
Moderate Elevation of Homocysteine Induces Endothelial Dysfunction through Adaptive UPR Activation and Metabolic Rewiring


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Chemical Treatments
2.2. Zebrafish Maintenance and Morpholino Microinjection
- CBSa MO: 5′-GGGACTGAAGGCATTATTCCTCAAT-3′
- CBSb MO: 5′-CTGGCATGGTTTACCCTGACTATCA-3′
- CBS SC MO: 5′-GGCACTCAAGGCAATATACGTCAAT-3′
- CGL MO: 5′-CTGCATCTCTGAAGACATGAGTGC-3′
- CGL SC MO: 5′-TATGCATATCTGAACACATCAGTCC-3′
2.3. Lysate Preparation
2.4. Western Blotting
2.5. RNA Extraction and qPCR
2.6. Homocysteine Measurement
2.7. Trypan Blue Exclusion Assay
2.8. Tube Formation Assay
2.9. Scratch Wound Assay
2.10. BrdU Incorporation Assay
2.11. Measurement of Mitochondrial Membrane Potential
2.12. Measurement of Cellular Reactive Oxygen Species (ROS)
2.13. Generation of mitoGFP Transduced Cells
2.14. Confocal Imaging of mitoGFP Transduced Cells
2.15. Mitochondrial Length Measurement
2.16. Rhodamine Phalloidin Staining
2.17. Zebrafish Imaging and Heartbeat Measurement
2.18. Glucose Uptake Assay
2.19. Extracellular Flux Analysis
2.20. Metabolite Extraction and Targeted Metabolomics
2.21. Analysis of Available Microarray Dataset
2.22. Statistical Analysis
3. Results
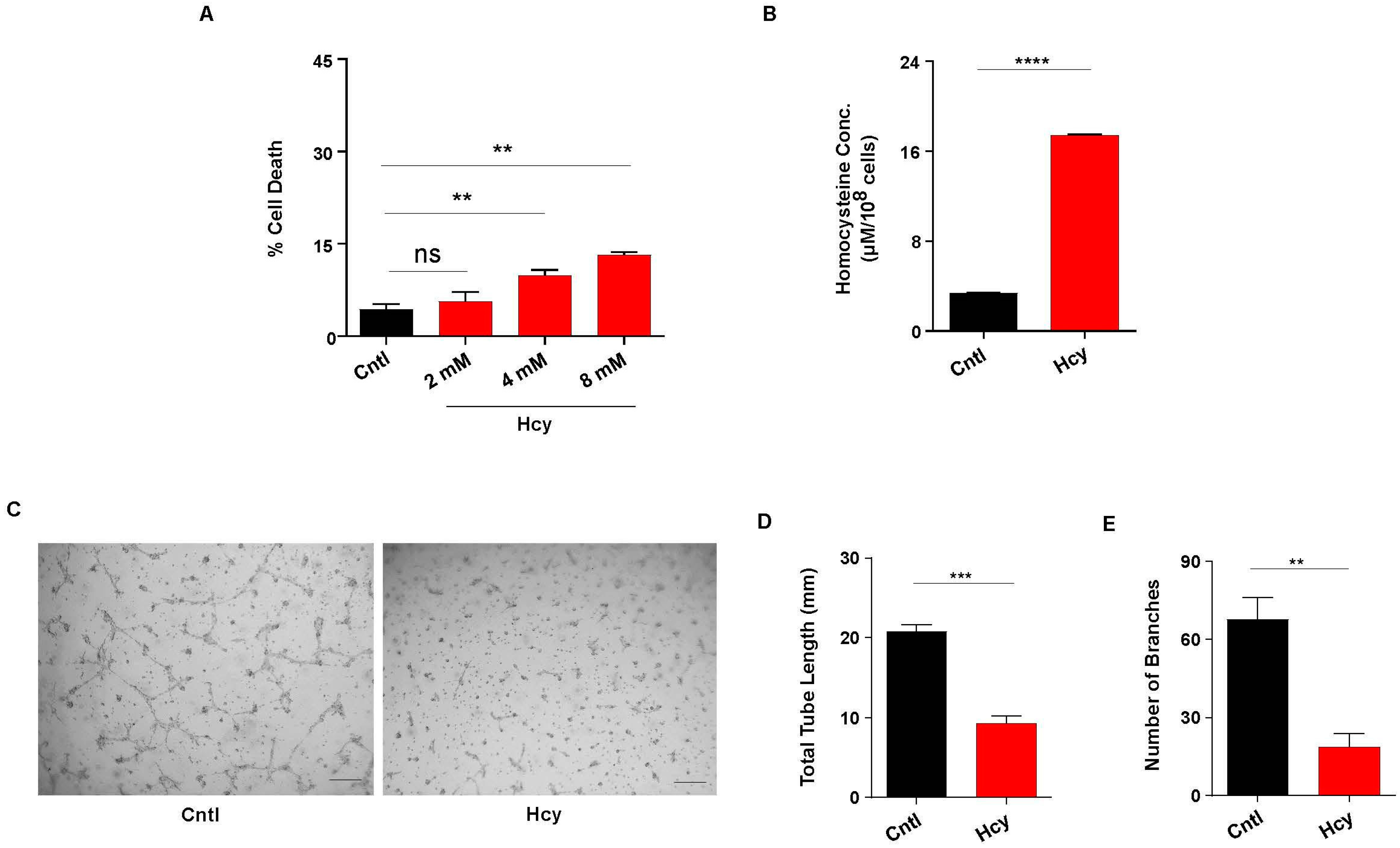
3.1. Moderate Increase in Hcy Levels Causes Endothelial Dysfunction
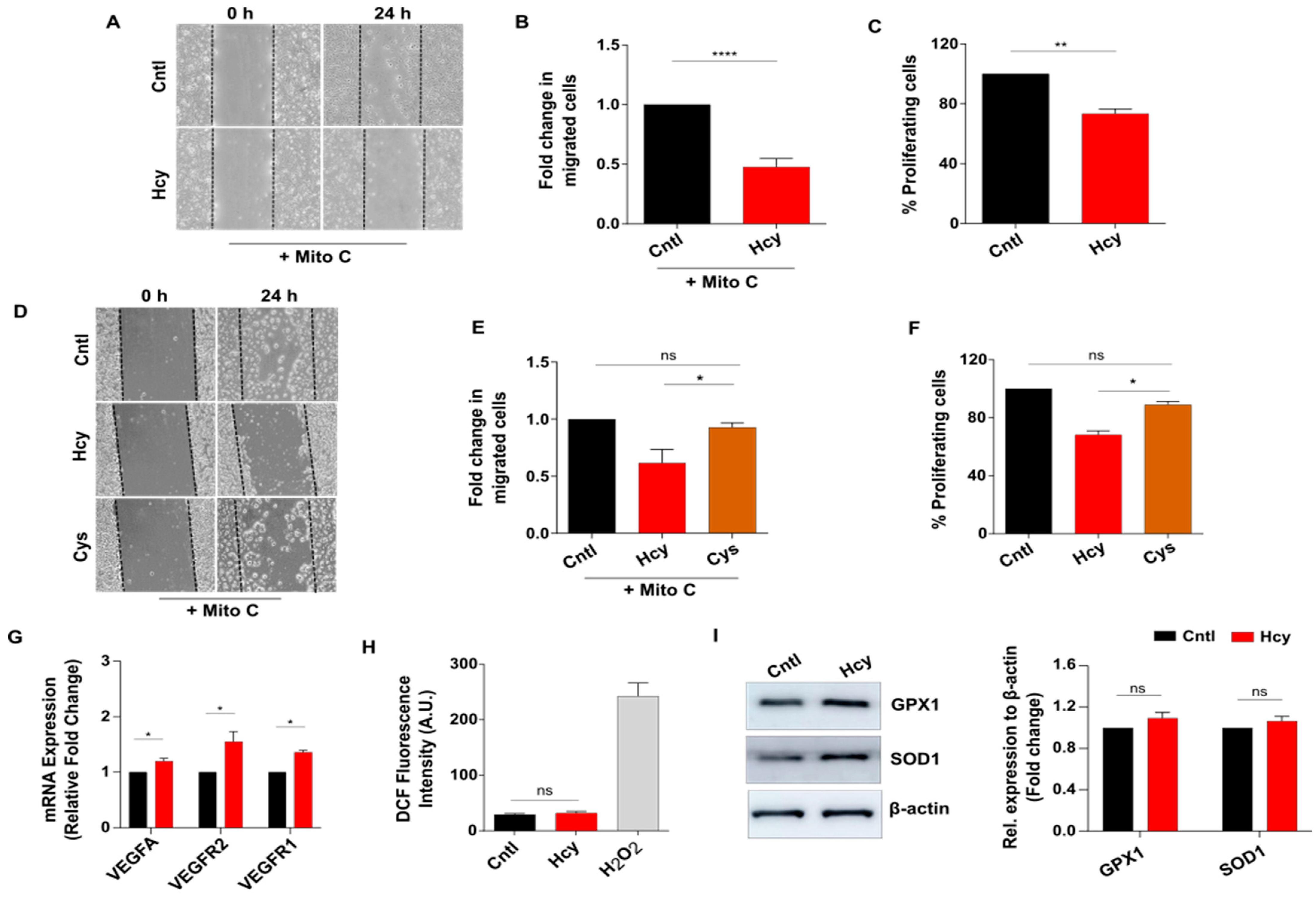
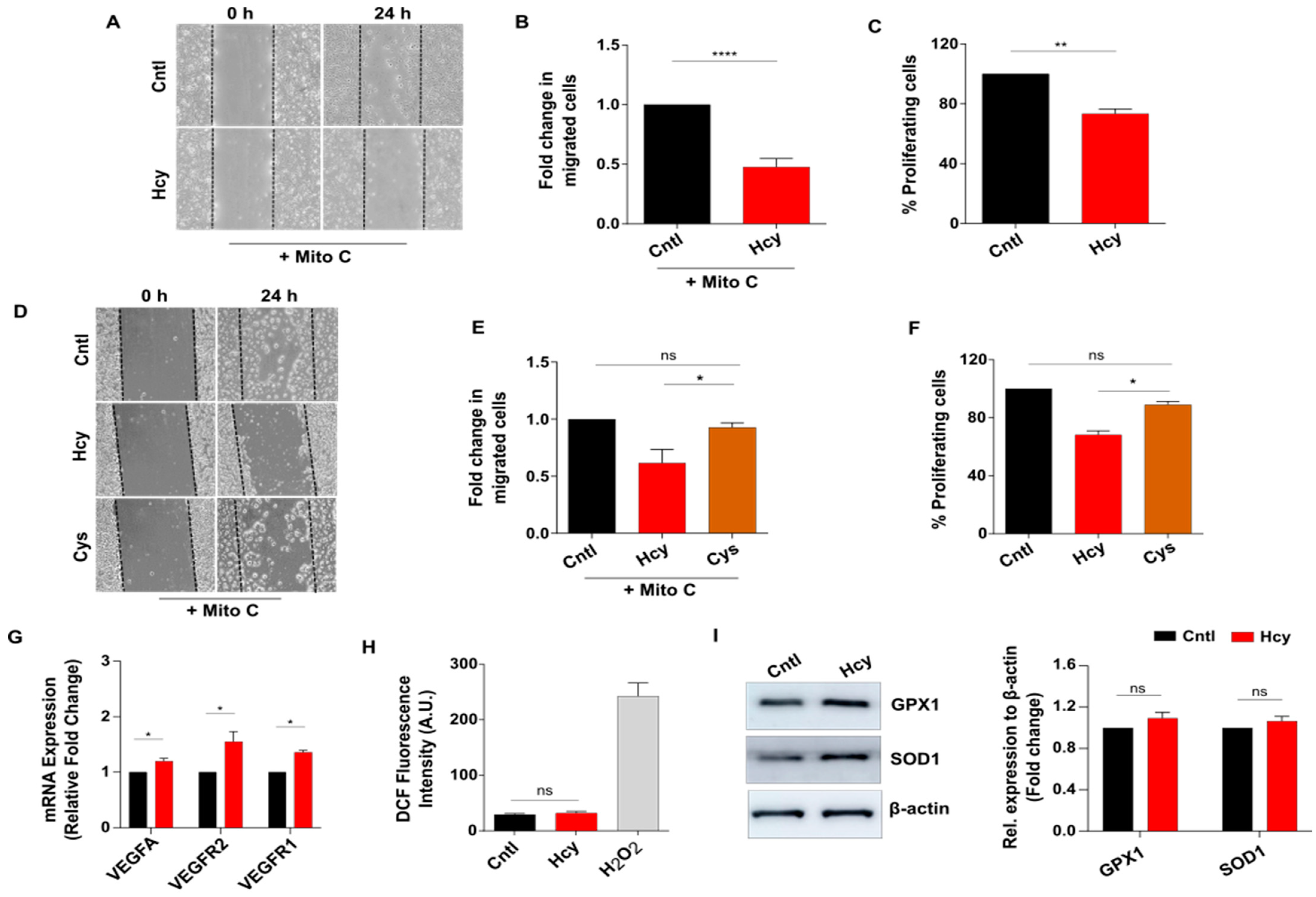
3.2. Moderate HHcy Impairs Endothelial Migration and Proliferation without Suppressing the Expression of VEGF/VEGFR Transcripts and ROS Level Change
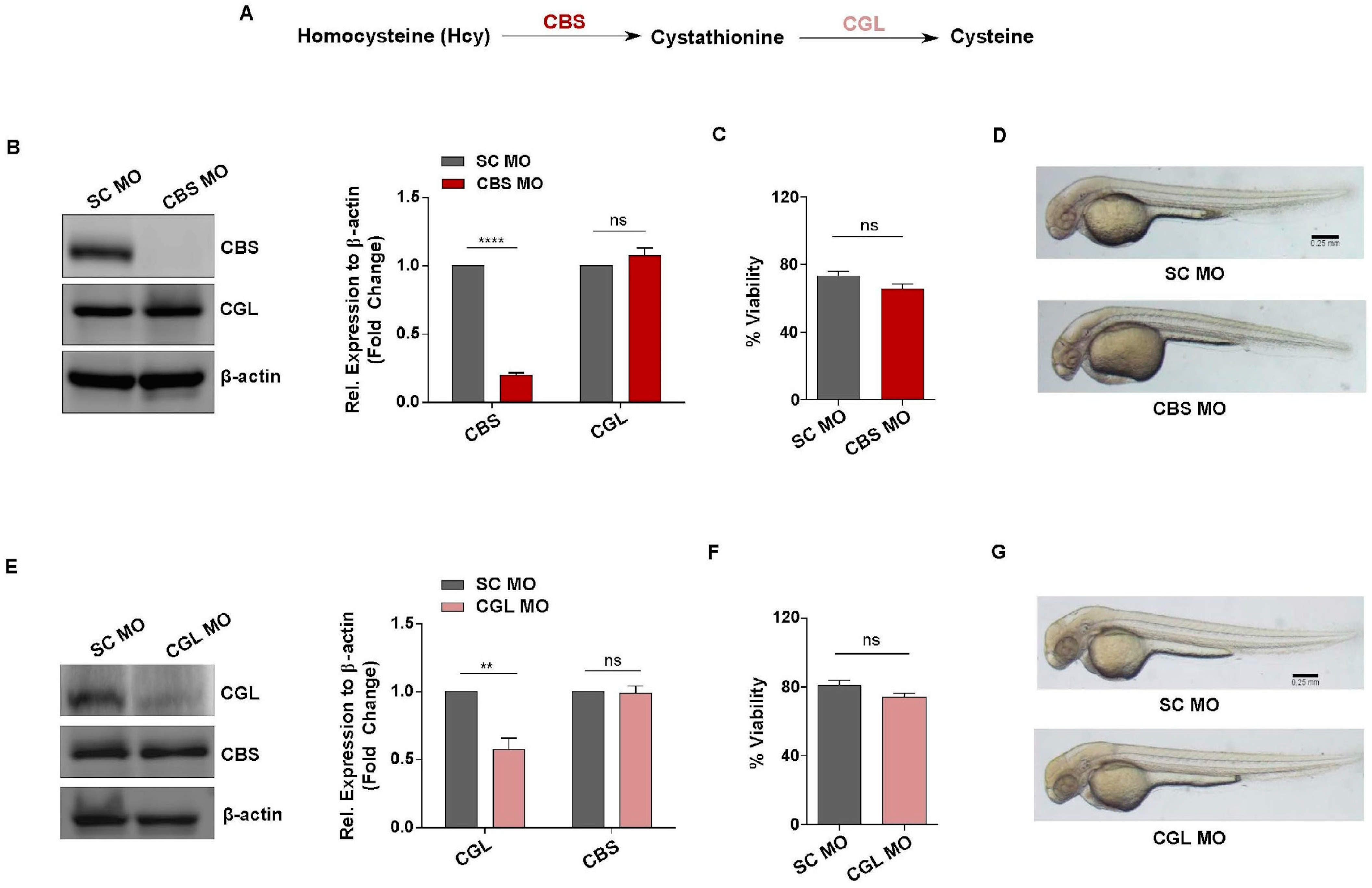
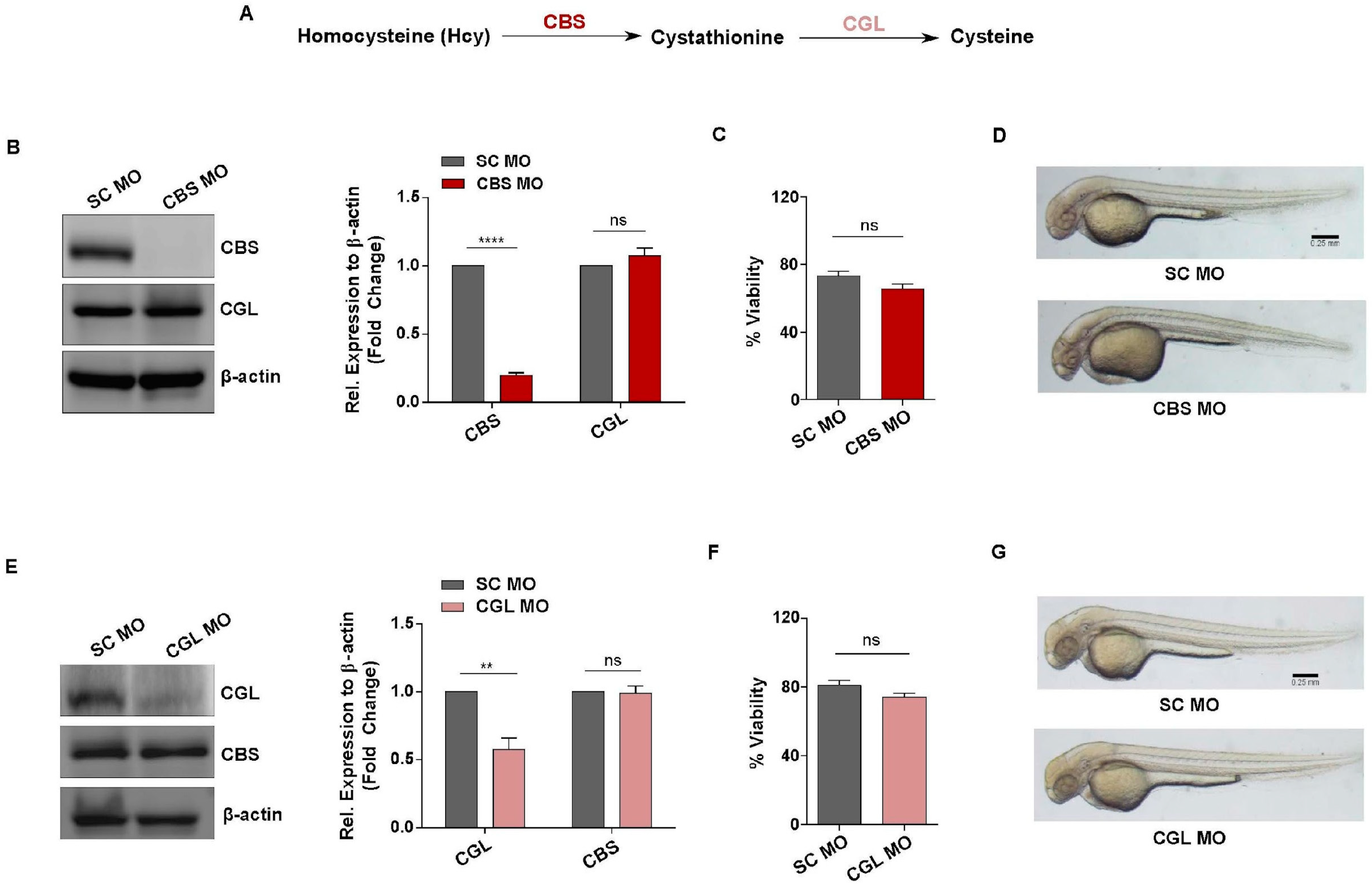
3.3. Generation of In Vivo Knockdown Models of CBS and CGL, Transsulfuration Pathway Regulators Involved in Hcy Catabolism
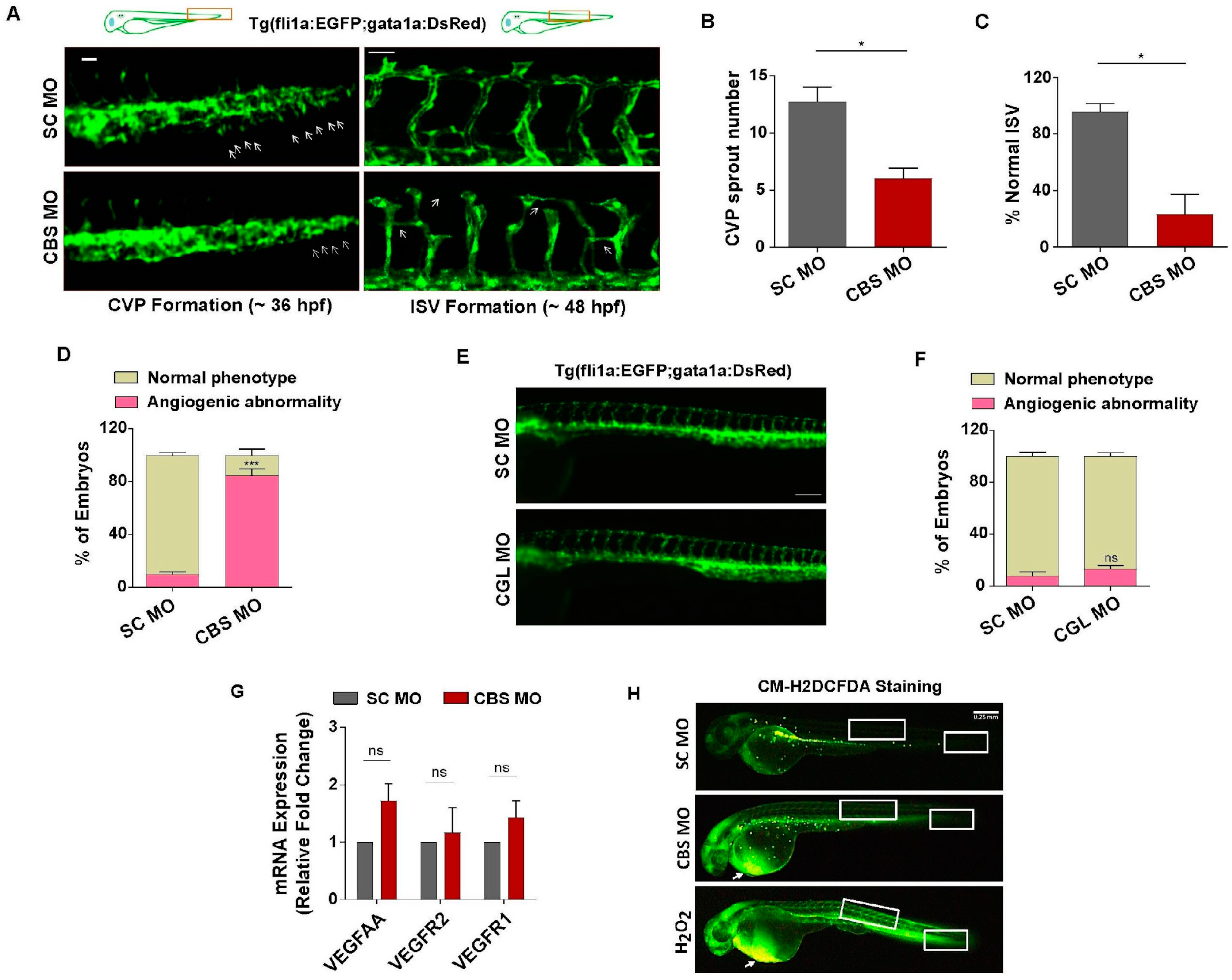
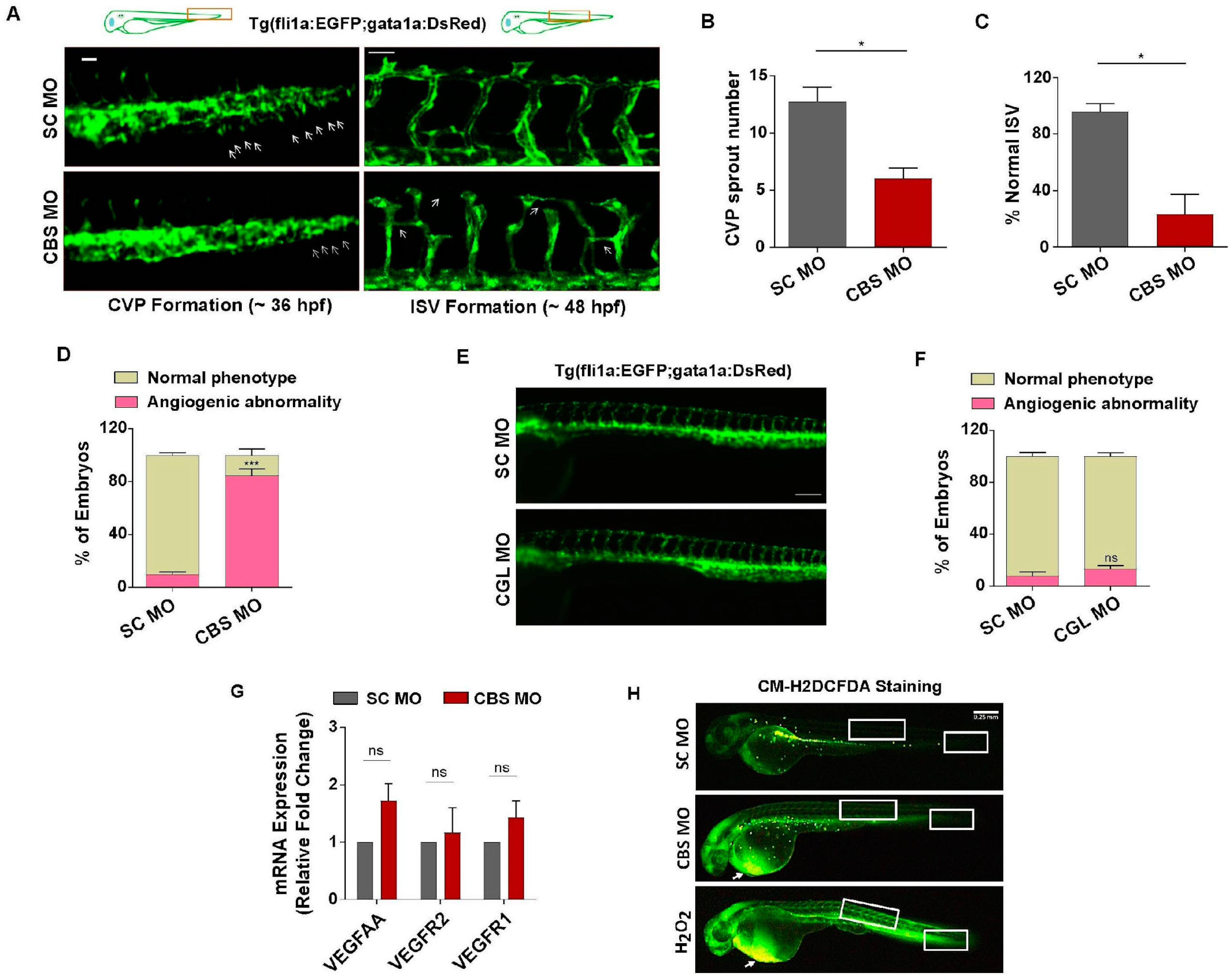
3.4. Moderate HHcy Induces Vascular Abnormality In Vivo without Suppressing the Expression of VEGF/VEGFR Transcripts and ROS Level Change
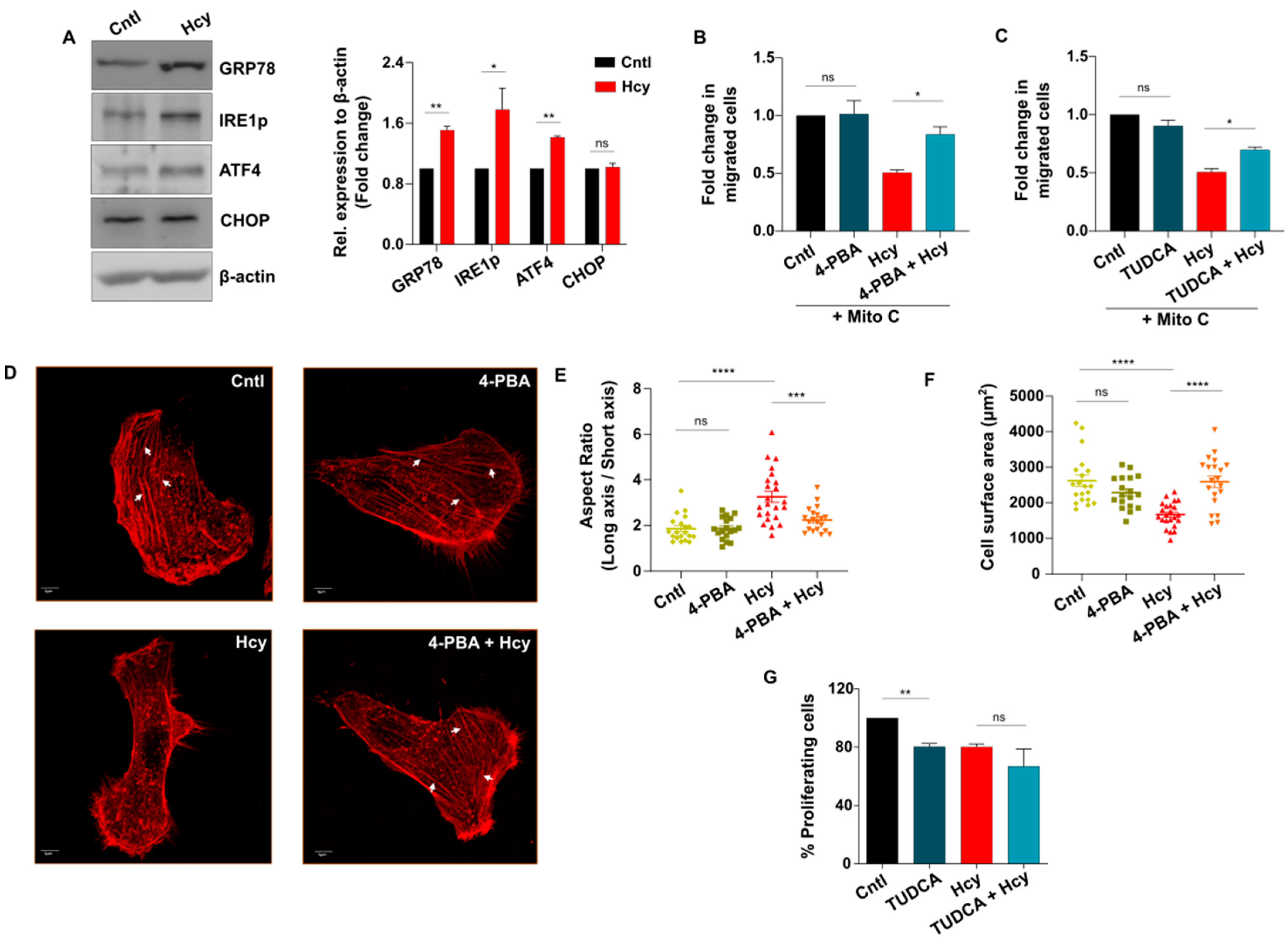
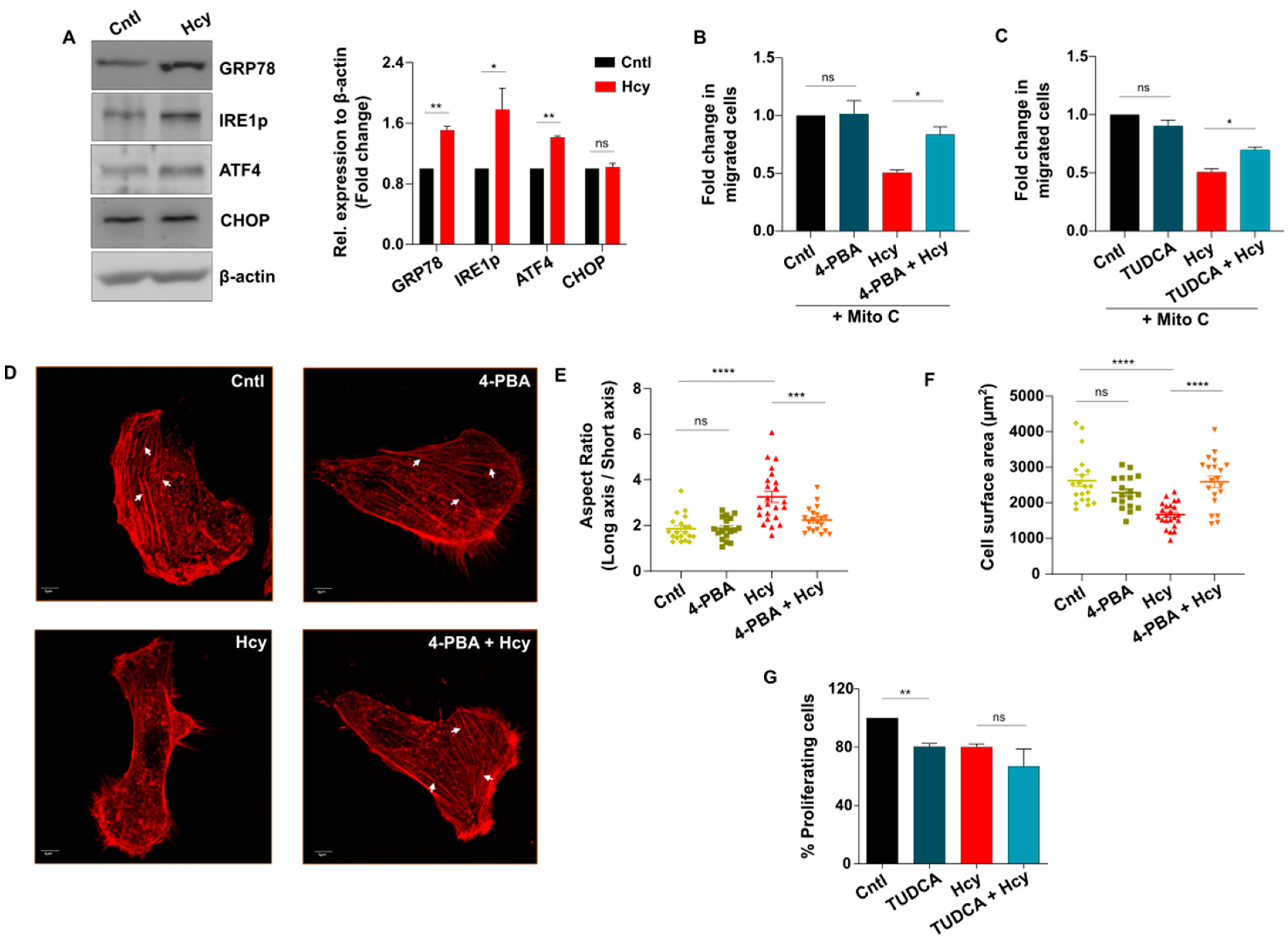
3.5. Adaptive UPR Activation Controls Endothelial Migration Defect in Moderate HHcy
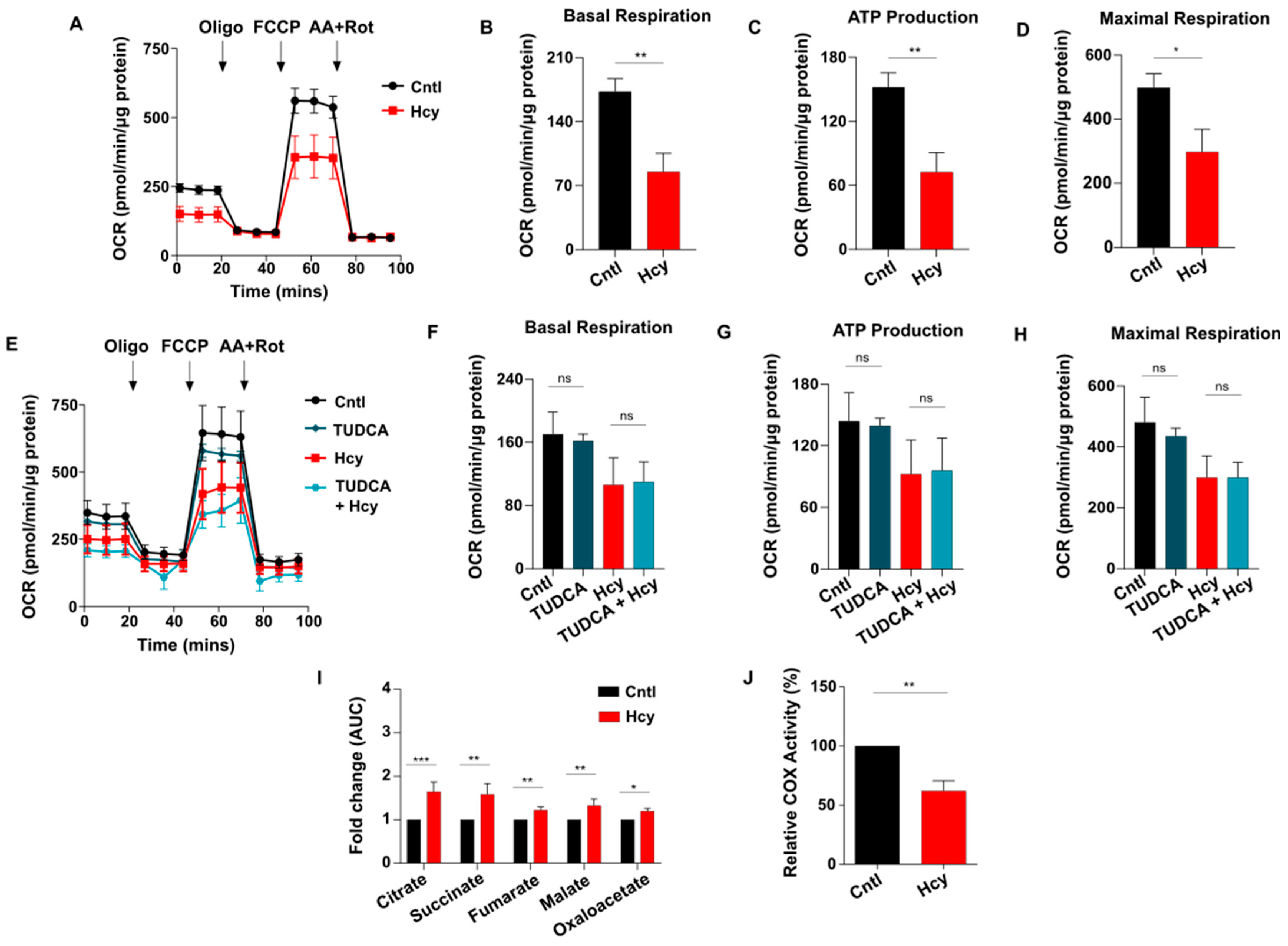
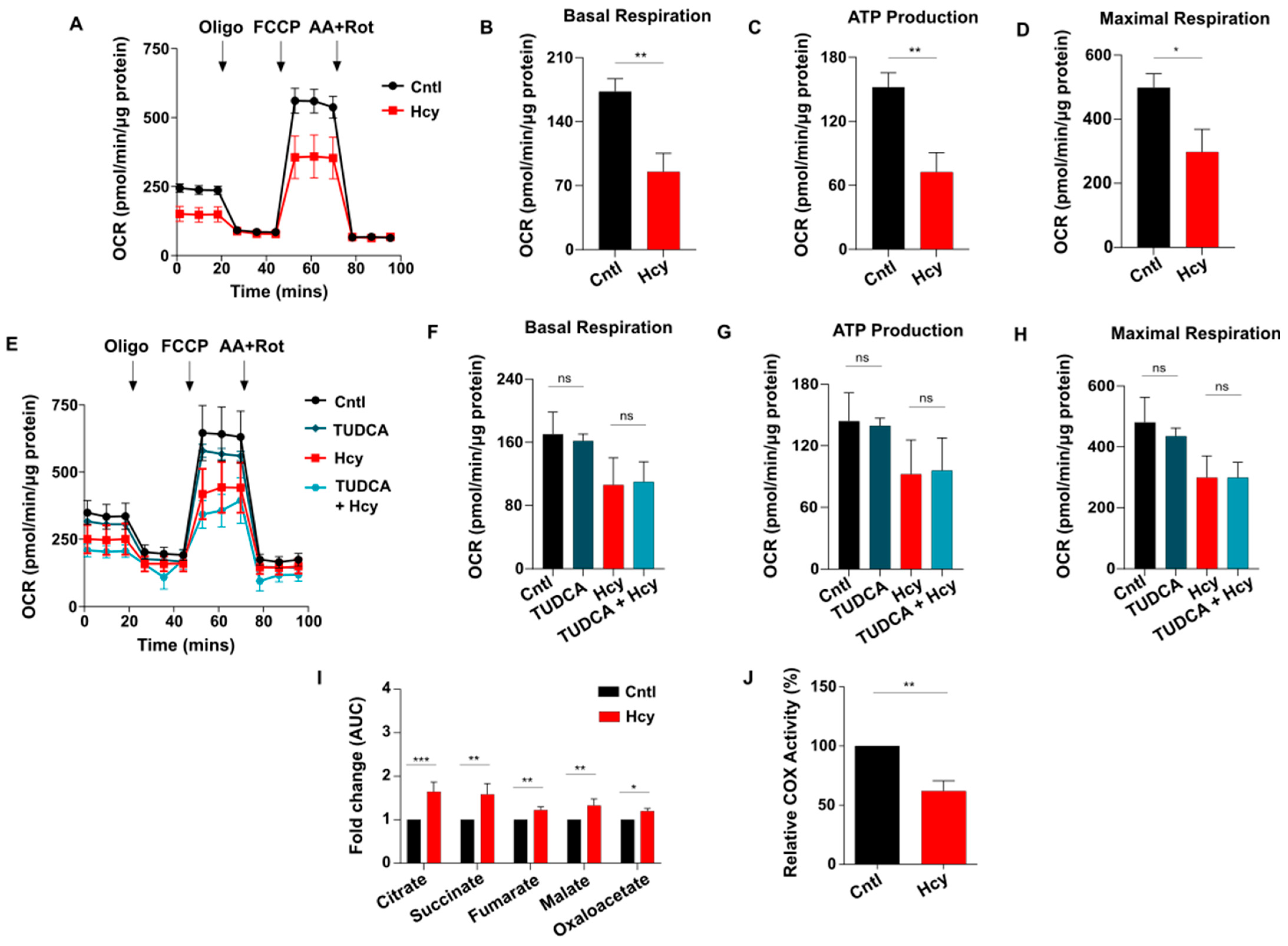
3.6. Sub-Lethal HHcy Linked Malfunctioned ETC Causes Reduction in Mitochondrial Respiration, a Crucial Regulator of Endothelial Proliferation
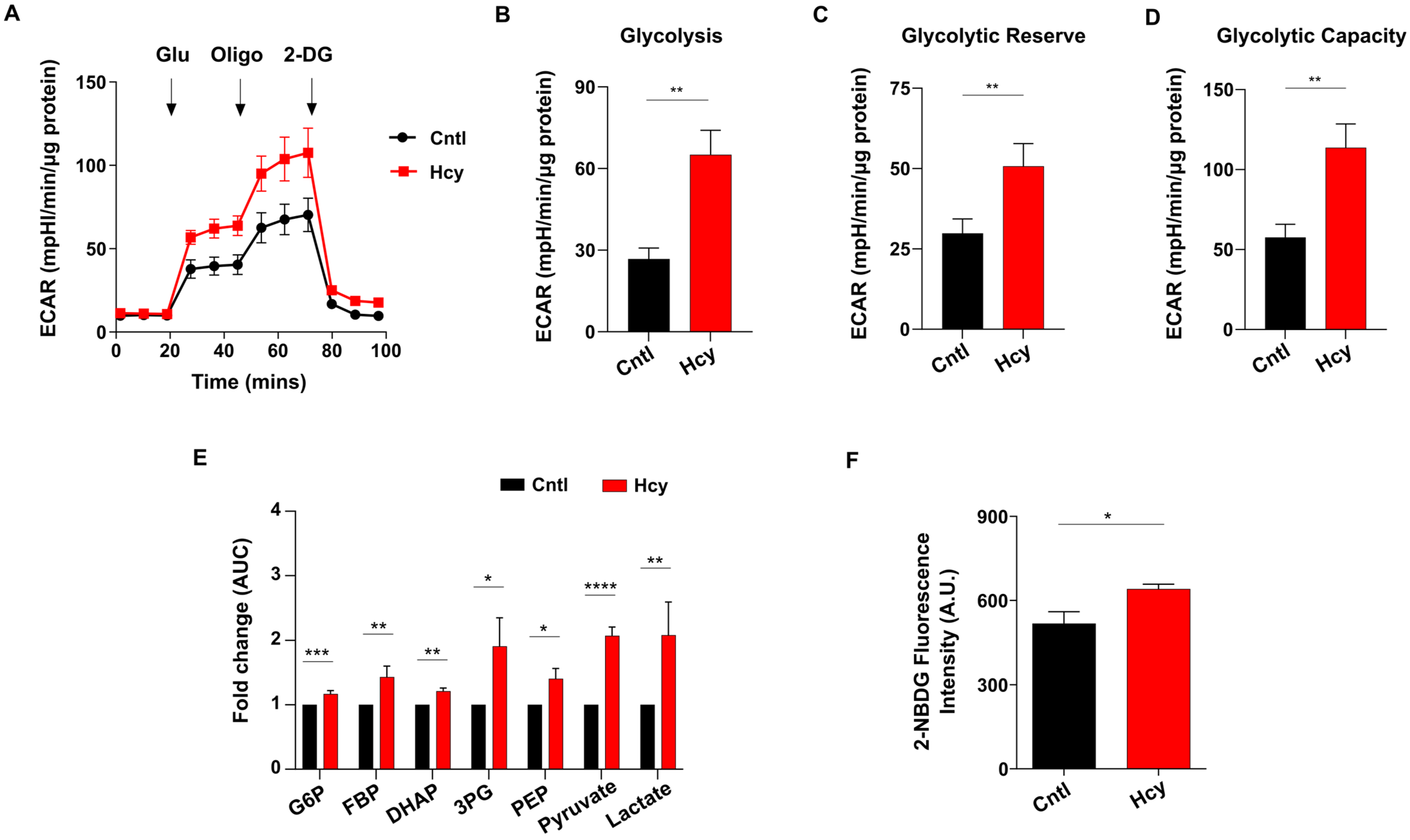
3.7. Endothelial Glycolysis Is Elevated upon Sub-Lethal HHcy Mediated Mitochondrial Respiration Defect
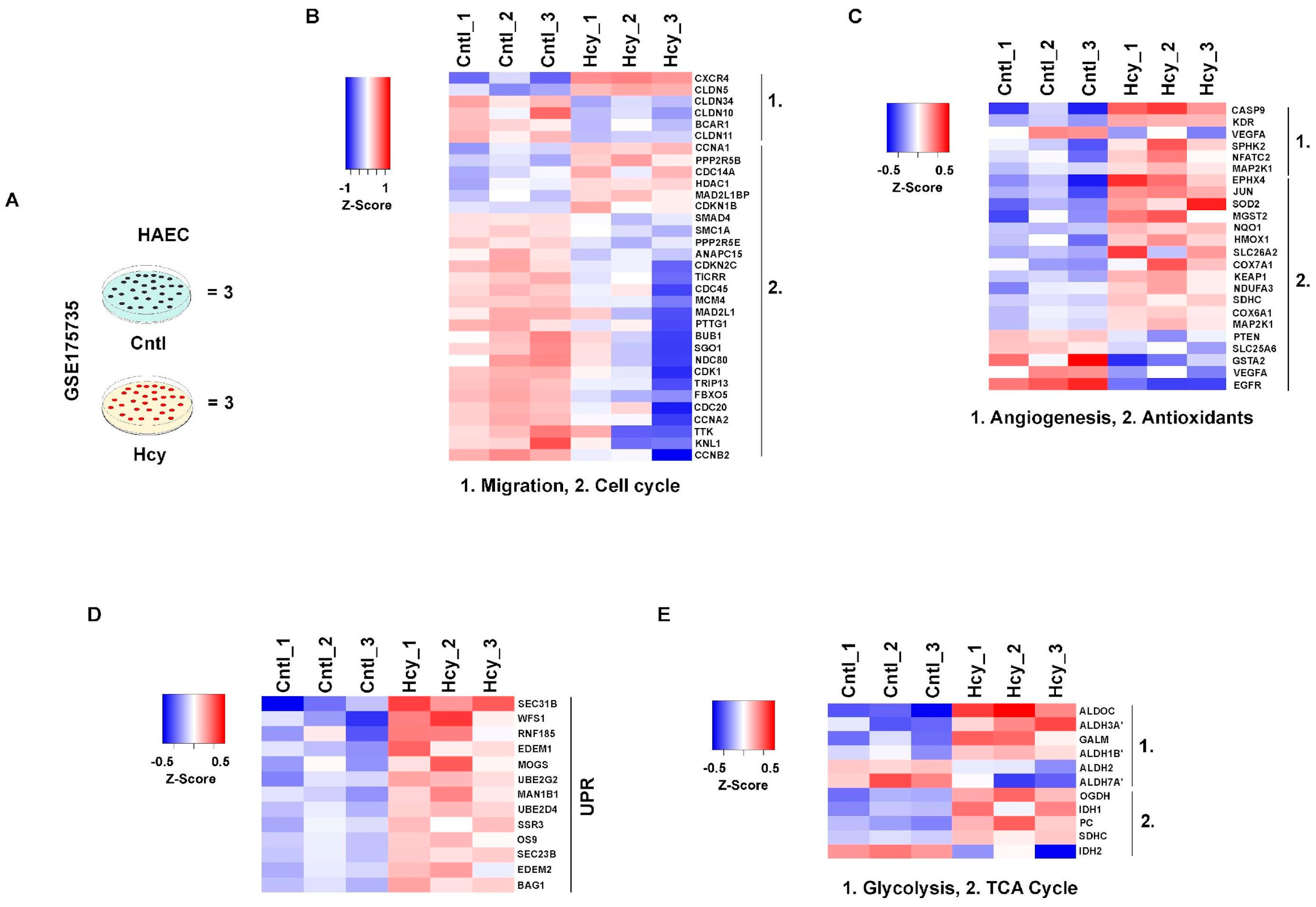
3.8. Mechanistic Signatures of Moderate HHcy Are Conserved in Adult Endothelial Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zaric, B.L.; Obradovic, M.; Bajic, V.; Haidara, M.A.; Jovanovic, M.; Isenovic, E.R. Homocysteine and Hyperhomocysteinaemia. Curr. Med. Chem. 2019, 26, 2948–2961. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, P.; Alam, S.F. Role of homocysteine in the development of cardiovascular disease. Nutr. J. 2015, 14, 6. [Google Scholar] [CrossRef] [PubMed]
- Troen, A.M. The central nervous system in animal models of hyperhomocysteinemia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 1140–1151. [Google Scholar] [CrossRef]
- Tinelli, C.; Di Pino, A.; Ficulle, E.; Marcelli, S.; Feligioni, M. Hyperhomocysteinemia as a Risk Factor and Potential Nutraceutical Target for Certain Pathologies. Front. Nutr. 2019, 6, 49. [Google Scholar] [CrossRef]
- McCully, K.S. Vascular pathology of homocysteinemia: Implications for the pathogenesis of arteriosclerosis. Am. J. Pathol. 1969, 56, 111–128. [Google Scholar]
- Kruger, W.D. Cystathionine β-synthase deficiency: Of mice and men. Mol. Genet. Metab. 2017, 121, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Li, W.; Wang, P.; Lv, X.; Gao, Y.; Huang, G. Folic acid inhibits homocysteine-induced cell apoptosis in human umbilical vein endothelial cells. Mol. Cell. Biochem. 2017, 444, 77–86. [Google Scholar] [CrossRef]
- Suhara, T.; Fukuo, K.; Yasuda, O.; Tsubakimoto, M.; Takemura, Y.; Kawamoto, H.; Yokoi, T.; Mogi, M.; Kaimoto, T.; Ogihara, T.; et al. Homocysteine Enhances Endothelial Apoptosis via Upregulation of Fas-Mediated Pathways. Hypertension 2004, 43, 1208–1213. [Google Scholar] [CrossRef]
- Tian, X.; Zhao, L.; Song, X.; Yan, Y.; Liu, N.; Li, T.; Yan, B.; Liu, B. HSP27 Inhibits Homocysteine-Induced Endothelial Apoptosis by Modulation of ROS Production and Mitochondrial Caspase-Dependent Apoptotic Pathway. BioMed Res. Int. 2016, 2016, 4847874. [Google Scholar] [CrossRef]
- Liu, S.; Sun, Z.; Chu, P.; Li, H.; Ahsan, A.; Zhou, Z.; Zhang, Z.; Sun, B.; Wu, J.; Xi, Y.; et al. EGCG protects against homocysteine-induced human umbilical vein endothelial cells apoptosis by modulating mitochondrial-dependent apoptotic signaling and PI3K/Akt/eNOS signaling pathways. Apoptosis 2017, 22, 672–680. [Google Scholar] [CrossRef]
- Gomez, J.; Sanchez-Roman, I.; Gomez, A.; Sanchez, C.; Suarez, H.; Lopez-Torres, M.; Barja, G. Methionine and homocysteine modulate the rate of ROS generation of isolated mitochondria in vitro. J. Bioenerg. Biomembr. 2011, 43, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Outinen, P.A.; Sood, S.K.; Liaw, P.C.Y.; Sarge, K.D.; Maeda, N.; Hirsh, J.; Ribau, J.; Podor, T.J.; Weitz, J.I.; Austin, R.C. Characterization of the stress-inducing effects of homocysteine. Biochem. J. 1998, 332 Pt 1, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wei, C.; Zhou, Y.; Yan, T.; Wang, Z.; Li, W.; Zhao, L. Homocysteine Induces Apoptosis of Human Umbilical Vein Endothelial Cells via Mitochondrial Dysfunction and Endoplasmic Reticulum Stress. Oxidative Med. Cell. Longev. 2017, 2017, 5736506. [Google Scholar] [CrossRef] [PubMed]
- Kil, J.-S.; Jeong, S.-O.; Chung, H.-T.; Pae, H.-O. Piceatannol attenuates homocysteine-induced endoplasmic reticulum stress and endothelial cell damage via heme oxygenase-1 expression. Amino Acids 2016, 49, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Wang, C.; Jin, Y.; Meng, Q.; Liu, Q.; Liu, Z.; Liu, K.; Liu, X.; Sun, H. Catalpol Inhibits Homocysteine-induced Oxidation and Inflammation via Inhibiting Nox4/NF-κB and GRP78/PERK Pathways in Human Aorta Endothelial Cells. Inflammation 2019, 42, 64–80. [Google Scholar] [CrossRef]
- Ji, C.; Yi, H.; Huang, J.; Zhang, W.; Zheng, M. Propofol alleviates inflammation and apoptosis in HCY-induced HUVECs by inhibiting endoplasmic reticulum stress. Mol. Med. Rep. 2021, 23, 333. [Google Scholar] [CrossRef] [PubMed]
- Guéant, J.-L.; Guéant-Rodriguez, R.-M.; Oussalah, A.; Zuily, S.; Rosenberg, I. Hyperhomocysteinemia in Cardiovascular Diseases: Revisiting Observational Studies and Clinical Trials. Thromb. Haemost. 2023, 123, 270–282. [Google Scholar] [CrossRef]
- Guieu, R.; Ruf, J.; Mottola, G. Hyperhomocysteinemia and cardiovascular diseases. Ann. Biol. Clin. 2022, 80, 7–14. [Google Scholar] [CrossRef]
- Abhinand, C.S.; Raju, R.; Soumya, S.J.; Arya, P.S.; Sudhakaran, P.R. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J. Cell Commun. Signal. 2016, 10, 347–354. [Google Scholar] [CrossRef]
- Nagai, Y.; Tasaki, H.; Takatsu, H.; Nihei, S.I.; Yamashita, K.; Toyokawa, T.; Nakashima, Y. Homocysteine Inhibits Angiogenesis in Vitro and in Vivo. Biochem. Biophys. Res. Commun. 2001, 281, 726–731. [Google Scholar] [CrossRef]
- Duan, J.; Murohara, T.; Ikeda, H.; Sasaki, K.-I.; Shintani, S.; Akita, T.; Shimada, T.; Imaizumi, T. Hyperhomocysteinemia Impairs Angiogenesis in Response to Hindlimb Ischemia. Arter. Thromb. Vasc. Biol. 2000, 20, 2579–2585. [Google Scholar] [CrossRef]
- Chang, P.-Y.; Lu, S.-C.; Lee, C.-M.; Chen, Y.-J.; Dugan, T.A.; Huang, W.-H.; Chang, S.-F.; Liao, W.S.; Chen, C.-H.; Lee, Y.-T.; et al. Homocysteine Inhibits Arterial Endothelial Cell Growth Through Transcriptional Downregulation of Fibroblast Growth Factor-2 Involving G Protein and DNA Methylation. Circ. Res. 2008, 102, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, L.; Wu, X.; Zhao, R.; Meng, Z.; Wang, K.; Wang, B.; Wang, H.; Shi, Z.; Li, G. Homocysteine inhibits the viability and migration ability of human umbilical vein endothelial cells by downregulating the expression of vascular endothelial growth factor. Exp. Ther. Med. 2019, 18, 3913–3919. [Google Scholar] [CrossRef]
- Rodríguez-Nieto, S.; Chavarría, T.; Martínez-Poveda, B.; Sánchez-Jiménez, F.; Rodríguez Quesada, A.; Medina, M.A. Anti-angiogenic effects of homocysteine on cultured endothelial cells. Biochem. Biophys. Res. Commun. 2002, 293, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Chakraborty, P.K.; Xiong, X.; Dwivedi, S.K.D.; Mustafi, S.B.; Leigh, N.R.; Ramchandran, R.; Mukherjee, P.; Bhattacharya, R. Cystathionine β-synthase regulates endothelial function via protein S-sulfhydration. FASEB J. 2016, 30, 441–456. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.-M.; Zhang, Q.; Li, Q.; Chen, Y.; Huang, X.; Yang, I.H.; Cao, L.; Wu, W.-K. Homocysteine-impaired angiogenesis is associated with VEGF VEGFR inhibition. Front. Biosci. Elite 2012, 4, 2525–2535. [Google Scholar] [CrossRef]
- Shastry, S.; Tyagi, N.; Hayden, M.R.; Tyagi, S.C. Proteomic analysis of homocysteine inhibition of microvascular endothelial cell angiogenesis. Cell. Mol. Biol. 2004, 50, 931–937. [Google Scholar] [PubMed]
- Roybal, C.N.; Yang, S.; Sun, C.-W.; Hurtado, D.; Jagt, D.L.V.; Townes, T.M.; Abcouwer, S.F. Homocysteine Increases the Expression of Vascular Endothelial Growth Factor by a Mechanism Involving Endoplasmic Reticulum Stress and Transcription Factor ATF4. J. Biol. Chem. 2004, 279, 14844–14852. [Google Scholar] [CrossRef]
- Tawfik, A.; Markand, S.; Al-Shabrawey, M.; Mayo, J.N.; Reynolds, J.; Bearden, S.E.; Ganapathy, V.; Smith, S.B. Alterations of retinal vasculature in cystathionine-β-synthase heterozygous mice: A model of mild to moderate hyperhomocysteinemia. Am. J. Pathol. 2014, 184, 2573–2585. [Google Scholar] [CrossRef]
- Leung, S.W.S.; Shi, Y. The glycolytic process in endothelial cells and its implications. Acta Pharmacol. Sin. 2022, 43, 251–259. [Google Scholar] [CrossRef]
- Bierhansl, L.; Conradi, L.-C.; Treps, L.; Dewerchin, M.; Carmeliet, P. Central Role of Metabolism in Endothelial Cell Function and Vascular Disease. Physiology 2017, 32, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Goveia, J.; Stapor, P.; Carmeliet, P. Principles of targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction in disease. EMBO Mol. Med. 2014, 6, 1105–1120. [Google Scholar] [CrossRef]
- Deng, J.; Lü, S.; Liu, H.; Liu, B.; Jiang, C.; Xu, Q.; Feng, J.; Wang, X. Homocysteine Activates B Cells via Regulating PKM2-Dependent Metabolic Reprogramming. J. Immunol. 2017, 198, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Diebold, L.P.; Gil, H.J.; Gao, P.; Martinez, C.A.; Weinberg, S.E.; Chandel, N.S. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat. Metab. 2019, 1, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- McCully, K.S. Homocysteine and vascular disease. Nat. Med. 1996, 2, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Lalwani, M.K.; Sharma, M.; Singh, A.R.; Chauhan, R.K.; Patowary, A.; Singh, N.; Scaria, V.; Sivasubbu, S. Reverse Genetics Screen in Zebrafish Identifies a Role of miR-142a-3p in Vascular Development and Integrity. PLoS ONE 2012, 7, e52588. [Google Scholar] [CrossRef]
- Rai, A.; Chatterjee, B.; Bhowmick, S.; Sagar, S.; Roy, S.S. Beclin 1 controls pigmentation by changing the nuclear localization of melanogenic factor MITF. Biochem. Biophys. Res. Commun. 2020, 528, 719–725. [Google Scholar] [CrossRef]
- Carpentier, G.; Berndt, S.; Ferratge, S.; Rasband, W.; Cuendet, M.; Uzan, G.; Albanese, P. Angiogenesis Analyzer for ImageJ—A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Sci. Rep. 2020, 10, 11568. [Google Scholar] [CrossRef]
- Sagar, S.; Faizan, I.; Chaudhary, N.; Singh, V.; Singh, P.; Gheware, A.; Sharma, K.; Azmi, I.; Singh, V.P.; Kharya, G.; et al. Obesity impairs cardiolipin-dependent mitophagy and therapeutic intercellular mitochondrial transfer ability of mesenchymal stem cells. Cell Death Dis. 2023, 14, 324. [Google Scholar] [CrossRef]
- Faizan, I.; Chaudhuri, R.; Sagar, S.; Albogami, S.; Chaudhary, N.; Azmi, I.; Akhtar, A.; Ali, S.M.; Kumar, R.; Iqbal, J.; et al. NSP4 and ORF9b of SARS-CoV-2 Induce Pro-Inflammatory Mitochondrial DNA Release in Inner Membrane-Derived Vesicles. Cells 2022, 11, 2969. [Google Scholar] [CrossRef]
- Jan, M.; Cueto, R.; Jiang, X.; Lu, L.; Sardy, J.; Xiong, X.; Yu, J.E.; Pham, H.; Khan, M.; Qin, X.; et al. Molecular processes mediating hyperhomocysteinemia-induced metabolic reprogramming, redox regulation and growth inhibition in endothelial cells. Redox Biol. 2021, 45, 102018. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Outinen, P.A.; Sood, S.K.; Pfeifer, S.I.; Pamidi, S.; Podor, T.J.; Li, J.; Weitz, J.I.; Austin, R.C. Homocysteine-Induced Endoplasmic Reticulum Stress and Growth Arrest Leads to Specific Changes in Gene Expression in Human Vascular Endothelial Cells. Blood 1999, 94, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Selhub, J. Homocysteine metabolism. Annu. Rev. Nutr. 1999, 19, 217–246. [Google Scholar] [CrossRef] [PubMed]
- Mudd, S.H.; Skovby, F.; Levy, H.L.; Pettigrew, K.D.; Wilcken, B.; E Pyeritz, R.; Andria, G.; Boers, G.H.; Bromberg, I.L.; Cerone, R. The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am. J. Hum. Genet. 1985, 37, 1–31. [Google Scholar]
- Kaur, B.; Sharma, P.K.; Chatterjee, B.; Bissa, B.; Nattarayan, V.; Ramasamy, S.; Bhat, A.; Lal, M.; Samaddar, S.; Banerjee, S.; et al. Defective quality control autophagy in Hyperhomocysteinemia promotes ER stress and consequent neuronal apoptosis through proteotoxicity. Cell Commun. Signal. 2023, 21, 258. [Google Scholar] [CrossRef] [PubMed]
- A Pascal, T.; E Gaull, G.; Beratis, N.G.; Gillam, B.M.; Tallan, H.H. Cystathionase Deficiency: Evidence for Genetic Heterogeneity in Primary Cystathioninuria. Pediatr. Res. 1978, 12, 125–133. [Google Scholar] [CrossRef]
- Gore, A.V.; Monzo, K.; Cha, Y.R.; Pan, W.; Weinstein, B.M. Vascular Development in the Zebrafish. Cold Spring Harb. Perspect. Med. 2012, 2, a006684. [Google Scholar] [CrossRef]
- Wang, X.-C.; Sun, W.-T.; Yu, C.-M.; Pun, S.-H.; Underwood, M.J.; He, G.-W.; Yang, Q. ER stress mediates homocysteine-induced endothelial dysfunction: Modulation of IKCa and SKCa channels. Atherosclerosis 2015, 242, 191–198. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, L.; Miao, Y.; Yang, J.; Wang, X.; Wang, C.-C.; Feng, J.; Wang, L. Homocysteine causes vascular endothelial dysfunction by disrupting endoplasmic reticulum redox homeostasis. Redox Biol. 2019, 20, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Lv, J.; Zhu, Y.; Duan, L.; Ma, L. Homocysteine Inhibits Hepatocyte Proliferation via Endoplasmic Reticulum Stress. PLoS ONE 2013, 8, e54265. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zhou, Y.; Xue, H.; Hou, H.; He, G.; Yang, Q. Endoplasmic reticulum stress mediates homocysteine-induced hypertrophy of cardiac cells through activation of cyclic nucleotide phosphodiesterase 1C. Acta Biochim. Biophys. Sin. 2022, 54, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Minerva Anestesiol. 1998, 12, 982–995. [Google Scholar] [CrossRef] [PubMed]
- Özcan, U.; Yilmaz, E.; Özcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical Chaperones Reduce ER Stress and Restore Glucose Homeostasis in a Mouse Model of Type 2 Diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]
- Schaks, M.; Giannone, G.; Rottner, K. Actin dynamics in cell migration. Essays Biochem. 2019, 63, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Lamalice, L.; Le Boeuf, F.; Huot, J. Endothelial Cell Migration During Angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef]
- Townsend, L.K.; Brunetta, H.S.; Mori, M.A.S. Mitochondria-associated ER membranes in glucose homeostasis and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E1053–E1060. [Google Scholar] [CrossRef]
- Markovinovic, A.; Greig, J.; Martín-Guerrero, S.M.; Salam, S.; Paillusson, S. Endoplasmic reticulum–mitochondria signaling in neurons and neurodegenerative diseases. J. Cell Sci. 2022, 135, jcs248534. [Google Scholar] [CrossRef]
- Quesada, A.R.; Muñoz-Chápuli, R.; Medina, M. Angiogenesis and signal transduction in endothelial cells. Cell. Mol. Life Sci. 2004, 61, 2224–2243. [Google Scholar] [CrossRef]
- Wierzbicki, A.S. Homocysteine and cardiovascular disease: A review of the evidence. Diabetes Vasc. Dis. Res. 2007, 4, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Cleophas, T.J.; Hornstra, N.; van Hoogstraten, B.; van der Meulen, J. Homocysteine, a risk factor for coronary artery disease or not? A meta-analysis. Am. J. Cardiol. 2000, 86, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Smulders, Y.M.; Blom, H.J. The homocysteine controversy. J. Inherit. Metab. Dis. 2011, 34, 93–99. [Google Scholar] [CrossRef]
- Toole, J.F.; Malinow, M.R.; Chambless, L.E.; Spence, J.D.; Pettigrew, L.C.; Howard, V.J. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: The Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA 2004, 291, 565–575. [Google Scholar] [CrossRef]
- Bønaa, K.H.; Njølstad, I.; Ueland, P.M.; Schirmer, H.; Tverdal, A.; Steigen, T.; Wang, H.; Nordrehaug, J.E.; Arnesen, E.; Rasmussen, K. Homocysteine Lowering and Cardiovascular Events after Acute Myocardial Infarction. N. Engl. J. Med. 2006, 354, 1578–1588. [Google Scholar] [CrossRef]
- Zou, T.; Liu, W.-J.; Li, S.-D.; Zhou, W.; Yang, J.-F.; Zou, C.-G. TRB3 mediates homocysteine-induced inhibition of endothelial cell proliferation. J. Cell. Physiol. 2010, 226, 2782–2789. [Google Scholar] [CrossRef]
- Eguchi, R.; Kawabe, J.-I.; Wakabayashi, I. VEGF-Independent Angiogenic Factors: Beyond VEGF/VEGFR2 Signaling. J. Vasc. Res. 2022, 59, 78–89. [Google Scholar] [CrossRef]
- Christensen, J.; Anderes, K. Beyond VEGF: Targeting tumor growth and angiogenesis via alternative mechanisms. Adv. Exp. Med. Biol. 2008, 610, 43–53. [Google Scholar]
- Muniyandi, A.; Hartman, G.D.; Song, Y.; Mijit, M.; Kelley, M.R.; Corson, T.W. Beyond VEGF: Targeting Inflammation and Other Pathways for Treatment of Retinal Disease. J. Pharmacol. Exp. Ther. 2023, 386, 15–25. [Google Scholar] [CrossRef]
- Dayal, S.; Lentz, S.R. Murine Models of Hyperhomocysteinemia and Their Vascular Phenotypes. Arter. Thromb. Vasc. Biol. 2008, 28, 1596–1605. [Google Scholar] [CrossRef]
- Tan, H.; Jiang, X.; Yang, F.; Li, Z.; Liao, D.; Trial, J.; Magera, M.J.; Durante, W.; Yang, X.; Wang, H. Hyperhomocysteinemia inhibits post-injury reendothelialization in mice. Cardiovasc. Res. 2006, 69, 253–262. [Google Scholar] [CrossRef]
- Morita, H.; Kurihara, H.; Yoshida, S.; Saito, Y.; Shindo, T.; Oh-Hashi, Y.; Kurihara, Y.; Yazaki, Y.; Nagai, R. Diet-Induced Hyperhomocysteinemia Exacerbates Neointima Formation in Rat Carotid Arteries After Balloon Injury. Circulation 2001, 103, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ma, J.; Xia, M.; Zhu, H.; Ling, W. Mild Hyperhomocysteinemia Induced by Feeding Rats Diets Rich in Methionine or Deficient in Folate Promotes Early Atherosclerotic Inflammatory Processes. J. Nutr. 2004, 134, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Böger, R.H.; Bode-Böger, S.M.; Sydow, K.; Heistad, D.D.; Lentz, S.R. Plasma Concentration of Asymmetric Dimethylarginine, an Endogenous Inhibitor of Nitric Oxide Synthase, Is Elevated in Monkeys With Hyperhomocyst(e)inemia or Hypercholesterolemia. Arter. Thromb. Vasc. Biol. 2000, 20, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Bowley, G.; Kugler, E.; Wilkinson, R.; Lawrie, A.; van Eeden, F.; Chico, T.J.A.; Evans, P.C.; Noël, E.S.; Serbanovic-Canic, J. Zebrafish as a tractable model of human cardiovascular disease. Br. J. Pharmacol. 2021, 179, 900–917. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Park, S.H.; Chung, J.F.; Choi, W.; Huh, H.K. Homocysteine-induced peripheral microcirculation dysfunction in zebrafish and its attenuation by L-arginine. Oncotarget 2017, 8, 58264–58271. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Prabhudesai, S.; Zhang, Y.; Rao, G.; Thirugnanam, K.; Hossen, N.; Dwivedi, S.K.D.; Ramchandran, R.; Mukherjee, P.; Bhattacharya, R. Cystathione β-synthase regulates HIF-1α stability through persulfidation of PHD2. Sci. Adv. 2020, 6, eaaz8534. [Google Scholar] [CrossRef]
- Ishii, I.; Akahoshi, N.; Yamada, H.; Nakano, S.; Izumi, T.; Suematsu, M. Cystathionine gamma-Lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J. Biol. Chem. 2010, 285, 26358–26368. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.-A. Sodium Phenylbutyrate and Ursodoxicoltaurine: First Approval. CNS Drugs 2022, 36, 1007–1013. [Google Scholar] [CrossRef]
- Huang, K.; Deng, R.; Liu, T.-C.; Gremida, A.; Deepak, P.; Chen, C.-H.; Davidson, N.; Kaufman, R.; Ciorba, M. A translational phase i study of tauroursodeoxycholic acid (tudca) to reduce symptoms and er stress in active ulcerative colitis. Inflamm. Bowel Dis. 2021, 27, S5–S6. [Google Scholar] [CrossRef]
- Stroka, K.M.; Vaitkus, J.A.; Aranda-Espinoza, H. Endothelial cells undergo morphological, biomechanical, and dynamic changes in response to tumor necrosis factor-α. Eur. Biophys. J. 2012, 41, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Henriquez, D.R.; Cánovas, J.; Villarroel-Campos, D.; Carreras-Sureda, A.; Pulgar, E.; Molina, E.; Hazari, Y.M.; Limia, C.M.; Alvarez-Rojas, S.; et al. IRE1α governs cytoskeleton remodelling and cell migration through a direct interaction with filamin A. Nat. Cell Biol. 2018, 20, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Pasini, S.; Liu, J.; Corona, C.; Peze-Heidsieck, E.; Shelanski, M.; Greene, L.A. Activating Transcription Factor 4 (ATF4) modulates Rho GTPase levels and function via regulation of RhoGDIα. Sci. Rep. 2016, 6, 36952. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Missiaen, R.; Morales-Rodriguez, F.; Eelen, G.; Carmeliet, P. Targeting endothelial metabolism for anti-angiogenesis therapy: A pharmacological perspective. Vasc. Pharmacol. 2017, 90, 8–18. [Google Scholar] [CrossRef]
- Potente, M.; Carmeliet, P. The Link Between Angiogenesis and Endothelial Metabolism. Annu. Rev. Physiol. 2017, 79, 43–66. [Google Scholar] [CrossRef]
- Annunziata, I.; Sano, R.; D’azzo, A. Mitochondria-associated ER membranes (MAMs) and lysosomal storage diseases. Cell Death Dis. 2018, 9, 328. [Google Scholar] [CrossRef]
- Inigo, M.; Deja, S.; Burgess, S.C. Ins and Outs of the TCA Cycle: The Central Role of Anaplerosis. Annu. Rev. Nutr. 2021, 41, 19–47. [Google Scholar] [CrossRef]
- Merchan, J.R.; Kovács, K.; Railsback, J.W.; Kurtoglu, M.; Jing, Y.; Piña, Y.; Gao, N.; Murray, T.G.; Lehrman, M.A.; Lampidis, T.J.; et al. Antiangiogenic activity of 2-deoxy-D-glucose. PLoS ONE 2010, 5, e13699. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatterjee, B.; Fatima, F.; Seth, S.; Sinha Roy, S. Moderate Elevation of Homocysteine Induces Endothelial Dysfunction through Adaptive UPR Activation and Metabolic Rewiring. Cells 2024, 13, 214. https://doi.org/10.3390/cells13030214
Chatterjee B, Fatima F, Seth S, Sinha Roy S. Moderate Elevation of Homocysteine Induces Endothelial Dysfunction through Adaptive UPR Activation and Metabolic Rewiring. Cells. 2024; 13(3):214. https://doi.org/10.3390/cells13030214
Chicago/Turabian StyleChatterjee, Barun, Fabeha Fatima, Surabhi Seth, and Soumya Sinha Roy. 2024. "Moderate Elevation of Homocysteine Induces Endothelial Dysfunction through Adaptive UPR Activation and Metabolic Rewiring" Cells 13, no. 3: 214. https://doi.org/10.3390/cells13030214
APA StyleChatterjee, B., Fatima, F., Seth, S., & Sinha Roy, S. (2024). Moderate Elevation of Homocysteine Induces Endothelial Dysfunction through Adaptive UPR Activation and Metabolic Rewiring. Cells, 13(3), 214. https://doi.org/10.3390/cells13030214