wilton-Clark, H.; Yokota, T. CRISPR-Cas9-mediated exon skipping as a cardioprotective strategy in Duchenne muscular dystrophy. Mol. Ther. Methods Clin. Dev. 2023, 30, 500–501. [Google Scholar] [CrossRef] [PubMed]
- Lyu, P.; Yoo, K.W.; Yadav, M.K.; Atala, A.; Aartsma-Rus, A.; Putten, M.V.; Duan, D.; Lu, B. Sensitive and reliable evaluation of single-cut sgRNAs to restore dystrophin by a GFP-reporter assay. PLoS ONE 2020, 15, e0239468. [Google Scholar] [CrossRef] [PubMed]
- Pickar-Oliver, A.; Gough, V.; Bohning, J.D.; Liu, S.; Robinson-Hamm, J.N.; Daniels, H.; Majoros, W.H.; Devlin, G.; Asokan, A.; Gersbach, C.A. Full-length dystrophin restoration via targeted exon integration by AAV-CRISPR in a humanized mouse model of Duchenne muscular dystrophy. Mol. Ther. 2021, 29, 3243–3257. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Chadwick, B.P. CRISPR mediated targeting of DUX4 distal regulatory element represses DUX4 target genes dysregulated in Facioscapulohumeral muscular dystrophy. Sci. Rep. 2021, 11, 12598. [Google Scholar] [CrossRef] [PubMed]
- Himeda, C.L.; Jones, T.I.; Jones, P.L. Targeted epigenetic repression by CRISPR/dSaCas9 suppresses pathogenic DUX4-fl expression in FSHD. Mol. Ther. Methods Clin. Dev. 2021, 20, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ma, X.; Gao, F.; Guo, Y. Off-target effects in CRISPR/Cas9 gene editing. Front. Bioeng. Biotechnol. 2023, 11, 1143157. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef]
- Malinin, N.L.; Lee, G.; Lazzarotto, C.R.; Li, Y.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Iafrate, A.J.; Le, L.P.; et al. Defining genome-wide CRISPR-Cas genome-editing nuclease activity with GUIDE-seq. Nat. Protoc. 2021, 16, 5592–5615. [Google Scholar] [CrossRef]
- Bosnakovski, D.; Oyler, D.; Mitanoska, A.; Douglas, M.; Ener, E.T.; Shams, A.S.; Kyba, M. Persistent Fibroadipogenic Progenitor Expansion Following Transient DUX4 Expression Provokes a Profibrotic State in a Mouse Model for FSHD. Int. J. Mol. Sci. 2022, 23, 1983. [Google Scholar] [CrossRef] [PubMed]
- Ewaisha, R.; Anderson, K.S. Immunogenicity of CRISPR therapeutics-Critical considerations for clinical translation. Front. Bioeng. Biotechnol. 2023, 11, 1138596. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.; Vintersten-Nagy, K.; Nagy, A. Universal Stem Cells: Making the Unsafe Safe. Cell Stem Cell 2020, 27, 198–199. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Monetti, C.; Shutova, M.V.; Neely, E.J.; Hacibekiroglu, S.; Yang, H.; Kim, C.; Zhang, P.; Li, C.; Nagy, K.; et al. Linking a cell-division gene and a suicide gene to define and improve cell therapy safety. Nature 2018, 563, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Y.; Suo, Y.; Li, C.; Chen, M.; Zheng, S.; Li, H.; Tang, C.; Fan, N.; Lan, T.; et al. Inducible caspase-9 suicide gene under control of endogenous oct4 to safeguard mouse and human pluripotent stem cell therapy. Mol. Ther. Methods Clin. Dev. 2022, 24, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Van Deutekom, J.C.; Fokkema, I.F.; Van Ommen, G.J.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Beggs, A.H.; Koenig, M.; Boyce, F.M.; Kunkel, L.M. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum. Genet. 1990, 86, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Liechti-Gallati, S.; Koenig, M.; Kunkel, L.M.; Frey, D.; Boltshauser, E.; Schneider, V.; Braga, S.; Moser, H. Molecular deletion patterns in Duchenne and Becker type muscular dystrophy. Hum. Genet. 1989, 81, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Speciale, A.A.; Ellerington, R.; Goedert, T.; Rinaldi, C. Modelling Neuromuscular Diseases in the Age of Precision Medicine. J. Pers. Med. 2020, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Afshar, M.E.; Abraha, H.Y.; Bakooshli, M.A.; Davoudi, S.; Thavandiran, N.; Tung, K.; Ahn, H.; Ginsberg, H.J.; Zandstra, P.W.; Gilbert, P.M. A 96-well culture platform enables longitudinal analyses of engineered human skeletal muscle microtissue strength. Sci. Rep. 2020, 10, 6918. [Google Scholar] [CrossRef]
- Jacques, E.; Kuang, Y.; Kann, A.P.; Le Grand, F.; Krauss, R.S.; Gilbert, P.M. The mini-IDLE 3D biomimetic culture assay enables interrogation of mechanisms governing muscle stem cell quiescence and niche repopulation. Elife 2022, 11, e81738. [Google Scholar] [CrossRef] [PubMed]
- Gulati, N.; Davoudi, S.; Xu, B.; Rjaibi, S.T.; Jacques, E.; Pham, J.; Fard, A.; McGuigan, A.P.; Gilbert, P.M. mini-MEndR: A miniaturized 96-well predictive assay to evaluate muscle stem cell mediated repair. bioRxiv 2023. [Google Scholar] [CrossRef]
- Han, J.J. FDA Modernization Act 2.0 allows for alternatives to animal testing. Artif. Organs 2023, 47, 449–450. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.S.; Luttrell, S.M.; Dupont, J.B.; Gray, K.; Lih, D.; Fleming, J.W.; Cunningham, N.J.; Jepson, S.; Hesson, J.; Mathieu, J.; et al. High-throughput, real-time monitoring of engineered skeletal muscle function using magnetic sensing. J. Tissue Eng. 2022, 13, 20417314221122127. [Google Scholar] [CrossRef] [PubMed]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef]
- Ryder-Cook, A.S.; Sicinski, P.; Thomas, K.; Davies, K.E.; Worton, R.G.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. Localization of the mdx mutation within the mouse dystrophin gene. EMBO J. 1988, 7, 3017–3021. [Google Scholar] [CrossRef] [PubMed]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef] [PubMed]
- Stedman, H.H.; Sweeney, H.L.; Shrager, J.B.; Maguire, H.C.; Panettieri, R.A.; Petrof, B.; Narusawa, M.; Leferovich, J.M.; Sladky, J.T.; Kelly, A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 1991, 352, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Pastoret, C.; Sebille, A. mdx mice show progressive weakness and muscle deterioration with age. J. Neurol. Sci. 1995, 129, 97–105. [Google Scholar] [CrossRef]
- Lefaucheur, J.P.; Pastoret, C.; Sebille, A. Phenotype of dystrophinopathy in old mdx mice. Anat. Rec. 1995, 242, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.M.; Blake, D.J.; Roche, A.; Fairbrother, U.; Riss, J.; Byth, B.C.; Knight, A.E.; Kendrick-Jones, J.; Suthers, G.K.; Love, D.R.; et al. Primary structure of dystrophin-related protein. Nature 1992, 360, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.; Nalbantoglu, J.; Petrof, B.J.; Ebihara, S.; Guibinga, G.H.; Tinsley, J.M.; Kamen, A.; Massie, B.; Davies, K.E.; Karpati, G. Adenovirus-mediated utrophin gene transfer mitigates the dystrophic phenotype of mdx mouse muscles. Hum. Gene Ther. 1999, 10, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Grady, R.M.; Teng, H.; Nichol, M.C.; Cunningham, J.C.; Wilkinson, R.S.; Sanes, J.R. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: A model for Duchenne muscular dystrophy. Cell 1997, 90, 729–738. [Google Scholar] [CrossRef]
- Deconinck, A.E.; Rafael, J.A.; Skinner, J.A.; Brown, S.C.; Potter, A.C.; Metzinger, L.; Watt, D.J.; Dickson, J.G.; Tinsley, J.M.; Davies, K.E. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 1997, 90, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Chapman, V.M.; Miller, D.R.; Armstrong, D.; Caskey, C.T. Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc. Natl. Acad. Sci. USA 1989, 86, 1292–1296. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.A.; Phelps, S.F.; Chapman, V.M.; Chamberlain, J.S. New mdx mutation disrupts expression of muscle and nonmuscle isoforms of dystrophin. Nat. Genet. 1993, 4, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Araki, E.; Nakamura, K.; Nakao, K.; Kameya, S.; Kobayashi, O.; Nonaka, I.; Kobayashi, T.; Katsuki, M. Targeted disruption of exon 52 in the mouse dystrophin gene induced muscle degeneration similar to that observed in Duchenne muscular dystrophy. Biochem. Biophys. Res. Commun. 1997, 238, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Young, C.S.; Mokhonova, E.; Quinonez, M.; Pyle, A.D.; Spencer, M.J. Creation of a Novel Humanized Dystrophic Mouse Model of Duchenne Muscular Dystrophy and Application of a CRISPR/Cas9 Gene Editing Therapy. J. Neuromuscul. Dis. 2017, 4, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Deenen, J.C.; Arnts, H.; van der Maarel, S.M.; Padberg, G.W.; Verschuuren, J.J.; Bakker, E.; Weinreich, S.S.; Verbeek, A.L.; van Engelen, B.G. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology 2014, 83, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Statland, J.M.; Tawil, R. Facioscapulohumeral Muscular Dystrophy. Contin. Lifelong Learn. Neurol. 2016, 22, 1916–1931. [Google Scholar] [CrossRef] [PubMed]
- Tihaya, M.S.; Mul, K.; Balog, J.; de Greef, J.C.; Tapscott, S.J.; Tawil, R.; Statland, J.M.; van der Maarel, S.M. Facioscapulohumeral muscular dystrophy: The road to targeted therapies. Nat. Rev. Neurol. 2023, 19, 91–108. [Google Scholar] [CrossRef] [PubMed]
- Wijmenga, C.; Frants, R.R.; Brouwer, O.F.; Moerer, P.; Weber, J.L.; Padberg, G.W. Location of facioscapulohumeral muscular dystrophy gene on chromosome 4. Lancet 1990, 336, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Winokur, S.T.; Bengtsson, U.; Feddersen, J.; Mathews, K.D.; Weiffenbach, B.; Bailey, H.; Markovich, R.P.; Murray, J.C.; Wasmuth, J.J.; Altherr, M.R.; et al. The DNA rearrangement associated with facioscapulohumeral muscular dystrophy involves a heterochromatin-associated repetitive element: Implications for a role of chromatin structure in the pathogenesis of the disease. Chromosome Res. 1994, 2, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Gabriels, J.; Beckers, M.C.; Ding, H.; De Vriese, A.; Plaisance, S.; van der Maarel, S.M.; Padberg, G.W.; Frants, R.R.; Hewitt, J.E.; Collen, D.; et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 1999, 236, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Himeda, C.L.; Jones, P.L. The Genetics and Epigenetics of Facioscapulohumeral Muscular Dystrophy. Annu. Rev. Genomics Hum. Genet. 2019, 20, 265–291. [Google Scholar] [CrossRef]
- Lemmers, R.J.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.; Amell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012, 44, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
- van den Boogaard, M.L.; Lemmers, R.; Balog, J.; Wohlgemuth, M.; Auranen, M.; Mitsuhashi, S.; van der Vliet, P.J.; Straasheijm, K.R.; van den Akker, R.F.P.; Kriek, M.; et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am. J. Hum. Genet. 2016, 98, 1020–1029. [Google Scholar] [CrossRef]
- Leidenroth, A.; Clapp, J.; Mitchell, L.M.; Coneyworth, D.; Dearden, F.L.; Iannuzzi, L.; Hewitt, J.E. Evolution of DUX gene macrosatellites in placental mammals. Chromosoma 2012, 121, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Eidahl, J.O.; Giesige, C.R.; Domire, J.S.; Wallace, L.M.; Fowler, A.M.; Guckes, S.M.; Garwick-Coppens, S.E.; Labhart, P.; Harper, S.Q. Mouse Dux is myotoxic and shares partial functional homology with its human paralog DUX4. Hum. Mol. Genet. 2016, 25, 4577–4589. [Google Scholar] [CrossRef] [PubMed]
- Bosnakovski, D.; Chan, S.S.K.; Recht, O.O.; Hartweck, L.M.; Gustafson, C.J.; Athman, L.L.; Lowe, D.A.; Kyba, M. Muscle pathology from stochastic low level DUX4 expression in an FSHD mouse model. Nat. Commun. 2017, 8, 550. [Google Scholar] [CrossRef] [PubMed]
- Bosnakovski, D.; Shams, A.S.; Yuan, C.; da Silva, M.T.; Ener, E.T.; Baumann, C.W.; Lindsay, A.J.; Verma, M.; Asakura, A.; Lowe, D.A.; et al. Transcriptional and cytopathological hallmarks of FSHD in chronic DUX4-expressing mice. J. Clin. Investig. 2020, 130, 2465–2477. [Google Scholar] [CrossRef]
- Giesige, C.R.; Wallace, L.M.; Heller, K.N.; Eidahl, J.O.; Saad, N.Y.; Fowler, A.M.; Pyne, N.K.; Al-Kharsan, M.; Rashnonejad, A.; Chermahini, G.A.; et al. AAV-mediated follistatin gene therapy improves functional outcomes in the TIC-DUX4 mouse model of FSHD. JCI Insight 2018, 3, e123538. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.J.; Srikuea, R.; Kirby, T.J.; Peterson, C.A.; Esser, K.A. Inducible Cre transgenic mouse strain for skeletal muscle-specific gene targeting. Skelet. Muscle 2012, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.; Jones, P.L. A cre-inducible DUX4 transgenic mouse model for investigating facioscapulohumeral muscular dystrophy. PLoS ONE 2018, 13, e0192657. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.I.; Chew, G.L.; Barraza-Flores, P.; Schreier, S.; Ramirez, M.; Wuebbles, R.D.; Burkin, D.J.; Bradley, R.K.; Jones, P.L. Transgenic mice expressing tunable levels of DUX4 develop characteristic facioscapulohumeral muscular dystrophy-like pathophysiology ranging in severity. Skelet. Muscle 2020, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Brais, B. Oculopharyngeal muscular dystrophy: A polyalanine myopathy. Curr. Neurol. Neurosci. Rep. 2009, 9, 76–82. [Google Scholar] [CrossRef]
- Davies, J.E.; Wang, L.; Garcia-Oroz, L.; Cook, L.J.; Vacher, C.; O’Donovan, D.G.; Rubinsztein, D.C. Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice. Nat. Med. 2005, 11, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Trollet, C.; Anvar, S.Y.; Venema, A.; Hargreaves, I.P.; Foster, K.; Vignaud, A.; Ferry, A.; Negroni, E.; Hourde, C.; Baraibar, M.A.; et al. Molecular and phenotypic characterization of a mouse model of oculopharyngeal muscular dystrophy reveals severe muscular atrophy restricted to fast glycolytic fibres. Hum. Mol. Genet. 2010, 19, 2191–2207. [Google Scholar] [CrossRef]
- Vest, K.E.; Phillips, B.L.; Banerjee, A.; Apponi, L.H.; Dammer, E.B.; Xu, W.; Zheng, D.; Yu, J.; Tian, B.; Pavlath, G.K.; et al. Novel mouse models of oculopharyngeal muscular dystrophy (OPMD) reveal early onset mitochondrial defects and suggest loss of PABPN1 may contribute to pathology. Hum. Mol. Genet. 2017, 26, 3235–3252. [Google Scholar] [CrossRef] [PubMed]
- van Putten, M.; Lloyd, E.M.; de Greef, J.C.; Raz, V.; Willmann, R.; Grounds, M.D. Mouse models for muscular dystrophies: An overview. Dis. Model. Mech. 2020, 13, dmm043562. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.E.; Statland, J.M. The Limb-Girdle Muscular Dystrophies. Contin. Lifelong Learn. Neurol. 2022, 28, 1698–1714. [Google Scholar] [CrossRef] [PubMed]
- Weller, A.H.; Magliato, S.A.; Bell, K.P.; Rosenberg, N.L. Spontaneous myopathy in the SJL/J mouse: Pathology and strength loss. Muscle Nerve 1997, 20, 72–82. [Google Scholar] [CrossRef]
- Bittner, R.E.; Anderson, L.V.; Burkhardt, E.; Bashir, R.; Vafiadaki, E.; Ivanova, S.; Raffelsberger, T.; Maerk, I.; Hoger, H.; Jung, M.; et al. Dysferlin deletion in SJL mice (SJL-Dysf) defines a natural model for limb girdle muscular dystrophy 2B. Nat. Genet. 1999, 23, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Richard, I.; Roudaut, C.; Marchand, S.; Baghdiguian, S.; Herasse, M.; Stockholm, D.; Ono, Y.; Suel, L.; Bourg, N.; Sorimachi, H.; et al. Loss of calpain 3 proteolytic activity leads to muscular dystrophy and to apoptosis-associated IkappaBalpha/nuclear factor kappaB pathway perturbation in mice. J. Cell Biol. 2000, 151, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, F.; Engelman, J.A.; Volonte, D.; Zhang, X.L.; Minetti, C.; Li, M.; Hou, H., Jr.; Kneitz, B.; Edelmann, W.; Lisanti, M.P. Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J. Biol. Chem. 2001, 276, 21425–21433. [Google Scholar] [CrossRef] [PubMed]
- Hack, A.A.; Ly, C.T.; Jiang, F.; Clendenin, C.J.; Sigrist, K.S.; Wollmann, R.L.; McNally, E.M. Gamma-sarcoglycan deficiency leads to muscle membrane defects and apoptosis independent of dystrophin. J. Cell Biol. 1998, 142, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Duclos, F.; Straub, V.; Moore, S.A.; Venzke, D.P.; Hrstka, R.F.; Crosbie, R.H.; Durbeej, M.; Lebakken, C.S.; Ettinger, A.J.; van der Meulen, J.; et al. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J. Cell Biol. 1998, 142, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Coral-Vazquez, R.; Cohn, R.D.; Moore, S.A.; Hill, J.A.; Weiss, R.M.; Davisson, R.L.; Straub, V.; Barresi, R.; Bansal, D.; Hrstka, R.F.; et al. Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: A novel mechanism for cardiomyopathy and muscular dystrophy. Cell 1999, 98, 465–474. [Google Scholar] [CrossRef]
- Hack, A.A.; Lam, M.Y.; Cordier, L.; Shoturma, D.I.; Ly, C.T.; Hadhazy, M.A.; Hadhazy, M.R.; Sweeney, H.L.; McNally, E.M. Differential requirement for individual sarcoglycans and dystrophin in the assembly and function of the dystrophin-glycoprotein complex. J. Cell Sci. 2000, 113 Pt 14, 2535–2544. [Google Scholar] [CrossRef] [PubMed]
- Durbeej, M.; Cohn, R.D.; Hrstka, R.F.; Moore, S.A.; Allamand, V.; Davidson, B.L.; Williamson, R.A.; Campbell, K.P. Disruption of the beta-sarcoglycan gene reveals pathogenetic complexity of limb-girdle muscular dystrophy type 2E. Mol. Cell 2000, 5, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Sasaoka, T.; Imamura, M.; Araishi, K.; Noguchi, S.; Mizuno, Y.; Takagoshi, N.; Hama, H.; Wakabayashi-Takai, E.; Yoshimoto-Matsuda, Y.; Nonaka, I.; et al. Pathological analysis of muscle hypertrophy and degeneration in muscular dystrophy in gamma-sarcoglycan-deficient mice. Neuromuscul. Disord. 2003, 13, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Udd, B.; Krahe, R. The myotonic dystrophies: Molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012, 11, 891–905. [Google Scholar] [CrossRef] [PubMed]
- Braz, S.O.; Acquaire, J.; Gourdon, G.; Gomes-Pereira, M. Of Mice and Men: Advances in the Understanding of Neuromuscular Aspects of Myotonic Dystrophy. Front. Neurol. 2018, 9, 519. [Google Scholar] [CrossRef] [PubMed]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Taneja, K.L.; McCurrach, M.; Schalling, M.; Housman, D.; Singer, R.H. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol. 1995, 128, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Cleary, J.D.; Liu, Y.; Banez-Coronel, M.; Bubenik, J.L.; Ayhan, F.; Ashizawa, T.; Xia, G.; Clark, H.B.; Yachnis, A.T.; et al. RAN Translation Regulated by Muscleblind Proteins in Myotonic Dystrophy Type 2. Neuron 2017, 95, 1292–1305. [Google Scholar] [CrossRef] [PubMed]
- van den Broek, W.J.; Wansink, D.G.; Wieringa, B. Somatic CTG*CAG repeat instability in a mouse model for myotonic dystrophy type 1 is associated with changes in cell nuclearity and DNA ploidy. BMC Mol. Biol. 2007, 8, 61. [Google Scholar] [CrossRef]
- van den Broek, W.J.; Nelen, M.R.; Wansink, D.G.; Coerwinkel, M.M.; te Riele, H.; Groenen, P.J.; Wieringa, B. Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum. Mol. Genet. 2002, 11, 191–198. [Google Scholar] [CrossRef]
- Seznec, H.; Lia-Baldini, A.S.; Duros, C.; Fouquet, C.; Lacroix, C.; Hofmann-Radvanyi, H.; Junien, C.; Gourdon, G. Transgenic mice carrying large human genomic sequences with expanded CTG repeat mimic closely the DM CTG repeat intergenerational and somatic instability. Hum. Mol. Genet. 2000, 9, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Pereira, M.; Foiry, L.; Nicole, A.; Huguet, A.; Junien, C.; Munnich, A.; Gourdon, G. CTG trinucleotide repeat “big jumps”: Large expansions, small mice. PLoS Genet. 2007, 3, e52. [Google Scholar] [CrossRef] [PubMed]
- Huguet, A.; Medja, F.; Nicole, A.; Vignaud, A.; Guiraud-Dogan, C.; Ferry, A.; Decostre, V.; Hogrel, J.Y.; Metzger, F.; Hoeflich, A.; et al. Molecular, physiological, and motor performance defects in DMSXL mice carrying >1,000 CTG repeats from the human DM1 locus. PLoS Genet. 2012, 8, e1003043. [Google Scholar] [CrossRef] [PubMed]
- Kanadia, R.N.; Johnstone, K.A.; Mankodi, A.; Lungu, C.; Thornton, C.A.; Esson, D.; Timmers, A.M.; Hauswirth, W.W.; Swanson, M.S. A muscleblind knockout model for myotonic dystrophy. Science 2003, 302, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Li, M.; Manchanda, M.; Batra, R.; Charizanis, K.; Mohan, A.; Warren, S.A.; Chamberlain, C.M.; Finn, D.; Hong, H.; et al. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med. 2013, 5, 1887–1900. [Google Scholar] [CrossRef] [PubMed]
- Koshelev, M.; Sarma, S.; Price, R.E.; Wehrens, X.H.; Cooper, T.A. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum. Mol. Genet. 2010, 19, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; van Eeden, F.J.; Schach, U.; Trowe, T.; Brand, M.; Furutani-Seiki, M.; Haffter, P.; Hammerschmidt, M.; Heisenberg, C.P.; Jiang, Y.J.; et al. Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development 1996, 123, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Larcher, T.; Lafoux, A.; Tesson, L.; Remy, S.; Thepenier, V.; Francois, V.; Le Guiner, C.; Goubin, H.; Dutilleul, M.; Guigand, L.; et al. Characterization of dystrophin deficient rats: A new model for Duchenne muscular dystrophy. PLoS ONE 2014, 9, e110371. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Fujii, W.; Tsuboi, M.; Tanihata, J.; Teramoto, N.; Takeuchi, S.; Naito, K.; Yamanouchi, K.; Nishihara, M. Generation of muscular dystrophy model rats with a CRISPR/Cas system. Sci. Rep. 2014, 4, 5635. [Google Scholar] [CrossRef] [PubMed]
- Kornegay, J.N. The golden retriever model of Duchenne muscular dystrophy. Skelet. Muscle 2017, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Mata Lopez, S.; Hammond, J.J.; Rigsby, M.B.; Balog-Alvarez, C.J.; Kornegay, J.N.; Nghiem, P.P. A novel canine model for Duchenne muscular dystrophy (DMD): Single nucleotide deletion in DMD gene exon 20. Skelet. Muscle 2018, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Shimatsu, Y.; Katagiri, K.; Furuta, T.; Nakura, M.; Tanioka, Y.; Yuasa, K.; Tomohiro, M.; Kornegay, J.N.; Nonaka, I.; Takeda, S. Canine X-linked muscular dystrophy in Japan (CXMDJ). Exp. Anim. 2003, 52, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Stirm, M.; Fonteyne, L.M.; Shashikadze, B.; Lindner, M.; Chirivi, M.; Lange, A.; Kaufhold, C.; Mayer, C.; Medugorac, I.; Kessler, B.; et al. A scalable, clinically severe pig model for Duchenne muscular dystrophy. Dis. Model. Mech. 2021, 14, dmm049285. [Google Scholar] [CrossRef] [PubMed]
- Nip, Y.; Bennett, S.R.; Smith, A.A.; Jones, T.I.; Jones, P.L.; Tapscott, S.J. Human DUX4 and porcine DUXC activate similar early embryonic programs in pig muscle cells: Implications for preclinical models of FSHD. Hum. Mol. Genet. 2023, 32, 1864–1874. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zheng, Y.; Kang, Y.; Yang, W.; Niu, Y.; Guo, X.; Tu, Z.; Si, C.; Wang, H.; Xing, R.; et al. Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum. Mol. Genet. 2015, 24, 3764–3774. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.P. ‘Nude’, a new hairless gene with pleiotropic effects in the mouse. Genet. Res. 1966, 8, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Bosma, G.C.; Custer, R.P.; Bosma, M.J. A severe combined immunodeficiency mutation in the mouse. Nature 1983, 301, 527–530. [Google Scholar] [CrossRef]
- Shultz, L.D.; Schweitzer, P.A.; Christianson, S.W.; Gott, B.; Schweitzer, I.B.; Tennent, B.; McKenna, S.; Mobraaten, L.; Rajan, T.V.; Greiner, D.L.; et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J. Immunol. 1995, 154, 180–191. [Google Scholar] [CrossRef]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Shores, E.W.; Hu-Li, J.; Anver, M.R.; Kelsall, B.L.; Russell, S.M.; Drago, J.; Noguchi, M.; Grinberg, A.; Bloom, E.T.; et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor gamma chain. Immunity 1995, 2, 223–238. [Google Scholar] [CrossRef]
- Wunderlich, M.; Mizukawa, B.; Chou, F.S.; Sexton, C.; Shrestha, M.; Saunthararajah, Y.; Mulloy, J.C. AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood 2013, 121, e90–e97. [Google Scholar] [CrossRef] [PubMed]
- Kollet, O.; Peled, A.; Byk, T.; Ben-Hur, H.; Greiner, D.; Shultz, L.; Lapidot, T. beta2 microglobulin-deficient (B2m(null)) NOD/SCID mice are excellent recipients for studying human stem cell function. Blood 2000, 95, 3102–3105. [Google Scholar] [CrossRef]
- Kamel-Reid, S.; Dick, J.E. Engraftment of immune-deficient mice with human hematopoietic stem cells. Science 1988, 242, 1706–1709. [Google Scholar] [CrossRef] [PubMed]
- Noto, F.K.; Adjan-Steffey, V.; Tong, M.; Ravichandran, K.; Zhang, W.; Arey, A.; McClain, C.B.; Ostertag, E.; Mazhar, S.; Sangodkar, J.; et al. Sprague Dawley Rag2-Null Rats Created from Engineered Spermatogonial Stem Cells Are Immunodeficient and Permissive to Human Xenografts. Mol. Cancer Ther. 2018, 17, 2481–2489. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Zhang, J.; Wu, W.; Yi, N.; He, W.; Lu, P.; Li, B.; Yang, N.; Wang, D.; Xue, Z.; et al. A novel immunodeficient rat model supports human lung cancer xenografts. FASEB J. 2019, 33, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Menoret, S.; Ouisse, L.H.; Tesson, L.; Delbos, F.; Garnier, D.; Remy, S.; Usal, C.; Concordet, J.P.; Giovannangeli, C.; Chenouard, V.; et al. Generation of Immunodeficient Rats With Rag1 and Il2rg Gene Deletions and Human Tissue Grafting Models. Transplantation 2018, 102, 1271–1278. [Google Scholar] [CrossRef]
- Arpke, R.W.; Darabi, R.; Mader, T.L.; Zhang, Y.; Toyama, A.; Lonetree, C.L.; Nash, N.; Lowe, D.A.; Perlingeiro, R.C.; Kyba, M. A new immuno-, dystrophin-deficient model, the NSG-mdx(4Cv) mouse, provides evidence for functional improvement following allogeneic satellite cell transplantation. Stem Cells 2013, 31, 1611–1620. [Google Scholar] [CrossRef]
- Chan, S.S.; Arpke, R.W.; Filareto, A.; Xie, N.; Pappas, M.P.; Penaloza, J.S.; Perlingeiro, R.C.R.; Kyba, M. Skeletal Muscle Stem Cells from PSC-Derived Teratomas Have Functional Regenerative Capacity. Cell Stem Cell 2018, 23, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Kannan, S.; Choi, I.Y.; Lim, H.; Zhang, H.; Chen, G.S.; Zhang, N.; Park, S.H.; Serra, C.; Iyer, S.R.; et al. Human pluripotent stem cell-derived myogenic progenitors undergo maturation to quiescent satellite cells upon engraftment. Cell Stem Cell 2022, 29, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Dhoke, N.R.; Kiley, J.; Mateos-Aierdi, A.J.; Tungtur, S.; Mondragon-Gonzalez, R.; Killeen, G.; Oliveira, V.K.P.; Lopez de Munain, A.; Perlingeiro, R.C.R. Gene Correction of LGMD2A Patient-Specific iPSCs for the Development of Targeted Autologous Cell Therapy. Mol. Ther. 2019, 27, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Diehl, R.; Ferrara, F.; Muller, C.; Dreyer, A.Y.; McLeod, D.D.; Fricke, S.; Boltze, J. Immunosuppression for in vivo research: State-of-the-art protocols and experimental approaches. Cell Mol. Immunol. 2017, 14, 146–179. [Google Scholar] [CrossRef] [PubMed]
- Sampaolesi, M.; Blot, S.; D’Antona, G.; Granger, N.; Tonlorenzi, R.; Innocenzi, A.; Mognol, P.; Thibaud, J.L.; Galvez, B.G.; Barthelemy, I.; et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 2006, 444, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Galvez, B.G.; Sampaolesi, M.; Brunelli, S.; Covarello, D.; Gavina, M.; Rossi, B.; Constantin, G.; Torrente, Y.; Cossu, G. Complete repair of dystrophic skeletal muscle by mesoangioblasts with enhanced migration ability. J. Cell Biol. 2006, 174, 231–243. [Google Scholar] [CrossRef]
- Cossu, G.; Previtali, S.C.; Napolitano, S.; Cicalese, M.P.; Tedesco, F.S.; Nicastro, F.; Noviello, M.; Roostalu, U.; Natali Sora, M.G.; Scarlato, M.; et al. Intra-arterial transplantation of HLA-matched donor mesoangioblasts in Duchenne muscular dystrophy. EMBO Mol. Med. 2016, 8, 1470–1471. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Ferrari, G.; Moyle, L.A.; Mackinlay, K.; Naouar, N.; Jalal, S.; Benedetti, S.; Wells, C.; Muntoni, F.; Tedesco, F.S. Assessing and enhancing migration of human myogenic progenitors using directed iPS cell differentiation and advanced tissue modelling. EMBO Mol. Med. 2022, 14, e14526. [Google Scholar] [CrossRef] [PubMed]
- Gerli, M.F.M.; Moyle, L.A.; Benedetti, S.; Ferrari, G.; Ucuncu, E.; Ragazzi, M.; Constantinou, C.; Louca, I.; Sakai, H.; Ala, P.; et al. Combined Notch and PDGF Signaling Enhances Migration and Expression of Stem Cell Markers while Inducing Perivascular Cell Features in Muscle Satellite Cells. Stem Cell Rep. 2019, 12, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Guidance for industry CGMP for Phase 1 Investigational Drugs; U.S. Department of Health and Human Services Food and Drug Administration: Silver Spring, MD, USA, 2008.
- Baghbaderani, B.A.; Tian, X.; Neo, B.H.; Burkall, A.; Dimezzo, T.; Sierra, G.; Zeng, X.; Warren, K.; Kovarcik, D.P.; Fellner, T.; et al. cGMP-Manufactured Human Induced Pluripotent Stem Cells Are Available for Pre-clinical and Clinical Applications. Stem Cell Rep. 2015, 5, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Eaker, S.; Abraham, E.; Allickson, J.; Brieva, T.A.; Baksh, D.; Heathman, T.R.J.; Mistry, B.; Zhang, N. Bioreactors for cell therapies: Current status and future advances. Cytotherapy 2017, 19, 9–18. [Google Scholar] [CrossRef]
- Hanley, P.J.; Mei, Z.; Durett, A.G.; Cabreira-Hansen Mda, G.; Klis, M.; Li, W.; Zhao, Y.; Yang, B.; Parsha, K.; Mir, O.; et al. Efficient manufacturing of therapeutic mesenchymal stromal cells with the use of the Quantum Cell Expansion System. Cytotherapy 2014, 16, 1048–1058. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.J.R.; Luquet, E.; Pletenka, J.; Leonard, A.; Warter, E.; Gurchenkov, B.; Carrere, J.; Rieu, C.; Hardouin, J.; Moncaubeig, F.; et al. Engineering 3D micro-compartments for highly efficient and scale-independent expansion of human pluripotent stem cells in bioreactors. Biomaterials 2023, 295, 122033. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.C.; Couture, S.M.; Ye, J.; Lin, Z.; Hua, G.; Huang, H.I.; Wu, J.; Hsu, D.; Carpenter, M.K.; Couture, L.A. Scalable GMP compliant suspension culture system for human ES cells. Stem Cell Res. 2012, 8, 388–402. [Google Scholar] [CrossRef]
- Lei, Y.; Schaffer, D.V. A fully defined and scalable 3D culture system for human pluripotent stem cell expansion and differentiation. Proc. Natl. Acad. Sci. USA 2013, 110, E5039–E5048. [Google Scholar] [CrossRef] [PubMed]
- Serra, M.; Correia, C.; Malpique, R.; Brito, C.; Jensen, J.; Bjorquist, P.; Carrondo, M.J.; Alves, P.M. Microencapsulation technology: A powerful tool for integrating expansion and cryopreservation of human embryonic stem cells. PLoS ONE 2011, 6, e23212. [Google Scholar] [CrossRef] [PubMed]
- Aguanno, S.; Petrelli, C.; Di Siena, S.; De Angelis, L.; Pellegrini, M.; Naro, F. A Three-Dimensional Culture Model of Reversibly Quiescent Myogenic Cells. Stem Cells Int. 2019, 2019, 7548160. [Google Scholar] [CrossRef] [PubMed]
- Simpson, A.; Hewitt, A.W.; Fairfax, K.A. Universal cell donor lines: A review of the current research. Stem Cell Rep. 2023, 18, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Lanza, R.; Russell, D.W.; Nagy, A. Engineering universal cells that evade immune detection. Nat. Rev. Immunol. 2019, 19, 723–733. [Google Scholar] [CrossRef]
- Bashor, C.J.; Hilton, I.B.; Bandukwala, H.; Smith, D.M.; Veiseh, O. Engineering the next generation of cell-based therapeutics. Nat. Rev. Drug Discov. 2022, 21, 655–675. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lu, Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, J.; Feghhi, M.; Etemadi, T. A review on exosomes application in clinical trials: Perspective, questions, and challenges. Cell Commun. Signal 2022, 20, 145. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Guo, S.; Ren, X.; Wu, Z.; Liu, S.; Yao, X. Current Strategies for Exosome Cargo Loading and Targeting Delivery. Cells 2023, 12, 1416. [Google Scholar] [CrossRef]
- Leng, L.; Dong, X.; Gao, X.; Ran, N.; Geng, M.; Zuo, B.; Wu, Y.; Li, W.; Yan, H.; Han, G.; et al. Exosome-mediated improvement in membrane integrity and muscle function in dystrophic mice. Mol. Ther. 2021, 29, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Yedigaryan, L.; Sampaolesi, M. Extracellular vesicles and Duchenne muscular dystrophy pathology: Modulators of disease progression. Front. Physiol. 2023, 14, 1130063. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; O’Rourke, J.R.; Sutherland, L.B.; Bezprozvannaya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature 2013, 499, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.E.; Goh, Q.; Kurosaka, M.; Gamage, D.G.; Petrany, M.J.; Prasad, V.; Millay, D.P. Myomerger induces fusion of non-fusogenic cells and is required for skeletal muscle development. Nat. Commun. 2017, 8, 15665. [Google Scholar] [CrossRef] [PubMed]
- Hindi, S.M.; Petrany, M.J.; Greenfeld, E.; Focke, L.C.; Cramer, A.A.W.; Whitt, M.A.; Khairallah, R.J.; Ward, C.W.; Chamberlain, J.S.; Prasad, V.; et al. Enveloped viruses pseudotyped with mammalian myogenic cell fusogens target skeletal muscle for gene delivery. Cell 2023, 186, 3520. [Google Scholar] [CrossRef]
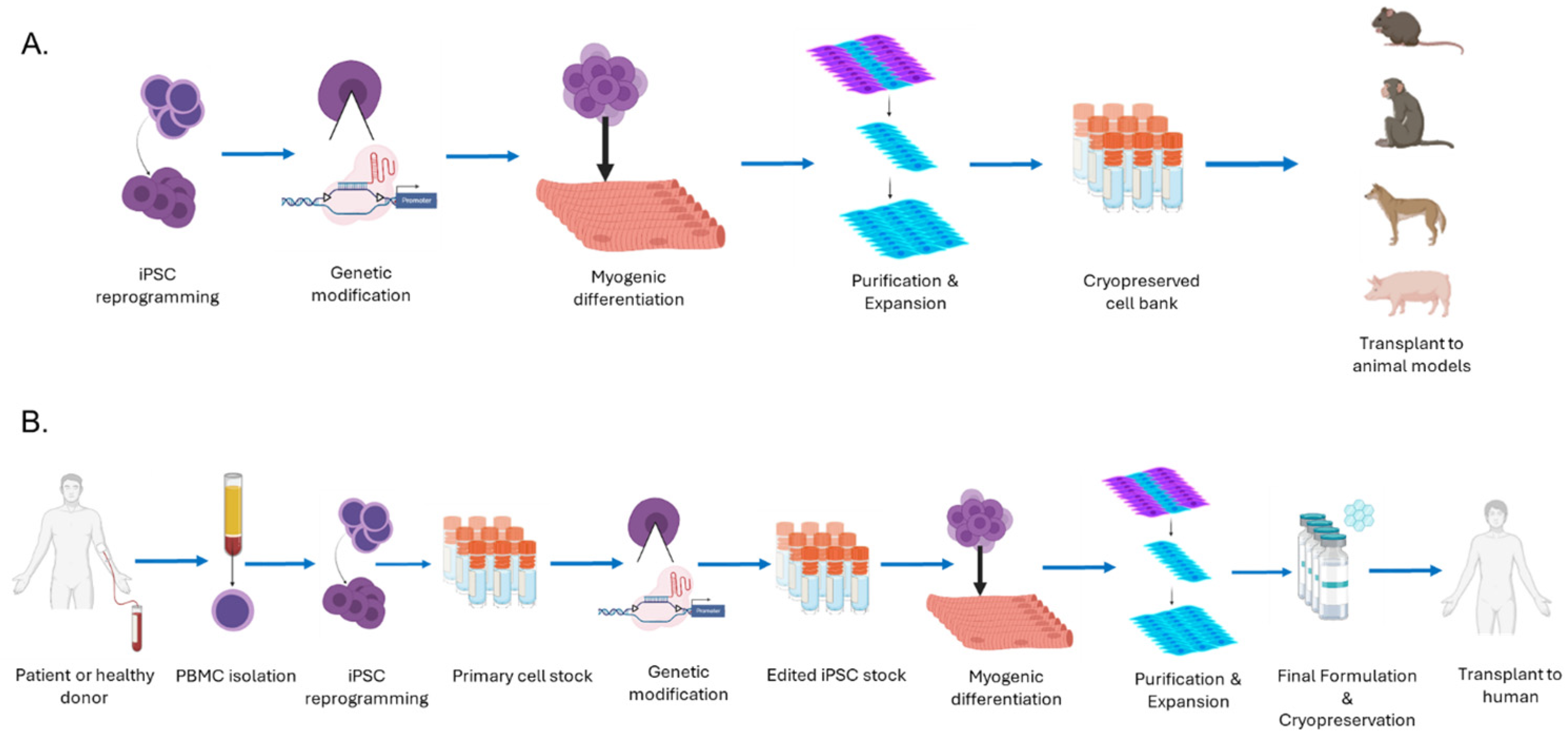

{kind=link}
| Study | Myogenic Progenitor Types | Post-Differentiation Day | Selection Marker | Purity after Selection |
|---|---|---|---|---|
| Magli et al., 2017 [38] | PAX7+ myogenic progenitor cells | Day 23 | CD54high+ | 90% PAX7+ cells |
| Hicks et al., 2018 [27] | Myogenic cells that can fuse the most | Day 50 | ERBB3+/NGFR+ | High PAX7 and MYF5 expression by QPCR |
| Wu et al., 2018 [39] | MYF5+ myogenic progenitor cells | Day 15 | CD10+/CD24- | Mainly MYF5+ cells |
| Choi et al., 2020 [40] | PAX7+ myogenic progenitor cells | Day 30 | CD271high+ | 85 ± 3.24% PAX7+ |
| Nalbandian et al., 2021 [28] | PAX7+ myogenic progenitor cells | Day 42 | FGFR4+ | 98.5% PAX7+ cells |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, C.; Serra, C.; Kalicharan, B.H.; Harding, J.; Rao, M. Challenges and Considerations of Preclinical Development for iPSC-Based Myogenic Cell Therapy. Cells 2024, 13, 596. https://doi.org/10.3390/cells13070596
Sun C, Serra C, Kalicharan BH, Harding J, Rao M. Challenges and Considerations of Preclinical Development for iPSC-Based Myogenic Cell Therapy. Cells. 2024; 13(7):596. https://doi.org/10.3390/cells13070596
Chicago/Turabian StyleSun, Congshan, Carlo Serra, Brianna Harley Kalicharan, Jeffrey Harding, and Mahendra Rao. 2024. "Challenges and Considerations of Preclinical Development for iPSC-Based Myogenic Cell Therapy" Cells 13, no. 7: 596. https://doi.org/10.3390/cells13070596
APA StyleSun, C., Serra, C., Kalicharan, B. H., Harding, J., & Rao, M. (2024). Challenges and Considerations of Preclinical Development for iPSC-Based Myogenic Cell Therapy. Cells, 13(7), 596. https://doi.org/10.3390/cells13070596