
Campylobacter jejuni Surface-Bound Protease HtrA, but Not the Secreted Protease nor Protease in Shed Membrane Vesicles, Disrupts Epithelial Cell-to-Cell Junctions

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Cultivation
2.2. Purification of Recombinant C. jejuni HtrA (rHtrA)
2.3. Preparation of Outer Membrane Vesicles (OMVs)
2.4. Cell Culture and Infection Assays
2.5. Transwell System and Transepithelial Electrical Resistance (TER) Measurement
2.6. Field Emission Scanning Electron Microscopy (FESEM)
2.7. Transmission Electron Microscopy for Negatively Stained Samples
2.8. Immunofluorescence Microscopy
2.9. SDS-PAGE and Western Blotting
2.10. Casein Zymography
2.11. Statistical Analyses
3. Results
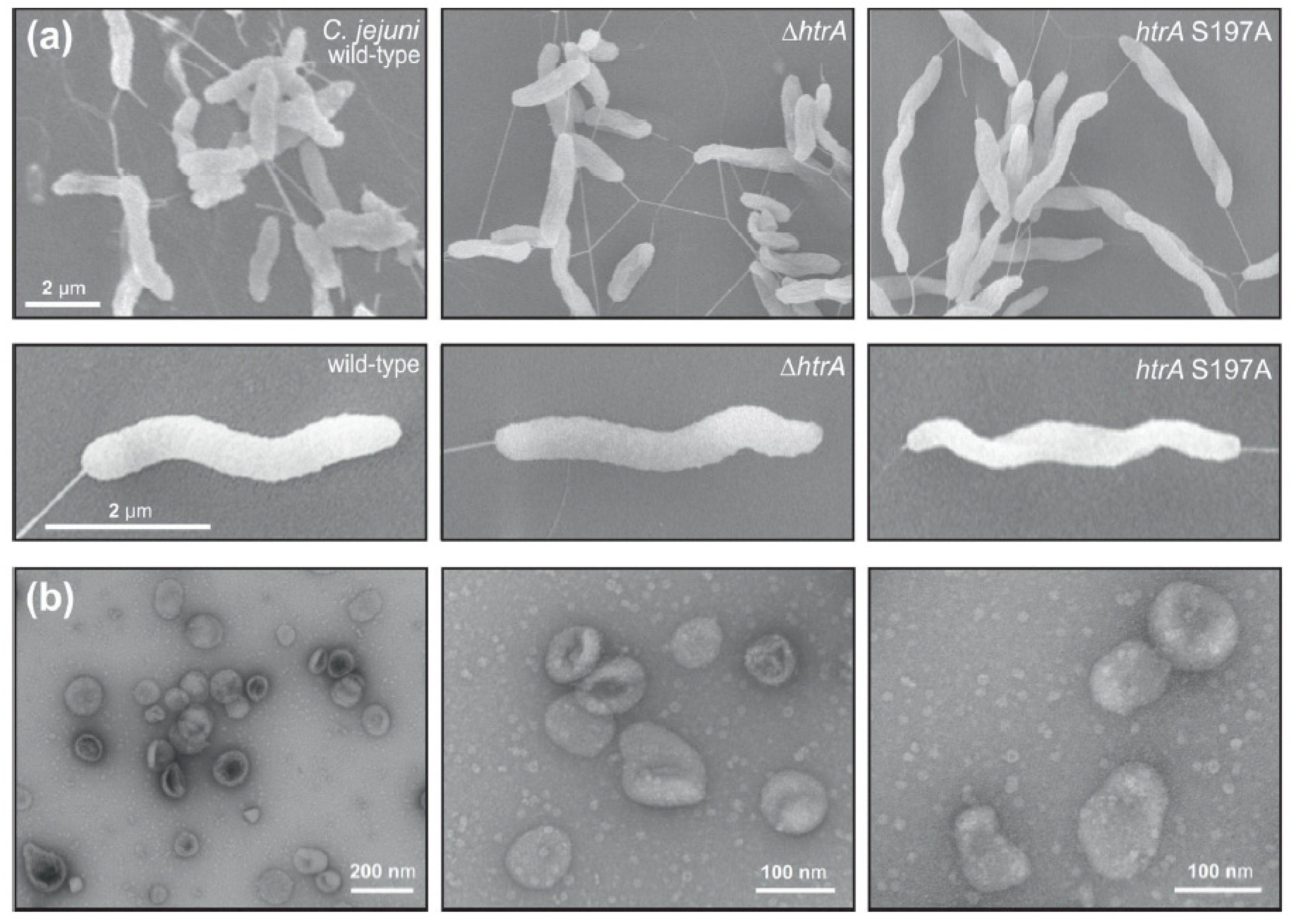
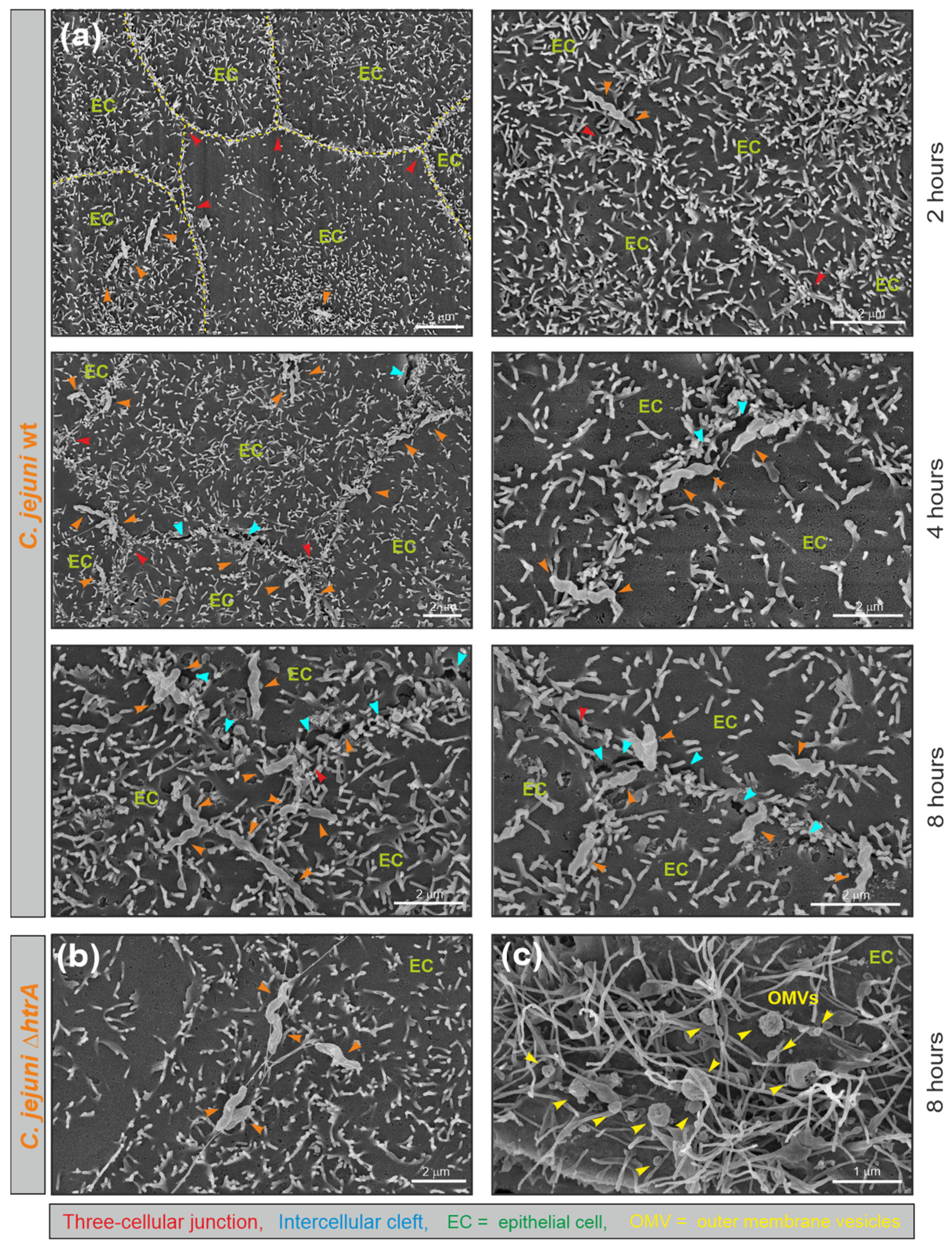
3.1. Electron Microscopic Analysis of C. jejuni and OMV Production
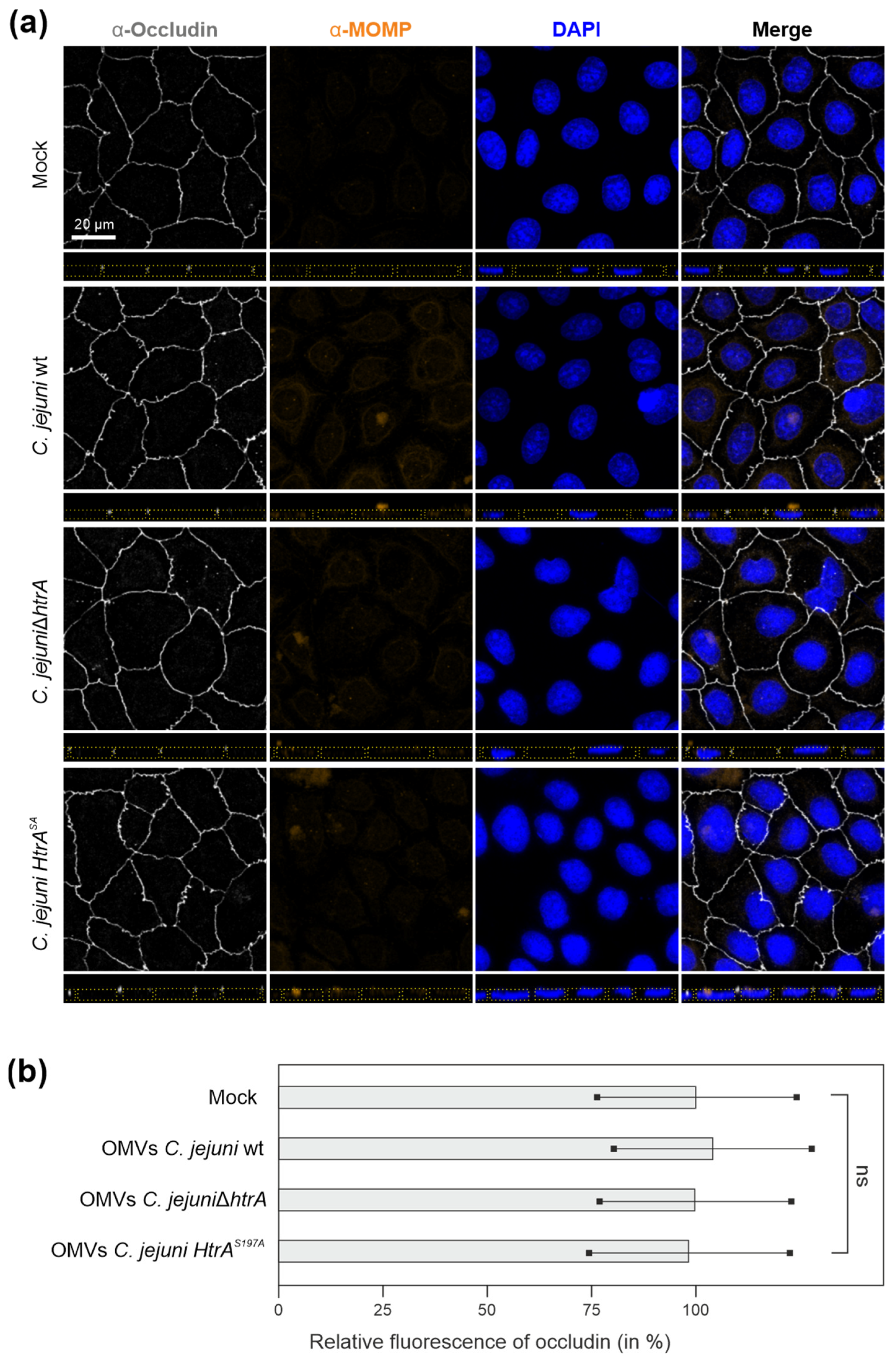
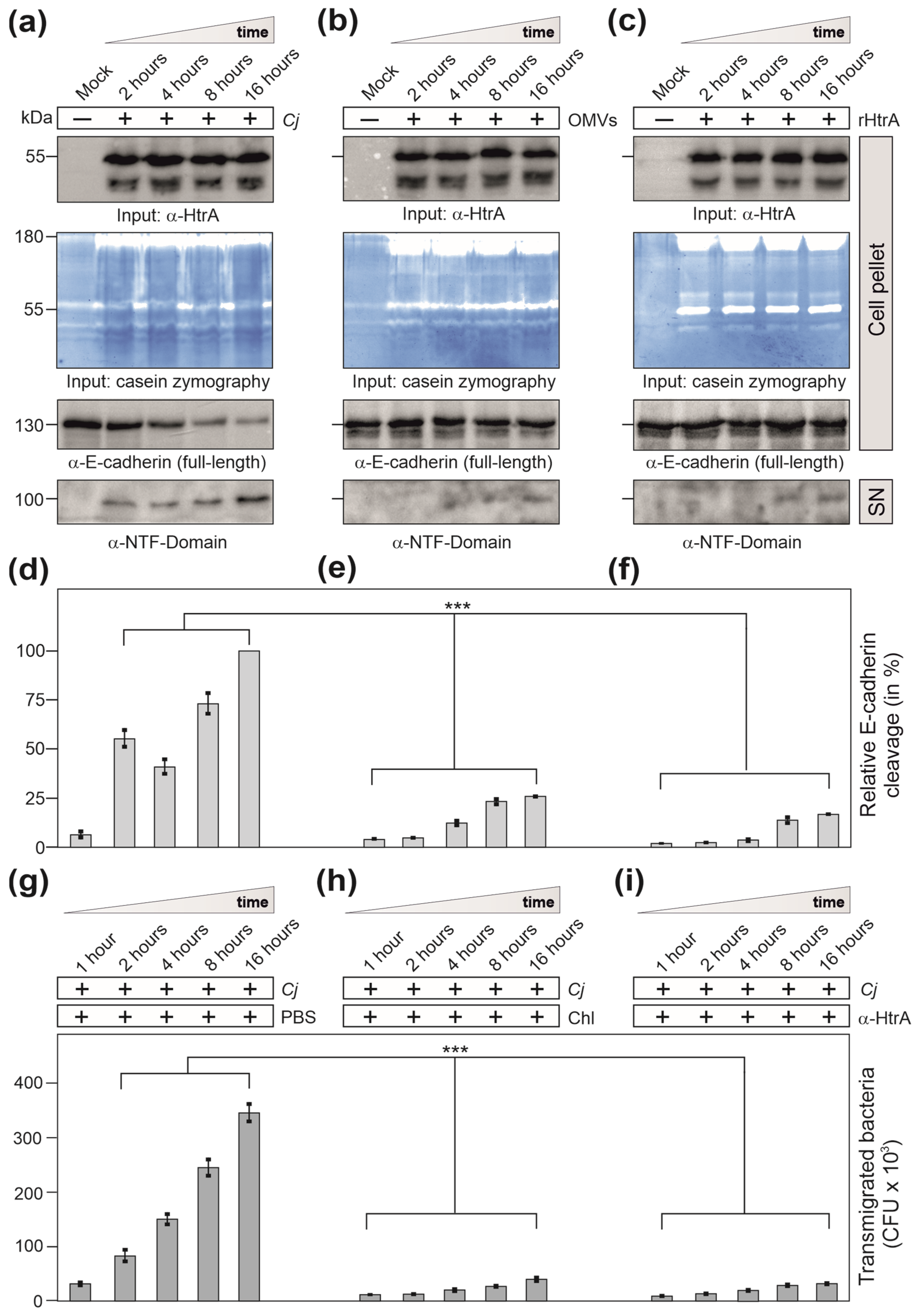
3.2. Are OMVs the Primary Route for HtrA Delivery in C. jejuni–Host Interactions?
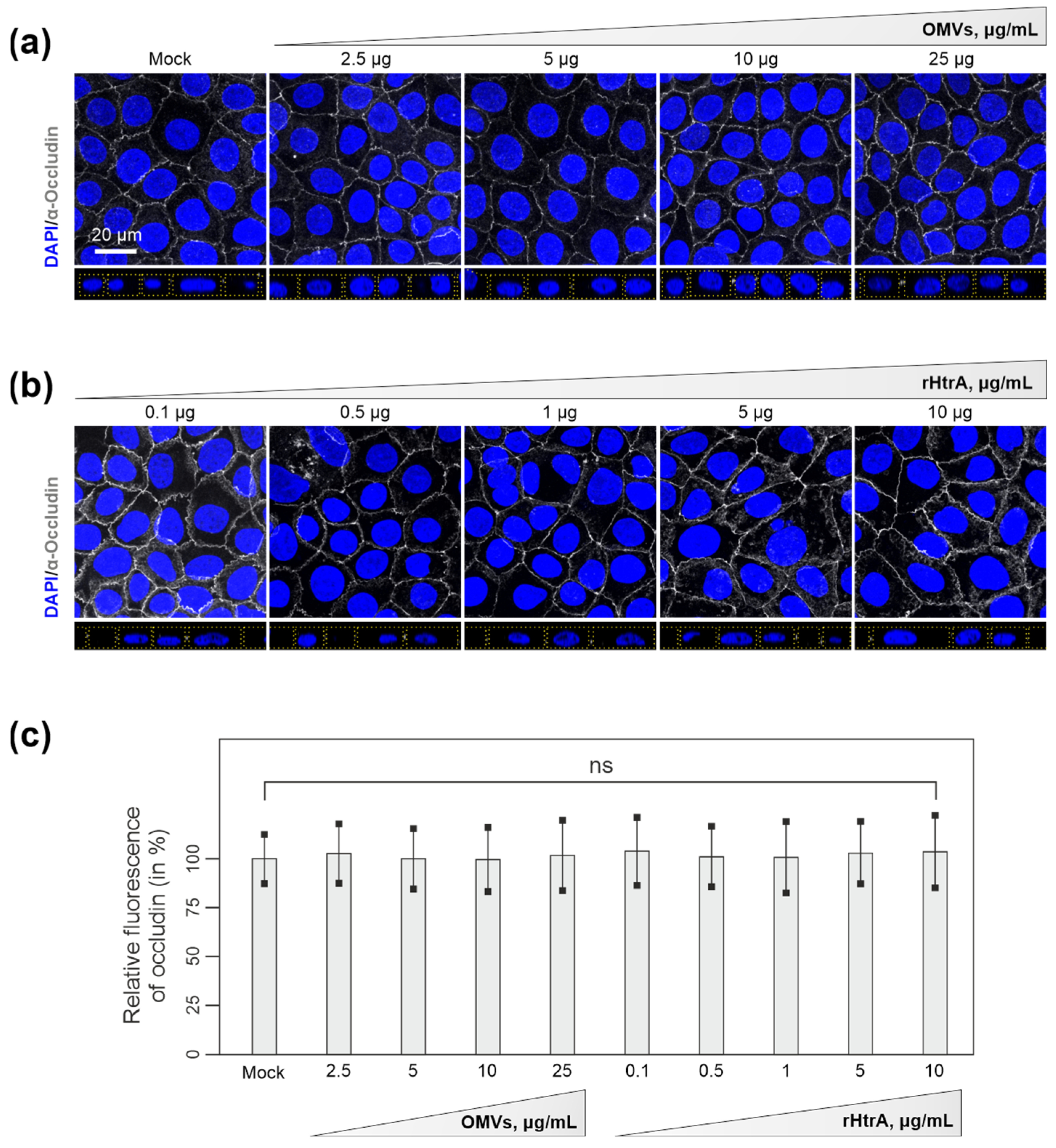
3.3. Efficiency of C. jejuni OMVs and Soluble rHtrA in Cleavage of Tight Junctions
3.4. C. jejuni Wt Infection of Epithelial Cells Results in Very Efficient Junctional Disruption
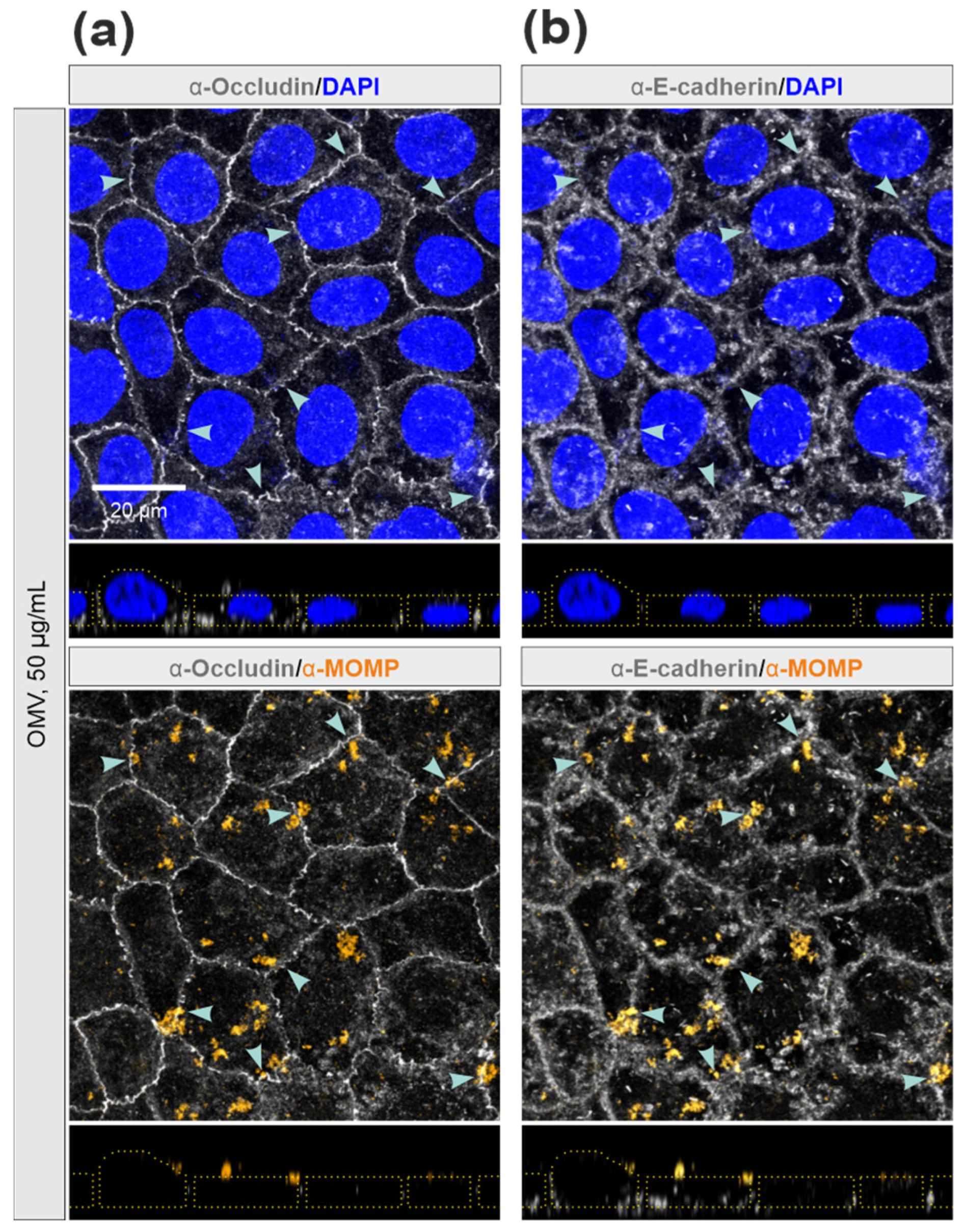
3.5. E-Cadherin was Disrupted by C. jejuni Wt Infection, but Not by Treatment with OMVs or rHtrA
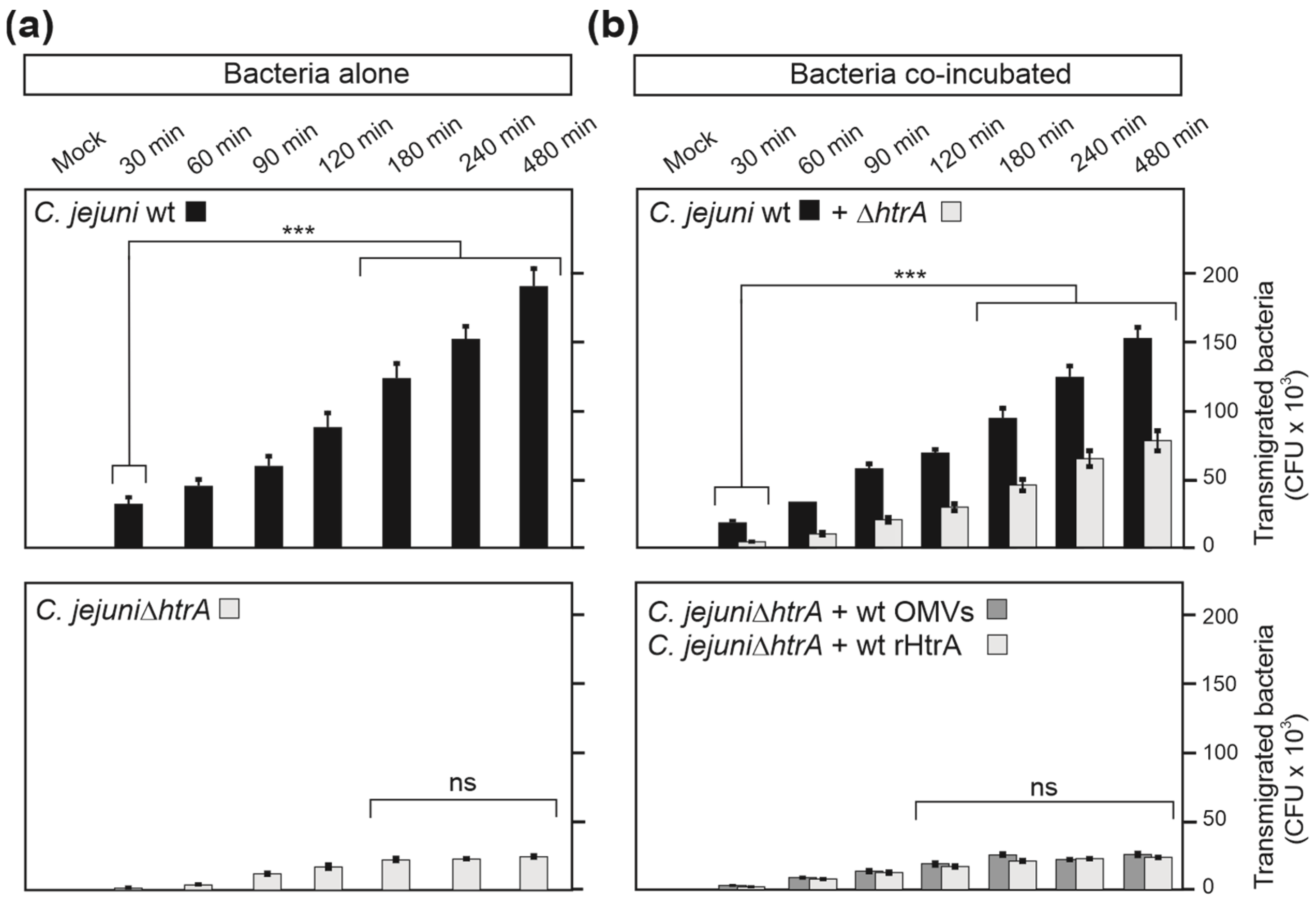
3.6. Inhibition of Protein Biosynthesis or Pre-Treatment with Anti-HtrA Antibodies Block C. jejuni Transmigration
3.7. HtrA-Expressing C. jejuni Promote Bacterial Transmigration
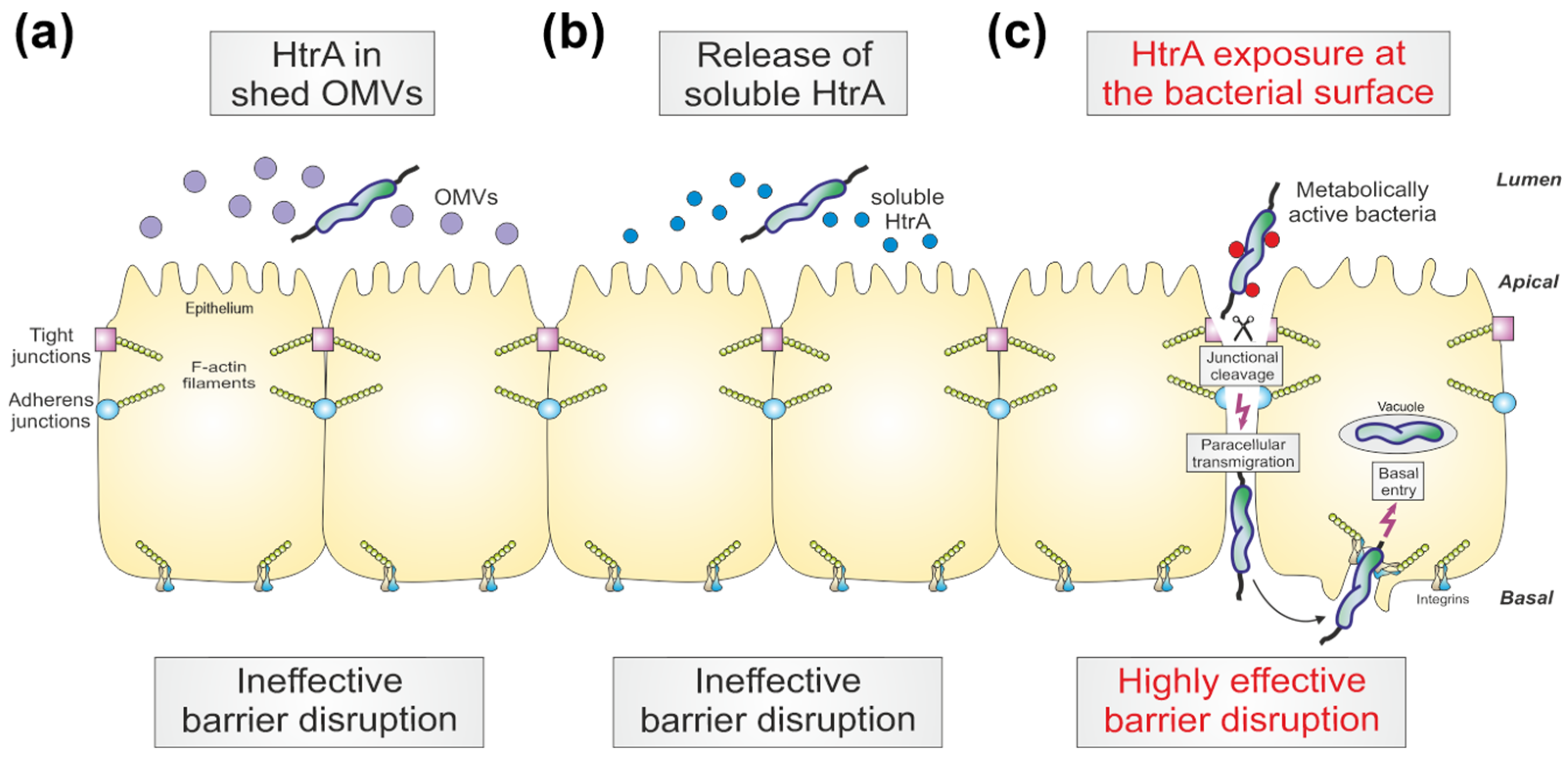
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Horowitz, A.; Chanez-Paredes, S.D.; Haest, X.; Turner, J.R. Paracellular permeability and tight junction regulation in gut health and disease. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 417–432. [Google Scholar] [CrossRef]
- Rusu, A.D.; Georgiou, M. The multifarious regulation of the apical junctional complex. Open Biol. 2020, 10, 190278. [Google Scholar] [CrossRef]
- Buckley, C.E.; St Johnston, D. Apical-basal polarity and the control of epithelial form and function. Nat. Rev. Mol. Cell Biol. 2022, 23, 559–577. [Google Scholar] [CrossRef]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef]
- Balda, M.S.; Matter, K. Tight junctions as regulators of tissue remodelling. Curr. Opin. Cell Biol. 2016, 42, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Vogelmann, R.; Amieva, M.R.; Falkow, S.; Nelson, W.J. Breaking into the epithelial apical-junctional complex—News from pathogen hackers. Curr. Opin. Cell Biol. 2004, 16, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Guttman, J.A.; Finlay, B.B. Tight junctions as targets of infectious agents. Biochim. Biophys. Acta-Biomembr. 2009, 1788, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.Y.; Yang, W.X.; Hu, Y.J. The role of epithelial tight junctions involved in pathogen infections. Mol. Biol. Rep. 2014, 41, 6591–6610. [Google Scholar] [CrossRef]
- Ruch, T.R.; Engel, J.N. Targeting the Mucosal Barrier: How Pathogens Modulate the Cellular Polarity Network. Cold Spring Harb. Perspect. Biol. 2017, 9, a027953. [Google Scholar] [CrossRef] [PubMed]
- Chegini, Z.; Noei, M.; Hemmati, J.; Arabestani, M.R.; Shariati, A. The destruction of mucosal barriers, epithelial remodeling, and impaired mucociliary clearance: Possible pathogenic mechanisms of Pseudomonas aeruginosa and Staphylococcus aureus in chronic rhinosinusitis. Cell Commun. Signal. 2023, 21, 306. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Boehm, M.; Wessler, S.; Tegtmeyer, N. Transmigration route of Campylobacter jejuni across polarized intestinal epithelial cells: Paracellular, transcellular or both? Cell Commun. Signal. 2013, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Burnham, P.M.; Hendrixson, D.R. Campylobacter jejuni: Collective components promoting a successful enteric lifestyle. Nat. Rev. Microbiol. 2018, 16, 551–565. [Google Scholar] [CrossRef]
- Ahmed, N.A.; Gulhan, T. Campylobacter in Wild Birds: Is It an Animal and Public Health Concern? Front. Microbiol. 2022, 12, 4180. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeyer, N.; Sharafutdinov, I.; Harrer, A.; Esmaeili, D.S.; Linz, B.; Backert, S. Campylobacter Virulence Factors and Molecular Host-Pathogen Interactions. Curr. Top. Microbiol. Immunol. 2021, 431, 169–202. [Google Scholar] [CrossRef]
- Püning, C.; Su, Y.L.; Lu, X.N.; Golz, G. Molecular Mechanisms of Campylobacter Biofilm Formation and Quorum Sensing. Curr. Top. Microbiol. Immunol. 2021, 431, 293–319. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Tegtmeyer, N.; Cróinín, T.Ó.; Boehm, M.; Heimesaat, M.M. Human campylobacteriosis. In Campylobacter Features, Detection, and Prevention of Foodborne Disease; Academic Press: Cambridge, MA, USA, 2017; pp. 1–25. [Google Scholar]
- Kemper, L.; Hensel, A. Campylobacter jejuni: Targeting host cells, adhesion, invasion, and survival. Appl. Microbiol. Biotechnol. 2023, 107, 2725–2754. [Google Scholar] [CrossRef]
- Kaakoush, N.O.; Castano-Rodriguez, N.; Mitchell, H.M.; Man, S.I.M. Global Epidemiology of Campylobacter Infection. Clin. Microbiol. Rev. 2015, 28, 687–720. [Google Scholar] [CrossRef]
- Al Othman, B.; Raabe, J.; Kini, A.; Lee, A.G. Update: The Miller Fisher variants of Guillain-Barré syndrome. Curr. Opin. Ophthalmol. 2019, 30, 462–466. [Google Scholar] [CrossRef]
- Finsterer, J. Triggers of Guillain-Barré Syndrome: Campylobacter jejuni Predominates. Int. J. Mol. Sci. 2022, 23, 14222. [Google Scholar] [CrossRef]
- Kalischuk, L.D.; Inglis, G.D.; Buret, A.G. Campylobacter jejuni induces transcellular translocation of commensal bacteria via lipid rafts. Gut Pathog. 2009, 1, 2. [Google Scholar] [CrossRef]
- Lamb-Rosteski, J.M.; Kalischuk, L.D.; Inglis, G.D.; Buret, A.G. Epidermal growth factor inhibits Campylobacter jejuni-induced claudin-4 disruption, loss of epithelial barrier function, and Escherichia coli translocation. Infect. Immun. 2008, 76, 3390–3398. [Google Scholar] [CrossRef] [PubMed]
- Foppa, C.; Rizkala, T.; Repici, A.; Hassan, C.; Spinelli, A. Microbiota and IBD: Current knowledge and future perspectives. Dig. Liver Dis. 2023. [CrossRef] [PubMed]
- Haag, L.M.; Fischer, A.; Otto, B.; Plickert, R.; Kuhl, A.A.; Gobel, U.B.; Bereswill, S.; Heimesaat, M.M. Intestinal Microbiota Shifts towards Elevated Commensal Escherichia coli Loads Abrogate Colonization Resistance against Campylobacter jejuni in Mice. PLoS ONE 2012, 7, e35988. [Google Scholar] [CrossRef] [PubMed]
- Ahlawat, S.; Kumar, P.; Mohan, H.; Goyal, S.; Sharma, K.K. Inflammatory bowel disease: Tri-directional relationship between microbiota, immune system and intestinal epithelium. Crit. Rev. Microbiol. 2021, 47, 254–273. [Google Scholar] [CrossRef] [PubMed]
- Sharafutdinov, I.; Tegtmeyer, N.; Musken, M.; Backert, S. Campylobacter jejuni Serine Protease HtrA Induces Paracellular Transmigration of Microbiota across Polarized Intestinal Epithelial Cells. Biomolecules 2022, 12, 521. [Google Scholar] [CrossRef] [PubMed]
- Boehm, M.; Simson, D.; Escher, U.; Schmidt, A.-M.; Bereswill, S.; Tegtmeyer, N.; Backert, S.; Heimesaat, M.M. Function of Serine Protease HtrA in the Lifecycle of the Foodborne Pathogen Campylobacter jejuni. Eur. J. Microbiol. Immunol. 2018, 8, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Bernegger, S.; Skórko-Glonek, J.; Wessler, S. Extracellular HtrA serine proteases: An emerging new strategy in bacterial pathogenesis. Cell. Microbiol. 2018, 20, e12845. [Google Scholar] [CrossRef]
- Skórko-Glonek, J.; Zurawa-Janicka, D.; Koper, T.; Jarzab, M.; Figaj, D.; Glaza, P.; Lipinska, B. HtrA Protease Family as Therapeutic Targets. Curr. Pharm. Des. 2013, 19, 977–1009. [Google Scholar] [CrossRef]
- Zarzecka, U.; Grinzato, A.; Kandiah, E.; Cysewski, D.; Berto, P.; Skórko-Glonek, J.; Zanotti, G.; Backert, S. Functional analysis and cryo-electron microscopy of Campylobacter jejuni serine protease HtrA. Gut Microbes 2020, 12, 1810532. [Google Scholar] [CrossRef]
- Brøndsted, L.; Andersen, M.T.; Parker, M.; Jorgensen, K.; Ingmer, H. The HtrA protease of Campylobacter jejuni is required for heat and oxygen tolerance and for optimal interaction with human epithelial cells. Appl. Environ. Microbiol. 2005, 71, 3205–3212. [Google Scholar] [CrossRef]
- Bæk, K.T.; Vegge, C.S.; Skórko-Glonek, J.; Brøndsted, L. Different Contributions of HtrA Protease and Chaperone Activities to Campylobacter jejuni Stress Tolerance and Physiology. Appl. Environ. Microbiol. 2011, 77, 57–66. [Google Scholar] [CrossRef]
- Heimesaat, M.M.; Alutis, M.; Grundmann, U.; Fischer, A.; Tegtmeyer, N.; Böhm, M.; Kuhl, A.A.; Gobel, U.B.; Backert, S.; Bereswill, S. The role of serine protease HtrA in acute ulcerative enterocolitis and extra-intestinal immune responses during Campylobacter jejuni infection of gnotobiotic IL-10 deficient mice. Front. Cell. Infect. Microbiol. 2014, 4, 77. [Google Scholar] [CrossRef]
- Heimesaat, M.M.; Fischer, A.; Alutis, M.; Grundmann, U.; Boehm, M.; Tegtmeyer, N.; Gobel, U.B.; Kuhl, A.A.; Bereswill, S.; Backert, S. The impact of serine protease HtrA in apoptosis, intestinal immune responses and extra-intestinal histopathology during Campylobacter jejuni infection of infant mice. Gut Pathog. 2014, 6, 16. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Escher, U.; Mousavi, S.; Boehm, M.; Backert, S.; Bereswill, S.; Heimesaat, M.M. Protease Activity of Campylobacter jejuni HtrA Modulates Distinct Intestinal and Systemic Immune Responses in Infected Secondary Abiotic IL-10 Deficient Mice. Front. Cell. Infect. Microbiol. 2019, 9, 79. [Google Scholar] [CrossRef] [PubMed]
- Bæk, K.T.; Vegge, C.S.; Brøndsted, L. HtrA chaperone activity contributes to host cell binding in Campylobacter jejuni. Gut Pathog. 2011, 3, 13. [Google Scholar] [CrossRef]
- Boehm, M.; Hoy, B.; Rohde, M.; Tegtmeyer, N.; Bæk, K.T.; Oyarzabal, O.A.; Brøndsted, L.; Wessler, S.; Backert, S. Rapid paracellular transmigration of Campylobacter jejuni across polarized epithelial cells without affecting TER: Role of proteolytic-active HtrA cleaving E-cadherin but not fibronectin. Gut Pathog. 2012, 4, 3. [Google Scholar] [CrossRef]
- Boehm, M.; Lind, J.; Backert, S.; Tegtmeyer, N. Campylobacter jejuni serine protease HtrA plays an important role in heat tolerance, oxygen resistance, host cell adhesion, invasion, and transmigration. Eur. J. Microbiol. Immunol. 2015, 5, 68–80. [Google Scholar] [CrossRef]
- Simson, D.; Boehm, M.; Backert, S. HtrA-dependent adherence and invasion of Campylobacter jejuni in human vs. avian cells. Lett. Appl. Microbiol. 2020, 70, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Harrer, A.; Bucker, R.; Boehm, M.; Zarzecka, U.; Tegtmeyer, N.; Sticht, H.; Schulzke, J.D.; Backert, S. Campylobacter jejuni enters gut epithelial cells and impairs intestinal barrier function through cleavage of occludin by serine protease HtrA. Gut Pathog. 2019, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Sharafutdinov, I.; Esmaeili, D.S.; Harrer, A.; Tegtmeyer, N.; Sticht, H.; Backert, S. Campylobacter jejuni Serine Protease HtrA Cleaves the Tight Junction Component Claudin-8. Front. Cell. Infect. Microbiol. 2020, 10, 590186. [Google Scholar] [CrossRef]
- Abfalter, C.M.; Schubert, M.; Götz, C.; Schmidt, T.P.; Posselt, G.; Wessler, S. HtrA-mediated E-cadherin cleavage is limited to DegP and DegQ homologs expressed by Gram-negative pathogens. Cell Commun. Signal. 2016, 14, 30. [Google Scholar] [CrossRef] [PubMed]
- Orench-Rivera, N.; Kuehn, M.J. Environmentally controlled bacterial vesicle-mediated export. Cell. Microbiol. 2016, 18, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Lindmark, B.; Rompikuntal, P.K.; Vaitkevicius, K.; Song, T.Y.; Mizunoe, Y.; Uhlin, B.E.; Guerry, P.; Wai, S.N. Outer membrane vesicle-mediated release of cytolethal distending toxin (CDT) from Campylobacter jejuni. BMC Microbiol. 2009, 9, 220. [Google Scholar] [CrossRef] [PubMed]
- Elmi, A.; Watson, E.; Sandu, P.; Gundogdu, O.; Mills, D.C.; Inglis, N.F.; Manson, E.; Imrie, L.; Bajaj-Elliott, M.; Wren, B.W.; et al. Campylobacter jejuni Outer Membrane Vesicles Play an Important Role in Bacterial Interactions with Human Intestinal Epithelial Cells. Infect. Immun. 2012, 80, 4089–4098. [Google Scholar] [CrossRef] [PubMed]
- Elmi, A.; Nasher, F.; Jagatia, H.; Gundogdu, O.; Bajaj-Elliott, M.; Wren, B.; Dorrell, N. Campylobacter jejuni outer membrane vesicle-associated proteolytic activity promotes bacterial invasion by mediating cleavage of intestinal epithelial cell E-cadherin and occludin. Cell. Microbiol. 2016, 18, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Neddermann, M.; Backert, S. Quantification of serine protease HtrA molecules secreted by the foodborne pathogen Campylobacter jejuni. Gut Pathog. 2019, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Löwer, M.; Weydig, C.; Metzler, D.; Reuter, A.; Starzinski-Powitz, A.; Wessler, S.; Schneider, G. Prediction of Extracellular Proteases of the Human Pathogen Helicobacter pylori Reveals Proteolytic Activity of the Hp1018/19 Protein HtrA. PLoS ONE 2008, 3, e3510. [Google Scholar] [CrossRef]
- Wai, S.N.; Lindmark, B.; Söderblom, T.; Takade, A.; Westermark, M.; Oscarsson, J.; Jass, J.; Richter-Dahlfors, A.; Mizunoe, Y.; Uhlin, B.E. Vesicle-mediated export and assembly of pore-forming oligomers of the enterobacterial ClyA cytotoxin. Cell 2003, 115, 25–35. [Google Scholar] [CrossRef]
- Olofsson, A.; Vallstrom, A.; Petzold, K.; Tegtmeyer, N.; Schleucher, J.; Carlsson, S.; Haas, R.; Backert, S.; Wai, S.N.; Grobner, G.; et al. Biochemical and functional characterization of Helicobacter pylori vesicles. Mol. Microbiol. 2010, 77, 1539–1555. [Google Scholar] [CrossRef]
- Schmidt, T.P.; Perna, A.M.; Fugmann, T.; Böhm, M.; Hiss, J.; Haller, S.; Gotz, C.; Tegtmeyer, N.; Hoy, B.; Rau, T.T.; et al. Identification of E-cadherin signature motifs functioning as cleavage sites for Helicobacter pylori HtrA. Sci. Rep. 2016, 6, 23264. [Google Scholar] [CrossRef]
- Negretti, N.M.; Konkel, M.E. Methods to Study Campylobacter jejuni Adherence to and Invasion of Host Epithelial Cells. Methods Mol Biol. 2017, 1512, 117–127. [Google Scholar] [CrossRef]
- Turkina, M.V.; Olofsson, A.; Magnusson, K.E.; Arnqvist, A.; Vikstrom, E. Helicobacter pylori vesicles carrying CagA localize in the vicinity of cell-cell contacts and induce histone H1 binding to ATP in epithelial cells. FEMS Microbiol. Lett. 2015, 362, fnv076. [Google Scholar] [CrossRef]
- Krause-Gruszczynska, M.; Rohde, M.; Hartig, R.; Genth, H.; Schmidt, G.; Keo, T.; Konig, W.; Miller, W.G.; Konkel, M.E.; Backert, S. Role of the small Rho GTPases Rac1 and Cdc42 in host cell invasion of Campylobacter jejuni. Cell. Microbiol. 2007, 9, 2431–2444. [Google Scholar] [CrossRef] [PubMed]
- Sharafutdinov, I.; Tegtmeyer, N.; Linz, B.; Rohde, M.; Vieth, M.; Tay, A.C.-Y.; Lamichhane, B.; Tuan, V.P.; Fauzia, K.A.; Sticht, H.; et al. A single-nucleotide polymorphism in Helicobacter pylori promotes gastric cancer development. Cell Host Microbe 2023, 31, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Moodley, Y.; Yamaoka, Y.; Pernitzsch, S.R.; Schmidt, V.; Traverso, F.R.; Schmidt, T.P.; Rad, R.; Yeoh, K.G.; Bow, H.; et al. Characterisation of worldwide Helicobacter pylori strains reveals genetic conservation and essentiality of serine protease HtrA. Mol. Microbiol. 2016, 99, 925–944. [Google Scholar] [CrossRef]
- Elmi, A.; Dorey, A.; Watson, E.; Jagatia, H.; Inglis, N.F.; Gundogdu, O.; Bajaj-Elliott, M.; Wren, B.W.; Smith, D.G.E.; Dorrell, N. The bile salt sodium taurocholate induces Campylobacter jejuni outer membrane vesicle production and increases OMV-associated proteolytic activity. Cell. Microbiol. 2018, 20, e12814. [Google Scholar] [CrossRef]
- Gabbert, A.D.; Mydosh, J.L.; Talukdar, P.K.; Gloss, L.M.; McDermott, J.E.; Cooper, K.K.; Clair, G.C.; Konkel, M.E. The Missing Pieces: The Role of Secretion Systems in Campylobacter jejuni Virulence. Biomolecules 2023, 13, 135. [Google Scholar] [CrossRef]
- Guerry, P.; Poly, F.; Riddle, M.; Maue, A.C.; Chen, Y.H.; Monteiro, M.A. Campylobacter polysaccharide capsules: Virulence and vaccines. Front. Cell Infect. Microbiol. 2012, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Frees, D.; Brondsted, L.; Ingmer, H. Bacterial proteases and virulence. Subcell. Biochem. 2013, 66, 161–192. [Google Scholar] [CrossRef]
- Backert, S.; Kwok, T.; Schmid, M.; Selbach, M.; Moese, S.; Peek, R.M.; Konig, W.; Meyer, T.F.; Jungblut, P.R. Subproteomes of soluble and structure-bound Helicobacter pylori proteins analyzed by two-dimensional gel electrophoresis and mass spectrometry. Proteomics 2005, 5, 1331–1345. [Google Scholar] [CrossRef] [PubMed]
- Jarzab, M.; Posselt, G.; Meisner-Kober, N.; Wessler, S. Helicobacter pylori-Derived Outer Membrane Vesicles (OMVs): Role in Bacterial Pathogenesis? Microorganisms 2020, 8, 1328. [Google Scholar] [CrossRef] [PubMed]
- Cuzic, S.; Antolic, M.; Ognjenovic, A.; Stupin-Polancec, D.; Grba, A.P.; Hrvacic, B.; Kramaric, M.D.; Musladin, S.; Pozgaj, L.; Zlatar, I.; et al. Claudins: Beyond Tight Junctions in Human IBD and Murine Models. Front. Pharmacol. 2021, 12, 682614. [Google Scholar] [CrossRef] [PubMed]
- Zeissig, S.; Burgel, N.; Gunzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharafutdinov, I.; Tegtmeyer, N.; Rohde, M.; Olofsson, A.; Rehman, Z.u.; Arnqvist, A.; Backert, S. Campylobacter jejuni Surface-Bound Protease HtrA, but Not the Secreted Protease nor Protease in Shed Membrane Vesicles, Disrupts Epithelial Cell-to-Cell Junctions. Cells 2024, 13, 224. https://doi.org/10.3390/cells13030224
Sharafutdinov I, Tegtmeyer N, Rohde M, Olofsson A, Rehman Zu, Arnqvist A, Backert S. Campylobacter jejuni Surface-Bound Protease HtrA, but Not the Secreted Protease nor Protease in Shed Membrane Vesicles, Disrupts Epithelial Cell-to-Cell Junctions. Cells. 2024; 13(3):224. https://doi.org/10.3390/cells13030224
Chicago/Turabian StyleSharafutdinov, Irshad, Nicole Tegtmeyer, Manfred Rohde, Annelie Olofsson, Zia ur Rehman, Anna Arnqvist, and Steffen Backert. 2024. "Campylobacter jejuni Surface-Bound Protease HtrA, but Not the Secreted Protease nor Protease in Shed Membrane Vesicles, Disrupts Epithelial Cell-to-Cell Junctions" Cells 13, no. 3: 224. https://doi.org/10.3390/cells13030224
APA StyleSharafutdinov, I., Tegtmeyer, N., Rohde, M., Olofsson, A., Rehman, Z. u., Arnqvist, A., & Backert, S. (2024). Campylobacter jejuni Surface-Bound Protease HtrA, but Not the Secreted Protease nor Protease in Shed Membrane Vesicles, Disrupts Epithelial Cell-to-Cell Junctions. Cells, 13(3), 224. https://doi.org/10.3390/cells13030224