Abstract
Integrin receptors are heterodimeric surface receptors that play multiple roles regarding cell–cell communication, signaling, and migration. The four members of the β2 integrin subfamily are composed of an alternative α (CD11a–d) subunit, which determines the specific receptor properties, and a constant β (CD18) subunit. This review aims to present insight into the multiple immunological roles of integrin receptors, with a focus on β2 integrins that are specifically expressed by leukocytes. The pathophysiological role of β2 integrins is confirmed by the drastic phenotype of patients suffering from leukocyte adhesion deficiencies, most often resulting in severe recurrent infections and, at the same time, a predisposition for autoimmune diseases. So far, studies on the role of β2 integrins in vivo employed mice with a constitutive knockout of all β2 integrins or either family member, respectively, which complicated the differentiation between the direct and indirect effects of β2 integrin deficiency for distinct cell types. The recent generation and characterization of transgenic mice with a cell-type-specific knockdown of β2 integrins by our group has enabled the dissection of cell-specific roles of β2 integrins. Further, integrin receptors have been recognized as target receptors for the treatment of inflammatory diseases as well as tumor therapy. However, whereas both agonistic and antagonistic agents yielded beneficial effects in animal models, the success of clinical trials was limited in most cases and was associated with unwanted side effects. This unfavorable outcome is most probably related to the systemic effects of the used compounds on all leukocytes, thereby emphasizing the need to develop formulations that target distinct types of leukocytes to modulate β2 integrin activity for therapeutic applications.
Keywords:
αβ integrins; leukocyte receptors; β2 integrins; LFA-1; MAC-1; LAD-1; migration; phagocytosis; NETosis; reactive oxygen species 1. Introduction
The αβ integrin receptor family is a large group of heterodimeric surface receptors that bind various cell surface receptors and components of the extracellular matrix (ECM) to support cell–cell interaction and migration and are involved in cell differentiation and signaling processes [1]. Within this family, the β2 integrin subfamily is characterized by leukocyte-restricted expression [2], and its four family members exert multiple regulatory functions within the innate and adaptive immune systems [3,4].
Integrins are a large family of evolutionarily conserved heterodimeric surface receptors that are composed of an α and a β chain [1]. Integrin αβ heterodimers are divided into four classes based on ligand specificity and evolutionary origin, namely leukocyte, collagen-binding, Arg-Gly-Asp (RGD)-binding, and laminin-binding integrins (Figure 1) [5]. In vertebrates, 18 different integrin α subunits and 8 β subunits can be expressed [1,6,7], whereas β1, β2, and αV-containing integrins constitute the largest three groups of this family [6,8]. To date, in total, 24 β integrin combinations are known to be expressed by humans [5].
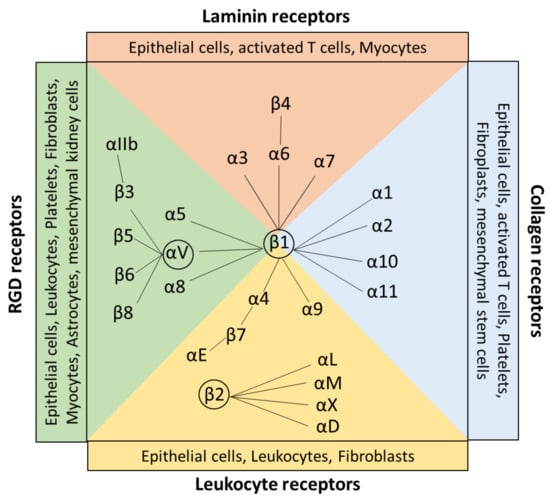
Figure 1.
Classification and expression pattern of the αβ integrin receptor family. Integrin heterodimers consist of different combinations of the α and β subunits. In general, integrins are classified by their ligand specificity in RGD-recognizing integrins (α5β1, αVβ1, αVβ3, αVβ5, αVβ6, αVβ8, and αllbβ3) shown in green, laminin-binding integrins (α3β1, α6β1, α7β1, and α6β4) shown in red, collagen-binding integrins (α1β1, α2β1, α10β1, and α11β1) shown in blue, and leukocyte integrins (α9β1, α4β1, αEβ7, αLβ2, αMβ2, αXβ2, and αDβ2) shown in yellow. The β1, β2, and αV containing integrins built the largest groups of the family. All integrins are expressed by a wide range of cells, including, for example, leukocytes, epithelial cells, and fibroblasts. Created with BioRender.com.
Usually, the ligand binding ability of αβ integrins is conferred by recognizing small peptide sequences of the ECM and both cell surface receptors and serum components [9]. Once they have bound to a target peptide, integrins provide adhesion and initial signaling, allowing cells to respond to mechanical or chemical factors [9].
Target sequences are the tripeptide motif Arg-Gly-Asp (RGD), triple-helical GFOGER peptides, two homologous sequences Leu-Asp-Val (LDV), and Ile-Asp-Ala-Pro (IDAP) [7,10]. RGD binds to αvβ1, αvβ3, αvβ5, αvβ6, αvβ8, α5β1, α8β1, and αIIbβ3. RGD receptors are expressed by a wide range of cell types, including, for example, astrocytes, leukocytes, and fibroblasts [11]. GFOGER =0binding integrins are collagen receptors, including α1β1, α2β1, α10β1, and α11β1 integrins, which are expressed by fibroblasts, stem cells, and epithelial cells. The tripeptide LDV is recognized by the leukocyte receptors α4β1, α4β7, and β2 subunits, which are expressed mainly on leukocytes and epithelial cells [11] (Figure 1). The target sequence IDAP is, besides LDV and RGD, a peptide sequence apparent in fibronectin and is a recognition site for α4β1 [10].
This review aims to provide an overview of the role of αβ integrins on immune cells, with a focus on β2 integrins for the maintenance of immune homeostasis and the induction and course of immune responses, including infections, autoimmunity, and tumorigenesis. The importance of integrins on malignant cells for tumorigenesis has recently been well reviewed in [12]. The pathophysiological importance of β2 integrins is confirmed by the phenotype of patients lacking integrin activity who suffer from recurrent severe infections but are also prone to developing autoimmune diseases. Similar defects have been recapitulated in appropriate mouse models used to thoroughly delineate the immunological functions of β2 integrins in health and disease. In accordance, a number of agonists and antagonists of integrin receptors have been developed and evaluated for therapeutic applications.
2. αβ Integrin Immune Functions and Targeted Therapies
2.1. RGD-Binding Integrins
The RGD subfamily comprises a large group of integrins that interact with many different ligands, as reviewed in [13], including ECM components such as collagen, fibronectin, vitronectin, and tenascin [14] as well as surface receptors like E-cadherin. RGD receptors are essential for vascular smooth muscle cell migration, phagocytosis, and the activation of TGF-β, which regulates innate immunity and anti-inflammatory surveillance [11]. Most of the eight RGD-binding integrins are expressed in various tumor types, regulating pathophysiological processes like tumor invasion and metastasis formation [15]. However, they are also known to be involved in other diseases like sepsis [16] as a systemic response to infection that may be fatal due to organ failure [17], fibrosis [18] that is caused by inflammation-induced excessive release of fibronectin-forming fibrils to enable collagen deposition [19,20], and viral infections [21]. In this regard, integrin targeting is highly appealing for the delivery of therapeutics. For example, RGD-loaded exosomes were shown to yield therapeutic effects in breast cancer models after intravenous injections [13,22]. Furthermore, preclinical studies showed that RGD integrin antagonists, such as RGD-based cyclic peptides, efficiently suppressed tumor angiogenesis [23]. The most prominent example is cilengitide, an αvβ3/αvβ5 antagonist employed for the treatment of glioblastoma, which, however, failed its clinical endpoint [13]. Interestingly, the administration of low-dose cilengitide in vitro and in animal models revealed that it promoted tumor angiogenesis instead of inhibiting it [24]. So far, no successful RGD-targeted peptide therapy has been made available [13].
2.2. Laminin-Binding Integrins
Laminin receptors play critical roles in regulating cell adhesion, proliferation, migration, and survival, and they are important for organ development [11]. Similar to RGD-binding integrins, laminin receptor expression has also been shown to correlate with tumor progression [25]. Moreover, mutations in the receptor α6β4 resulted in junctional epidermolysis bullosa, a blistering skin disease [26]. So far, no specific therapy targeting laminin-binding integrins is available.
2.3. Collagen-Binding Integrins
The collagen receptor α1β1, also known as very late antigen 1 (VLA-1), was first identified in activated T cells [27]. VLA-1 acts as a promoter of inflammatory responses, and it facilitates monocyte transmigration by binding collagen XIII [11]. SAN-300 is a monoclonal antibody (mAb) directed against α1β1 integrin and was evaluated in a clinical phase II study for the treatment of patients with rheumatoid arthritis (RA) [11]. Furthermore, in animal disease models, inhibiting the interaction of α1β1, integrin, and collagen impaired T cell accumulation in the epidermis and consequently prevented psoriasis [28]. The integrin α2β1, also known as VLA-2, is expressed on platelets and mediates collagen binding at sites of inflammation. In this regard, vatelizumab, a mAb targeting the α2 subunit (CD49b) of VLA-2, was investigated in a clinical phase II study in multiple sclerosis (MS) patients [29]. After treatment with vatelizumab, higher frequencies of regulatory T cells (Treg) were observed in MS patients, which might have resulted from the inhibition of p38 mitogen-activated protein kinase (MAPK) signaling that is critically involved in the polarization of T helper (TH)17 cells and is activated by the α2 integrin cytoplasmic domain. Even though the clinical trial did not reach the end point, the blockade of VLA-2 may serve to shift the TH17/Treg polarization balance toward Treg [29].
2.4. Leukocyte Receptors
Leukocyte receptors play critical roles in leukocyte recruitment and (auto-)inflammatory diseases. The leukocyte receptor’s binding integrin subfamily comprises β2 integrins, which are discussed in more detail in the following section, and the integrins α4β1 (VLA-4; CD49d/CD29) and α4β7 [2]. The α4β1 integrin was shown to be necessary for leukocyte recruitment to the inflamed central nervous system (CNS) in experimental autoimmune encephalomyelitis (EAE), a murine MS model [30]. Its interaction with VCAM (vascular cell adhesion molecule)-1 mediated leukocyte adherence in the CNS. This observation prompted the development of Natalizumab, an IgG4 mAb directed against α4, which blocks α4β1 integrin and has been approved for MS treatment [31]. Hence, Natalizumab constitutes the first targeted therapy for the treatment of relapsing-remitting MS [2].
Furthermore, Vedolizumab and Etrolizumab also target α4β7 integrin [2,32]. Vedolizumab is a humanized IgG1 mAb that proved to be effective for the treatment of moderate-to-severe Crohn’s disease [2]. Etrolizumab is a humanized mAb that selectively binds the β7-subunit of α4β7 and αEβ7 integrin heterodimers, thereby antagonizing α4β7/Mucosal address cell adhesion molecule (MAdCAM-)1-mediated lymphocyte recruitment and αEβ7/E-cadherin interaction [33]. Etrolizumab yielded therapeutic effects in patients with severe inflammatory bowel disease (IBD) [34]. Moreover, Kunkel et al., using β7−/− mice, demonstrated that α4β7 integrin was involved in the adhesion of leukocytes to Peyer’s patch endothelial venules [35]. Treatment with β7- and MAdCAM-1-specific mAb relieved experimental murine chronic colitis [36].
In addition, the α4β1 integrin was shown to play a critical role in hematopoietic stem cell (HSC) homing. Its binding to VCAM-1 caused HSC retention in the bone marrow (BM) [9]. The importance of α4β1 integrin for HSC biology has been confirmed by Rettig et al., showing that α4 integrin knockout mice presented with elevated numbers of HSC in the bloodstream compared to wild-type littermates [37]. In accordance, treatment of wild-type mice with the VCAM-1 antagonist Bortezomib increased HSC migration [38]. Therefore, Bortezomib is of great interest to enrich and isolate HSC from the blood of healthy patients for use in transplantations [9].
3. The β2 Integrin Family
3.1. Family Members and Their Ligands
β2 integrins belong to the leukocyte receptor subfamily of αβ integrins, and their expression is confined to leukocytes [3,8]. These heterodimeric surface receptors are composed of a variable α (CD11a-CD11d) and a common β2 (CD18) subunit, thereby forming CD11a/CD18 (αLβ2, LFA-1), CD11b/CD18 (αMβ2, MAC-1, complement receptor 3 (CR3)), CD11c/CD18 (αXβ2, p150.95, and CR4), and CD11d/CD18 (αDβ2) (Figure 2). β2 integrins commonly bind ligands via the αI domain [4], and the α subunit of each β2 integrin determines the extracellular ligand specificity to different ligands. These comprise a heterogenous group, including, for example, intracellular adhesion molecules (ICAM), VCAM, and endothelial cell-specific molecules (ESMs), as well as other cell surface and serum molecules [3,5,39].
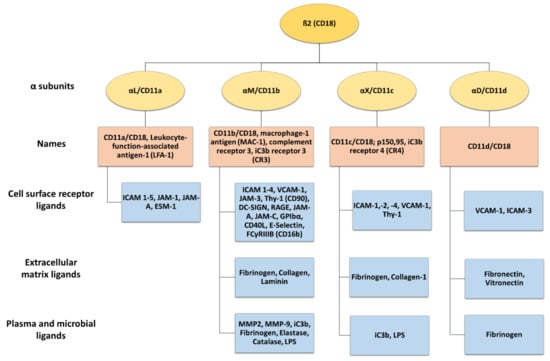
Figure 2.
The β2 integrin receptor subfamily and their ligands. The common β2 integrin subunit (CD18) can pair with one of four α subunits (αL-CD11a, αM-CD11b, αX-CD11c, and αD-CD11d), forming LFA-1, MAC-1, complement receptor 4 (150.95/CR4), and CD18/CD11d, respectively. LFA-1 is the only β2 integrin expressed on T cells, while CD11b/CD18, CD11c/CD18, and CD11d/CD18 are also expressed on myeloid cells. β2 integrins can interact with a wide range of ligands, including cell surface receptors, extracellular matrix ligands, plasma, and microbial ligands. Created with BioRender.com.
LFA-1 is expressed by all types of leukocytes [40] and in platelets [41]. Activated LFA-1 engages ICAM 1–5, junctional adhesion molecule (JAM-)1, JAM-A, and ESM-1 [8,42,43]. Macrophage-1 antigen (MAC-1) is predominantly expressed by polymorphonuclear neutrophils (PMNs), monocytes, macrophages, and dendritic cells (DCs) [8,44]. MAC-1 binds many ligands, including surface receptors like ICAM 1–4, VCAM-1, JAM-3, receptor for advanced glycation end products (RAGE), thymus cell antigen 1 (Thy-1, CD90), platelet glycoprotein Ibα, DC-specific ICAM-3-grabbing non integrin (DC-SIGN) [8,45], E-selectin, and many others [46,47,48,49,50,51] (Figure 2). In addition, MAC-1 engages several matrix proteins like fibronectin and collagen [52,53], plasma proteins including fibrinogen, elastase, matrix metalloproteinase-2 (MMP-2), and MMP-9, and microbial ligands like lipopolysaccharide (LPS) [46]. Furthermore, MAC-1 is also known as a complement receptor 3 (CR3) and binds iC3b, facilitating the phagocytosis of complement-opsonized pathogens [54,55].
CD11c is a classical marker for murine DC, but in humans, it is also expressed by other myeloid cell types like monocytes/macrophages and PMN [56]. CD11c engages with diverse ligands such as ICAM-1, -2, and -4 [57,58,59,60], bacterial components like LPS [61], iC3b, as well as fibrinogen and collagen [62,63]. In addition, CD11c strongly binds VCAM-1, and as shown for monocytes, this serves to stop leukocyte rolling on endothelia, enabling transmigration through inflamed endothelial cells of the aorta [60].
CD11d/CD18 is the most recently discovered β2 integrin and is expressed under basal conditions by a few types of leukocytes, but it is most abundant in macrophages. In humans, CD11d/CD18 is highly expressed on natural killer (NK) cells, B cells, and γδ T cells [8,64]. It binds to cellular receptors like VCAM-1 and ICAM-3 [65], and, like MAC-1m to matrix proteins including fibronectin, vitronectin, and fibrinogen [4].
The expression of all β2 integrins is often upregulated in the case of cell activation [8], but as outlined in Section 3.2, their ligand affinity is controlled by signaling-dependent conformational changes.
3.2. Structure and Activation of β2 Integrins
The various α (CD11a–d) and the common β subunit (CD18) of β2 integrins are each composed of a long extracellular, a transmembrane, and a cytoplasmic domain [66] (Figure 3A). In the following section, the structure and mode of activation of β2 integrins are presented in more detail, referring often to LFA-1 as the common β2 integrin of all leukocyte populations.
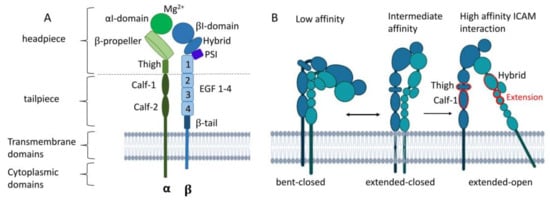
Figure 3.
Structure of LFA-1 and conformational changes during integrin activation. (A) LFA-1 can be divided into three domains: an extracellular domain with a headpiece and tailpiece, a transmembrane, and cytoplasmic domain. (B) LFA-1 undergoes three conformational changes from bent-closed (low ICAM affinity) toward extended-closed with intermediate ICAM affinity state toward extended-open with high ICAM affinity. Upon ligand binding, the headpiece swings out of the hybrid domain with intermediate affinity. The extension between the thigh and calf-1 domain of the α subunit and the hybrid and EGF1–2 of the β domain (shown in red) results in the extended-one conformation with high ligand affinity. PSI, plexin–semaphorin–integrin. Created with BioRender.com.
3.2.1. The α and β Subunit
The extracellular domain of the α subunit is comprised of a globular headpiece and a tailpiece. The former is composed of the αI unit, the β propeller, and the thigh domain. The lower tailpiece domain consists of the calf-1 and calf-2 regions [67,68]. By expanding the cleft between the β propeller and the αI domain, LFA-1 can interact with larger ligands. The top of the αI domain contains a metal ion-dependent adhesion site (MIDAS) that binds magnesium (Mg2+) to coordinate glutamic acid residues of ICAM-1 [69], which is required for conformational changes and thereby integrin activation [67,68].
The β subunit is connected to the cytoskeleton and transmits outside and inside signals [8]. The headpiece and the tailpiece of the β subunit consist of the βI domain, a so-called hybrid domain, a plexin–semaphorin–integrin (PSI) component, four epidermal growth factor (EGF)-like domains, and a β-tail. The βI domain also contains a MIDAS, which forms a bridge with a ligand or the glutamate residues of the αI domain [8,67,68].
3.2.2. Activation of LFA-1
β2 integrins in the bent-closed conformation display interactions between the integrin headpiece and tailpiece and display only a low ligand affinity (Figure 3B). Upon ligand binding, LFA-1 undergoes sequential conformational changes, comprising straightening of the headpiece and tailpiece, yielding an extended-closed state as an intermediate. The tailpiece extends and opens the headpiece, with the hybrid domain swinging out. The extension movements of the ecto-domains occur between the thigh and calf-1 of the α subunit and between I-EGF1 and I-EGF2 of the β subunit. When the αI domain engages ICAM-1, a glutamic acid residue in the linker region becomes available to bind the βI MIDAS region [67,68]. Remodeling of the MIDAS within the βI domain and a ‘swing-out’ of the hybrid domain result in the extended-open state with a high ligand affinity [68,70,71]. Inside the cell, β2 integrin-binding adapter proteins like Talin-1 and Kindlin-3 that are attached to the cytoplasmic domain of CD18 cause the acquisition of the high-affinity state of β2 integrins [11]. At that state, β2 integrins can cluster and contribute to the formation of adhesion complexes. Integrin engagement and activation lead to bidirectional mechanotransduction and signaling across the plasma membrane of the cells.
3.2.3. Inside-Out Signaling
The multistep activation process from bent-close to extended-open, including the process through which intracellular signals induce integrin activation that favors its extension, is termed ‘inside-out’ signaling [11,72]. Inside-out signaling is mediated, for example, by cytokine/chemokine stimulation and T cell receptor (TCR) activation, leading to downstream signaling and cytoskeletal rearrangements [73] (Figure 4A).
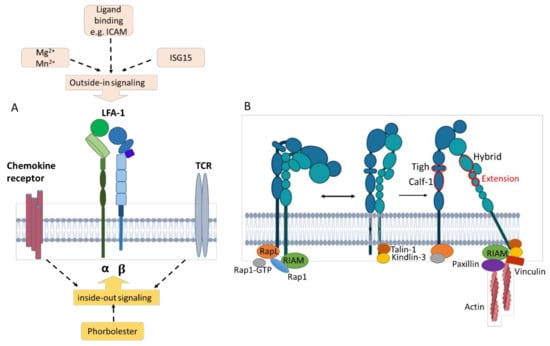
Figure 4.
Triggering bidirectional inside-out and outside-in signals of LFA-1. (A) Inside-out signaling is triggered, e.g., by chemokine/cytokine signaling and TCR stimulation from within the cell, resulting in LFA-1 activation. Outside-in signaling is induced by ligand binding such as ICAM-1 as well as interferon-stimulated gene 15 and divalent cations (Mg2+, Mn2+). (B) Inside-out signaling through chemokine or TCR stimulation recruits Rap1-GTP. RAPL engages CD11a and activates Rap-1. RIAM engages Rap-1 and the β subunit. The β subunit binds talin-1 and kindlin-3, stabilizing the intermediate- and high-affinity states. Additional binding of paxillin and vinculin forms a frame for interaction with cytoskeletal elements. Outside-in signaling activates adapter proteins such as talin-1, which mediate downstream signaling and mechanotransduction. Created with BioRender.com.
Inside-out triggering results in the recruitment of Rap1-GTP [8,72]. Rap-1 activation is mediated by the regulator of adhesion and polarization enriched in lymphocyte (RAPL) molecules that engage CD11a. RIAM (Rap-1-GTP interacting adaptor molecule) engages with both active Rap-1 and the integrin β subunit [74] and binds talin-1 and kindlin-3 [8,75], which stabilizes the intermediate and high-affinity receptor states by binding to different membrane motifs of the β subunit [11,75,76] (Figure 4B). Talin is the main intracellular binding protein that disrupts the interaction between the α and the β integrin subunits, thereby extending the tailpiece of the β subunit [77]. The interaction between talins and F-actin is important for anchoring LFA-1 to the actin cytoskeleton [76]. Additional binding of paxillin and vinculin forms a frame for interaction with cytoskeletal elements [75] since both constitute adaptor proteins with multiple binding sites for other adhesion components like focal adhesion kinase (FAK). FAK is a cytoplasmic tyrosine kinase that is activated by intramolecular interactions and, in turn, phosphorylates paxillin, promoting the exposure of additional protein docking sites that can regulate downstream processes such as migration and chemotaxis [11].
3.2.4. Outside-In Signaling
The rearrangement of the cytoskeleton and intracellular responses due to β2 integrin binding to ligands induce outside-in signaling [78] (Figure 4A). This leads to the acquisition of a high-affinity state [73,79]. Engagement of ICAM and binding of soluble extracellular interferon-stimulated gene 15 (ISG15), an ubiquitin-like secreted protein, but also divalent cations such as Mg2+ promote outside-in signaling by binding the αI domain [75,80]. Both ISG15 [80] and LFA-1 cross-linking [81] have been reported to induce IFN-γ production. Outside-in signaling modulates gene expression, cell proliferation, survival, and apoptosis [11,82].
In response to activation, cytoskeletal proteins and kinases, as well as key adapters, assemble at the cell membrane, forming adhesion complexes that transfer signals from the ECM to the cell surface [11]. Adapter proteins in LFA-1-mediated signal transduction pathways can be differentiated as structural, scaffolding, and catalytic adaptors. Structural adaptors such as talin-1 recruit the protein complex Arp2/3 via association with vinculin, and this protein complex facilitates cytoskeletal reorganization. This so-called ‘molecular clutch’ is crucial for the mechanotransduction and activation of the cell [11,78]. Scaffolding adaptors like adhesion and degranulation-promoting adapter proteins couple LFA-1 and regulate F-actin clustering, cell polarization, and T-cell motility. Catalytic adaptors facilitate specific reactions like targeting protein kinase A and protein kinase C (PKC) to the membrane, cytoskeleton, and filopodia of migrating T cells [78].
3.3. Multifacetted Functions of β2 Integrins
Leukocytes are characterized by a cell-type-specific expression pattern of β2 integrins that may be altered, for example, in response to activation [8] and aging [83]. Whereas β2 integrins exert comparable functions in distinct cell types, like rolling on endothelial cells and phagocytosis of opsonized pathogens, they exert other functions in a more cell-type-restricted manner, such as, for example, controlling reactive oxygen species (ROS) production and the release of neutrophil extracellular traps (NETs). In the following section, general as well as more cell-type-specific properties of β2 integrins are discussed.
3.3.1. Functions of β2 Integrins
Trafficking
Activated β2 integrins are crucial for the adhesion and trafficking of leukocytes from the bloodstream to sites of tissue inflammation by binding proteins of the ECM [5]. Leukocyte adhesion comprises multiple steps, including rolling along endothelia, adhesion, arrest, crawling, and finally, extravasation along a chemokine gradient to navigate to inflammatory sites [84]. Ligand binding to selectins and ICAMs induces β2 integrin clustering and the formation of multi-protein complexes that consist of intracellular signaling and adaptor proteins that connect β2 integrins with the cytoskeleton [85]. Leukocyte rolling is mediated by glycoproteins on the leukocyte cell surface, such as P-selectin glycoprotein ligand-1, which binds to endothelial selectins [9]. Chemotactic migration mediated by β2 integrins activates the p38 MAPK and phosphoinositide 3-kinase (PI3K) pathways. Chemokine stimulation enables β2 integrins to undergo a change in their conformation, thereby acquiring a high-affinity state. This leads to the binding of ICAMs on endothelial cells, resulting in a migration arrest [9,84,86]. LFA-1 and MAC-1 not only bind ICAM but also other surface receptors like RAGE and JAM receptors [45], and MAC-1 also engages CD90 [87], and all of these contacts contribute to trans-epithelial migration. Subsequently, integrin–ligand bonds are broken, reformed, and stabilized by cytoskeletal proteins such as talin and kindlin-3, which results in actin reorganization and spreading of the cells and, finally, leukocyte transmigration through the endothelium [3].
Regarding PMN migration, LFA-1 regulates adhesion to the endothelium, and MAC-1 facilitates intravascular crawling [88]. In the case of monocytes and lymphocytes, migration is mediated by LFA-1 [89]. Human monocytes, monocyte-derived macrophages, and monocyte-derived DC express both LFA-1 and MAC-1. Under physiological conditions, CD11c/CD18 was reported to be dominant, conferring adhesion to fibrinogen [56]. CD11d/CD18 and α4β1 were shown to be essential for leukocyte arrest during extravasation by binding to VCAM-1. Under steady-state conditions, CD11d/CD18 is not highly expressed and consequently might play a minor role in VCAM-1-mediated extravasation compared to α4β1 integrin. In contrast, under inflammatory conditions, CD11d/CD18 is highly expressed on human peripheral leukocytes and murine macrophage populations, which reflects its role in regulating, e.g., extravasation [4].
Phagocytosis
Phagocytosis is a unique form of cell endocytosis whereby cells internalize solid particles through vesicles, including microbial pathogens [90,91,92]. The complement receptors MAC-1 (CR3) and CD11c/CD18 (CR4) are essential for the phagocytosis of complement-opsonized pathogens by myeloid cell types via β2 integrin-induced Rho activity [3,93,94]. MAC-1 and CD11c/CD18 play important roles in the clearance of pathogens, cellular debris, and apoptotic cells opsonized with complementary factors, C3 and C4, respectively [8,95]. Moreover, apoptotic bodies derived from tumor cells can be taken up by MAC-1 on myeloid cells, NK cells, conventional (c)DC, and via CD36 by all DC populations, which in either case induces tolerance [96]. In addition, MAC-1 and CD11c/CD18 recognize iC3b-opsonized apoptotic cells, which attenuated pro-inflammatory cytokine production through nuclear factor kappa of activated B cells (NF-κB) inhibition [97].
Immunological Synapse
Besides their role in migration, LFA-1 and MAC-1 are also important constituents of the contact areas between antigen-presenting cells (APCs) and T cells required to confer T cell activation [98] and between immune effector cells and target cells to mediate cytotoxicity [99], generally termed the immunological synapse (IS). As previously shown by us, LFA-1 on CD4+ T cells is required for their polarization toward TH1 [100], but LFA-1 [101] and MAC-1 [102] on APC served as negative regulators of T cell activation.
Signaling
β2 integrins have been shown to regulate the responsiveness of leukocytes toward toll-like receptor (TLR) stimulation and have been implicated in various signaling processes [103], including, for example, calcium signaling [104], Src kinases [105,106], integrin-linked kinase [107], FAK [108], MAPK [109], Rho GTPases [110], and NF-κB [111].
3.3.2. Cell-Type-Specific Expression Pattern and Activities
Innate Immune Cells
- PMN
PMNs predominantly express LFA-1 and MAC-1 [8,112]. Heit et al. demonstrated that LFA-1 was important for chemotactic migration of PMN toward IL-8, whereas MAC-1 was required for chemoattraction toward N-Formyl-Met-Leu-Phe [113] and for transendothelial migration [114]. Moreover, sialyl Lewis x (sLex), a known ligand of E-selectin [115], was reported to play a major role in PMN rolling [116]. In that study, Zen and coworkers demonstrated that MAC-1 and LFA-1 were decorated with sLex-type glycans and recognized E-selectin. The removal of sLex completely interrupted Mac-1/E-selectin binding, and incubation with anti-sLex mAb significantly induced PMN degranulation, whereas transmigration through collagen-coated transwell filters and intestinal epithelial cell monolayers was inhibited.
Despite the pronounced role of LFA-1 and MAC-1 for PMN migration, we observed a larger infiltration of β2 integrin- [117] and MAC-1- [118] deficient PMN in mouse models of pulmonal aspergillosis, respectively, into the lung than that observed for the control animals. This observation underlined the fact that PMNs do not exclusively depend on β2 integrins for migration. In this regard, Mizgerd and coworkers identified VLA-4 as responsible for PMN infiltration into the lungs of CD18-deficient mice in acute bacterial pneumonia [119].
A key function of PMN is to kill pathogens intracellularly after MAC-1/CR3-conferred phagocytosis [120,121]. Moreover, MAC-1 physically interacts with FcγRIIA in human PMN by rearranging the cytoskeleton, enabling calcium-mediated signaling of FcγRIIA/B, resulting in phagocytosis and the release of pro-inflammatory cytokines [122]. In line with this, PMN of patients suffering from leukocyte adhesion deficiency (LAD)-1 due to impaired β2 integrin activity [123] (see Section 3.4.1) presented with impaired phagocytic activity that was mimicked by CD11b and CD18 blocking mAb, respectively [124].
Another anti-microbial mechanism of PMN is the release of granules, termed degranulation, of primary (containing pro-inflammatory and antimicrobial proteins) and secondary (e.g., NADPH oxidase and collagenase) granules into the phagosome, which prevents host tissue damage and kills pathogens [125]. Horie and Kita described human eosinophils involvement of MAC-1 during degranulation in vitro [126]. Hence, the mAb-mediated blockade of CD11b and CD18, respectively, inhibited eosinophil adhesion and degranulation, whereas the blockade of CD11a had no such effect.
Further, activated PMN may release NETs, which are composed of chromatin associated with antimicrobial proteins that bind pathogens [127]. This kind of regulated PMN cell death has been termed NETosis [128]. Behnen and coworkers demonstrated that the formation of immune complexes (IC) activated human PMN and consequently NET formation, mediated by FCγRIIIb as a binding partner of MAC-1 [129]. ICs are present in multiple organs and are composed of autoantibodies, which arise in the course of autoimmune diseases and infections [130]. Simultaneous mAb-mediated blockades of CD11b and CD18 decreased IC-induced NET release by PMN [129]. On the contrary, the blockade of CD11a had no effect on NET production, suggesting a unique role of MAC-1 in this regard. However, McDonald reported that NETosis required LFA-1-dependent PMN/platelet interactions [131]. The interruption of PMN/platelet interactions in mouse and human models decreased vascular NET formation. In addition, the blockade of CD11b resulted in complete inhibition of ROS-dependent NET release during cellular infection by A. fumigatus in vitro [132]. Likewise, A. fumigatus-induced NETosis required ROS generation by NOX2 (NADPH oxidase 2) as a downstream effector of MAC-1 since blockade of CD11b decreased NOX2 production and consequently NET release.
In agreement with the overall role of β2 integrins for the anti-microbial effector mechanisms of PMN [133,134], we demonstrated that a PMN-specific knockdown of β2 integrins yielded an aggravated course of invasive pulmonal aspergillosis in A. fumigatus-infected mice [117]. These mice presented with a higher fungal burden, despite a higher abundance of PMN in the pulmonary tissue and lower levels of pro-inflammatory cytokines, such as TNF-α. PMN with impaired β2 integrin expression also showed lower phagocytosis and NETosis capacities compared to their wild-type counterparts. These findings largely corroborate our previous findings in the same disease model using CD11b−/− mice [118]. The congruent finding of elevated PMN numbers in LAD-1 patients [123] as well as CD18−/− mice [135] and mice with PMN-restricted β2 integrin deficiency [117] may indicate a compensatory effect to counteract attenuated functional capacity. Altogether, it is conceivable that MAC-1 plays a dominant role in the main pathogen killing mechanisms of PMN that are crucial for the early control of infection.
Monocytes/Macrophages
Human monocytes were reported to express all β2 integrins, while mouse CD11c/CD18 was apparent only on alveolar macrophages [136], and the expression levels of CD11d/CD18 were low in all monocyte/macrophage populations [8]. Interestingly, LFA-1 was not expressed by murine microglia, which allowed us to distinguish between microglial and CNS-infiltrating immune cells [137].
LFA-1 strongly binds ICAM-3, which is upregulated in apoptotic cells [138], and silencing of LFA-1 in macrophages decreased the clearance of apoptotic cells [139]. Tang et al. showed that the stimulation of M1 macrophages with, e.g., IFN-γ, yielded stronger phagocytosis of tumor cells due to the elevated expression of LFA-1 and CD11c/CD18 [140]. Additionally, enhanced CD11c expression in macrophage-enriched tumors was found to correlate with elevated patient survival rates [140]. Yuan et al. demonstrated that macrophage-derived exosomes are densely decorated with LFA-1 and that their passage through the blood–brain barrier was increased upon brain inflammation due to upregulation of ICAM-1 in the inflamed tissue [141].
MAC-1 was shown to be crucial for the polarization of pro-inflammatory (M1) macrophages by inducing the expression of microRNA Let7a [142]. CD11b inhibition promoted macrophage polarization toward an anti-inflammatory (M2) state and accelerated tumor growth, while the activation of CD11b shifted polarization toward pro-inflammatory M1 macrophages, favoring tumor suppression in animal models of murine and human cancers.
In human monocytes and macrophages, CD11c/CD18 was shown to be more important than MAC-1 in mediating adhesion to fibrinogen [56]. Khalaji and coworkers reported that the adhesion of monocytes to collagen type I increased in the elderly due to the age-dependent elevated activation of CD11c/CD18 [143]. Further, CD11c+ macrophages were reported to be proangiogenic and to play an important role in experimental choroidal neovascularization, a destructive angiogenesis in age-related macular degeneration [144].
In vitro-polarized M1 macrophages were found to express much higher levels of CD11d than M2 macrophages [145]. High expression of CD11d enabled the migration of M1 macrophages to sites of inflammation, while moderate CD11d expression on M2 macrophages was important for migration to mesenchymal tissues [145]. Thus, the relative expression of both β2 integrins may determine their role in leukocyte extravasation [4,146]. On the contrary, Yakubenko and coworkers showed that elevated expression of CD11d/CD18 in macrophages increased their cell adhesiveness and inhibited migration in vitro [147]. These results were confirmed in vivo using an anti-CD11d blocking mAb.
mAb-mediated ligation of CD11b and CD11c on human monocytes, respectively, but not on CD11a, increased NF-κB activity, which in turn enhanced the expression of pro-inflammatory IL-8, macrophage inflammatory protein (MIP-)1α, and MIP-1β [111]. In line with this, MAC-1 clustering and activation initiated a gene program reflecting vascular inflammation [148]. In agreement, MAC-1 deficiency resulted in attenuated vessel wall inflammation after the induction of experimental angioplasty. Further, MAC-1 was demonstrated to trigger a toll/IL-1 receptor superfamily-like signaling cascade 1, which induced NF-κB activity. However, MAC-1/ICAM-1 interactions on human macrophages were reported to indirectly inhibit TLR signaling by promoting the expression of anti-inflammatory IL-10 [3,149,150], SOCS (suppressor of cytokine signaling) 3, and A20-binding inhibitor of NF-κB activation 3 [148].
Further, TLR stimulation activated PI3K, and RapL-mediated inside-out signaling activated β2 integrins [149,151]. In turn, integrin outside-in signaling activated Src/Syk, which resulted in the degradation of the signaling transducers MyD88 and TIR-domain-containing adapter-inducing IFN-β and thereby attenuated TLR signaling [149]. Further, Querrey and coworkers identified MAC-1 as a negative regulator of TLR4/MyD88 signaling in non-classical monocytes after lung transplantation in a murine model of primary graft dysfunction [152]. When lungs derived from CD11b−/− mice were transplanted to recipient wild-type mice, lung injuries were aggravated due to increased production of chemokine (C-X-C motif) ligand 2, which promoted PMN chemoattraction into the lungs. Yee and Hamerman [153] reported that CD18−/− murine BM-derived macrophages and DC were hypersensitive toward various TLR ligands through exacerbated production of pro-inflammatory cytokines like IL-12 and IL-6. These data show that in general, MAC-1 ligation generates a negative activation signal for monocytes and macrophages but also suggests an individual cell-specific role of β2 integrins regarding their effect on TLR-mediated stimulation, depending on the homeostatic versus inflammatory conditions.
DC
DCs, like all leukocytes, express LFA-1, but the expression of the other β2 integrin family members considerably differs between DC subtypes and species. In humans, MAC-1 and CD11c/CD18 are expressed on cDCs but are absent on pDCs [136]. In mice, CD11c/CD18 is expressed by all DC subtypes, and therefore, CD11c is often used as a pan DC marker [154]. MAC-1 is mainly expressed by murine cDC2 [155]. CD11d/CD18 is expressed at a low level by human and mouse DC [136].
In murine BM-derived DC, MAC-1 was shown to constitute a component of the receptor complex that regulates TLR4 internalization and has been identified as a positive regulator of TLR4-induced signaling [156]. In line with this, murine CD11b−/− DC displayed attenuated activation via TLR4 upon LPS stimulation, which in turn affected their T cell stimulatory capacity [156]. The activation of β2 integrins was inhibited in DC by CD18-binding cytohesin-1 and cytohesin-1-interacting protein (CYTIP) [8,157]. Cytohesin-1 upregulated RhoA activity in DC, which is important for chemokine-induced conformational changes of β2 integrins and consequently their activation [158]. We and others demonstrated that CYTIP sequestered cytohesin-1 in the cytoplasm, thereby limiting its interaction with β2 integrins [101,159]. Several studies have shown that various viral pathogens like Herpes simplex virus type 1 [160] and cytomegalovirus (CMV) [161] conferred degradation of CYTIP in DC, resulting in higher LFA-1 activity and thereby stronger cell adhesion.
Recently, we showed that DCs derived from mice with a DC-restricted β2 integrin knockdown overexpressed stimulation-induced cytokines as a result of enhanced activity of STAT (signal transducer and activator of transcription)-1, -3, and -5, and they concomitantly impaired expression of SOCS proteins [162]. In contrast, a gene enrichment analysis of DC with a CD11c-specific deletion of β2 integrins demonstrated reduced expression of genes involved in inflammatory pathways, including TNF-α signaling and IFN-γ responses [162]. Further, positive regulatory genes of TLR-induced signaling, like WD repeat and FYVE domain containing 1 [163], were upregulated, whereas COMM domain containing 2, an inhibitor of NF-κB-dependent gene expression [164], was significantly downregulated. In autoimmune EAE, these CD18-deficient DCs limited the TH1/TH17 auto-inflammatory response due to impaired induction of T-bet-expressing T cells, resulting in a delayed and milder course of disease. Altogether, these results suggest that β2 integrins on DC are responsible for the induction of inflammatory responses [162].
We demonstrated that, within the IS between APC and T cells, LFA-1 on the T cell side was important for T cell activation and TH1 polarization [100]. Of note, we also reported that active LFA-1 [101] and MAC-1 [102] on murine DCs inhibited T cell activation. This inhibitory effect of MAC-1 on T cell proliferation was also shown for human monocyte-derived DC [165], and furthermore, MAC-1 on APC can suppress antigen presentation and TH17 differentiation, leading to immune tolerance [3,166]. Interestingly, Nording et al. demonstrated that MAC-1 on DC is crucial for DC/platelet interaction by binding to the platelet glycoprotein Ibα [167]. Furthermore, activated platelets upregulate MAC-1 on DC. Moreover, the engulfment of apoptotic material via interactions of CD36 on apoptotic bodies with MAC-1 on DC suppressed their APC activity [96].
NK Cells
NK cells express mainly LFA-1 and MAC-1, and in humans, they have also been reported to express CD11c/CD18 [168,169] and CD11d/CD18 at high levels, especially upon infection and inflammation [170,171].
In resting human NK cells, LFA-1 had a low ligand binding affinity that was stimulated by engagement of the NK cell receptors 2B4 and CD16 [172]. The binding of complement C3a, known for its tumor-promoting and immunosuppressive functions, by LFA-1 has been demonstrated to induce acquisition of the high-affinity conformation of LFA-1 and decrease the infiltration of NK cells into the tumor microenvironment in various murine tumor models [173]. LFA-1 was reported to be important for the adhesion of NK cells to tumor cells [174], causing LFA-1 accumulation at the IS [175]. Pariani et al. proposed a Golgi-dependent trafficking pathway involving A kinase anchoring protein 350 (AKAP350) as crucial for NK cell cytolytic activity [176]. Perez and coworkers suggested in an in vitro study that LFA-1 could regulate the degranulation of human NK cells [177]. They showed that CD3−CD8+CD56+ NK cells contained a high amount of cytolytic granules, including perforin and granzyme A. Stimulation of LFA-1 on NK cells as well as their coculture with target cells increased the release of perforin and, accordingly, the cytolytic activity requiring LFA-1/ICAM-2 interactions. Further, phospho-epitope profiling revealed that LFA-1 activity was dependent on p44/42 MAPK pathway activation. In line with this, the inhibition of p44/42 MAPK decreased LFA-1-directed perforin release and, hence, the cytolytic activity of NK cells. March and Loing reported that the stimulation of LFA-1 by incubation with plate-bound ICAM-1 polarized granules in a Syk-, phospholipase C-, PKC-, and paxillin-dependent manner [178].
MAC-1 has been defined as a marker for NK cell maturation in mice [179]. NK cells obtained from CD18-deficient mice showed hyporesponsive properties in vitro as well as alterations of their developmental program in vivo [180]. They also presented with somewhat diminished and missing self-recognition. However, the early immune response to mouse CMV infection was unaffected.
In agreement with the role of LFA-1 and MAC-1 for the cytolytic function of NK cells, β2 integrin deficiency largely abrogated their responsiveness toward stimulation [170]. However, the role of CD11c/CD18 and CD11d/CD18 in human NK cells has scarcely been analyzed so far.
Adaptive Immune Cells
- B cells
B cells express mainly LFA-1 [181], but the expression of MAC-1 [182], CD11c/CD118 [183], and CD11d/CD18 [171] has also been observed in some subsets, as outlined in the following section. LFA-1 is necessary for the binding of B cells to ICAM-1 [184]. In human and mouse models, LFA-1 was found to be expressed at the highest level in memory B cells (MBCs), which consequently had a higher capacity to bind to ICAM-1 and VCAM-1 than naïve B cells [185].
The expression of MAC-1 was reported to be mainly restricted to CD27+ MBC and to contribute to migration, as evidenced by mAb-mediated blocking experiments [182]. Furthermore, MAC-1 attenuated B cell receptor (BCR) signaling to maintain autoreactive B cell tolerance and reduce TLR-3-dependent NK stimulation [8,186]. Further, B1 cells that differ from conventional B cells by producing polyreactive natural antibodies to counteract pathogens [187] have also been shown to express MAC-1 and were found to be enriched in systemic lupus erythematosus (SLE) patients [188]. Here, this B1 subpopulation expressed more CD86 and acted as an efficient APC. In CD11b−/− mice, BCR stimulation resulted in stronger B cell proliferation, survival, and autoantibody production, as observed for wild-type B cells, suggesting a role of MAC-1 in maintaining B cell tolerance [186].
Rubtsov et al. reported on murine CD11b+CD11c+ B cells, termed age-associated B cells (ABCs), that accumulated in old female mice and in young SLE-prone mice of either gender [189]. ABCs were also observed in older female patients suffering from autoimmune diseases. Altogether, these findings suggested a direct role of CD11b+CD11c+ B cells in autoimmunity [190]. Nagy-Baló et al. showed that human tonsillar MBC expressed CD11c/CD18 to a low extent, which strongly increased upon BCR stimulation [191]. CD11c/CD18 is important for the proliferation, migration, and adhesion of BCR-activated MBCs to fibrinogen-covered follicular dendritic cells (FDCs). Such contacts facilitate the binding of antigens stored by FDCs, thereby promoting B cell survival and proliferation [192]. CD11c+ MCBs were characterized by the strong expression of genes involved in B cell activation, differentiation, and antigen presentation, thereby constituting precursors of antibody-secreting cells [183]. Rincon-Arevalo and coworkers observed higher frequencies of CD11c+ B cells in the blood of SLE patients than in samples of healthy donors. Interestingly, in either donor group, CD11c+ B cells showed higher expression levels of CD19, CD45RO, CD45RA, CD69, and Ki-67 and diminished expression of CD21 and C-X-C motif chemokine receptor 5 than CD11c− B cells [193].
MAC-1 and CD11c/CD18 expression on B cells has also been observed in several B cell malignancies, such as chronic lymphocytic leukemia (CLL) [194]. Functional studies suggested a role for these receptors in the IL-10 production of CLL B cells in response to CpG stimulation [195]. Song et al. showed that CD11c-expressing B cells arise outside of germinal centers in cases of (auto-)inflammation and aging, depending on T follicular helper cells [196]. In cases of resolved infections, such B cells were apparent in the marginal zone of the spleen, depending on LFA-1 and VLA-4 activity, and constituted memory B cells readily activated to produce antibodies upon secondary infection. Human B cells also express CD11d, but its functional importance has not been thoroughly investigated [171].
T Cells
Of all β2 integrins, T cells express only LFA-1 [98]. In the case of APC/T cell interaction, LFA-1 is positioned in the peripheral supramolecular activation cluster (p-SMAC) of the IS. LFA-1 is linked to the intracellular proteins talin, kindlin-3, and Rap1, which stabilize the interaction between the TCR and major histocompatibility complex II (MHCII)-loaded peptide within the center of the (c-)SMAC [3,197,198]. On the T cell side, the c-SMAC is also enriched in TCR-associated co-receptors like CD3, CD4, and CD8 [8,199,200,201] as well as CD28, which engages costimulatory receptors (e.g., CD80, CD86) of the APC [202]. Upon activation and binding to ICAM-1, LFA-1 contributes to T cell activation and polarization of the T cell response [66,100].
Normally, T cells engage APC via multiple short-term contacts called kinapses [203] before long-lasting contacts are formed to achieve their full activation. The conformational change in LFA-1 can lead to a 10,000-fold increase in affinity for ICAM-1 on APC [66,204]. Bleijs and coworkers observed that low-affinity binding of ICAM-3 in combination with high-affinity binding to ICAM-1 induced the formation of large LFA-1 clusters [205]. Moreover, low-affinity LFA-1/ICAM-3 interactions contributed to stabilize LFA-1/ICAM-1-dependent cell–cell contact. The high-affinity adhesiveness of LFA-1 proved essential for full T cell activation [75]. Moreover, the LFA-1/ICAM interaction influenced human T cell polarization by modulating TH1 and TH2 responses [73,206]. LFA-1 cross-linking also phosphorylated the signal transducer and activator of transcription 3 (STAT3), which is associated with tubulin-depolymerizing stathmin, thereby controlling cytoskeletal reorganization in migrating human T cells (Figure 5, left part) [207].
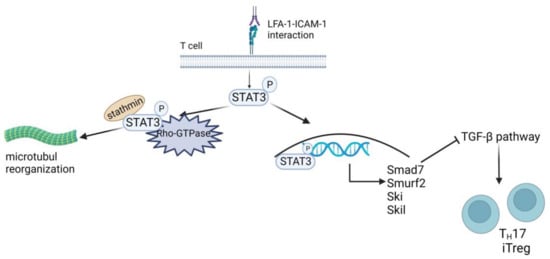
Figure 5.
Role of LFA-1-mediated contact for TH17/iTreg polarization. LFA-1 triggers STAT3 activation and interrupts TGF-β signaling. LFA-1 triggered STAT3 activation by phosphorylation (P), translocating STAT3 to the nucleus, and its interaction with stathmin and Rho GTPase controls microtubule dynamics. In the nucleus, STAT3 upregulates the expression of the TGF-β-inhibiting transcription factors SMAD7, SMURF2, SKI, and SKIL. These proteins hinder TGF-β-mediated inhibition of IL-2 secretion and T-cell differentiation into TH17 or iTreg cells. Created with BioRender.com.
Tregs are a specialized subgroup of CD4+ T cells that serve to maintain self-tolerance by inhibiting the activation of autoreactive T cells, and they can be divided into naturally occurring thymus-derived Foxp3+ (t)Treg and peripheral (p)Treg [208,209]. The latter originates from extrathymic naive CD4+Foxp3− T cells upon TCR stimulation in the presence of TGF-β [209,210] and upon antigen presentation in the absence of co-stimulation [211], respectively. LFA-1 has been shown to be required for Treg differentiation in vivo [212,213] and for Treg induction in vitro [214]. However, Verma and coworkers demonstrated that LFA-1-mediated activation of STAT3 induced expression of various genes associated with TGF-β pathway inhibition, including SMAD7 (mothers against decapentaplegic family member 7), SMURF2 (SMAD ubiquitination regulatory factor 2), SKI (Sloan-Kettering Institute proto-oncogene), and SKIL (SKI-like proto-oncogene) (Figure 5, right part) [206]. The induction of an LFA-1/ICAM-1 interaction-dependent refractory state in mouse and human CD4+ T cells toward TGF-β attenuated pTreg induction and polarization toward TH17 (Foxp3+RORγt+).
Tregs suppress T cell immune responses by various mechanisms, including anti-inflammatory cytokines (e.g., IL-10, TGF-β) [215,216], the depletion of IL-2 [217], the induction of apoptosis [218,219], metabolic pathways [220,221], and the modulation of the maturation and function of APC [215,222,223,224,225]. Concerning the latter, Tregs were reported to inhibit the upregulation of costimulatory receptors on DC via LFA-1 on the T cell side by binding ICAM on DC [226,227]. Further, Chen et al. demonstrated that strong LFA-1-dependent adhesions between Treg and DC led to cytoskeletal rearrangements in the latter, attenuating their ability to engage naïve T cells due to the Treg-focused displacement of cytoskeletal components [228]. In line with this, we observed a higher state of DC activation in mice with a Foxp3-specific knockdown of LFA-1 [229]. In accordance, in in vitro Treg/DC cocultures, LFA-1-deficient Treg interacted to a lower extent with DC, and the latter presented with a markedly activated immunophenotype. Moreover, LFA-1-deficient Tregs were characterized by the impaired expression of genes associated with suppressive activity, and the corresponding mice were characterized by systemic TH2-biased inflammation. Altogether, our findings suggested that LFA-1 was not only important for Treg induction but was essential for maintaining the tolerizing activity of Treg.
In addition to its role in T cell differentiation and activation, LFA-1 on activated CD8+ T cells is required to kill infected target cells by sealing the IS between the T cell and the target cell to direct cytolytic granules [3,66]. Furthermore, LFA-1 can function as a so-called mechanical gate to attract granules containing perforin and granzyme and trigger their fusion with the synaptic membrane [230]. Degranulation takes place mostly in regions of active force exertion within a cytotoxic synapse, suggesting that mechanosensing is involved in the release of cytolytic granules [230,231]. Similar to PMN, CD4+ and CD8+ T cells reportedly form T cell extracellular traps (TETs) [232], which are associated with skin diseases and leishmania infections [233]. It is unknown yet whether LFA-1 activation is involved in TET formation.
3.4. Leukocyte Adhesion Deficiencies
Leukocyte adhesion deficiencies (LADs) are rare hereditary disorders characterized by a defect in leukocyte trafficking. Decreased expression or activity of adhesion or adaptor proteins results in the manifestation of three different types of LADs, as summarized in Figure 6 [234].
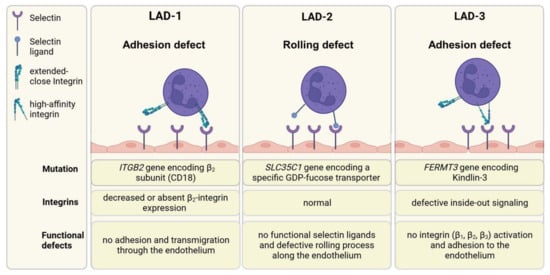
Figure 6.
Leukocyte adhesion deficiency syndromes. The trafficking of leukocytes is a multistep process, including rolling along endothelia, adhesion, arrest, crawling, and finally, extravasation. LAD-1 syndrome is characterized by decreased or absent β2 integrin expression, resulting in impairment of leukocyte firm adhesion and migration into tissue. LAD-2 patients have a deficiency in selectin ligand expression, resulting in insufficient rolling of leukocytes. Humans with LAD-3 syndrome have no functional kindlin-3, which is an important adaptor protein for correct integrin activation. β1, β2, and β3 integrins cannot switch to their high-affinity state and interact with their ligands. Consequently, leukocytes are not able to adhere to the endothelium. Created with BioRender.com.
3.4.1. LAD-1
The crucial role of β2 integrins in the immune system is emphasized by the phenotype of LAD-1 patients, who suffer mainly from frequent and severe infections [235]. LAD-1 affects about one per million people worldwide [236]. In LAD-1 patients, a mutation in the CD18 gene leads to the attenuated expression of functional heterodimeric β2 integrins, resulting in impaired leukocyte adhesion and migration into the tissue [237] and attenuated pathogen-killing functions, e.g., of PMN [238]. In some patients, CD18 expression differs between cell types, and accordingly, the severity of LAD-1 can be subdivided into mild (>30% of patients), moderate (2–30%), and severe forms (<2%) [239]. About 75% of patients with severe LAD-1 die by the age of 2 years. Patients with moderate LAD-1 survive childhood, but their mortality amounts to 50% by the age of 40 years [240].
The most prevalent symptoms are recurrent bacterial infections of the skin, periodontitis, and poor wound healing [134,241,242]. In addition to these immunodeficiency symptoms, LAD-1 patients may suffer from autoimmune phenomena like IBD, type-1 diabetes, and nephritis [243,244]. In accordance with this, it has been demonstrated that LAD-1 patients with IBD presented with a higher number of Foxp3+ Tregs, but these displayed reduced suppressive activity [227,245]. Until now, the only curative therapy was allogeneic hematopoietic stem cell transplantation [246,247].
3.4.2. LAD-2
LAD-2 results from a mutation in the SLC35C1 gene that encodes a specific GDP-fucose transporter [248]. This causes a rolling defect of leukocytes by the aberrant expression of Sialyl Lewis X (sLeX) and other selectin ligands. Selectin ligands are carbohydrates that share many structural features with SLeX, whereby fucose is an essential element of all selectin ligands [249]. LAD-2 patients lack these fucosylated glycoconjugates and may show similar symptoms as LAD-1 patients, but may also present with severe mental and growth retardations [250]. The fucosylated glycoconjugates are synthesized by fucosyltransferases from the donor substrate, GDP-fucose. Thus, the defect in fucose metabolism can be partially compensated by oral fucose substitution therapy [251]. By this fucose is taken up into the cells, GDP-fucose is synthesized and transferred into the Golgi apparatus for utilization by fucosyltransferases. Therefore, the lack of selectin ligands can be compensated, normalizing PMN counts and function. Interruption of this therapy leads to the loss of selectin ligands after several days. Of note, the metabolic pathway that causes the psychomotor and growth retardations is still unknown.
3.4.3. LAD-3
LAD-3 patients present with a mutation in the FERMT3 gene encoding kindlin-3, which is important for integrin activation on leukocytes and thrombocytes [252,253]. Affected patients suffer from leukocytosis and recurrent infections resembling symptoms of LAD-1. Kindlin-3 is a cytoskeletal adaptor protein that interacts with the cytoplasmic tails of β1, β2, and β3 integrins, thereby shifting the receptors to a high-affinity state via inside-out signaling [254]. Hence, kindlin-3 deficiency precludes integrin activation. As shown in FERMT3−/− mice, the adhesion of LFA-1 to kindlin-3 and conformational activation are impaired compared to wild-type mice [255]. Consequently, β1 and β2 integrin-mediated adhesion fail [254]. In addition, LAD-III patients have a bleeding tendency [256], as kindlin-3 is necessary to confer conformational activation of integrin α(IIb)β3 as a prerequisite to bind fibrinogen for platelet aggregation to initiate blood clotting [257]. Furthermore, osteoporosis-like bone defects, as observed in some LAD-3 patients [258], may result from integrin dysfunction of bone-resorbing osteoclasts, as observed in kindlin-3−/− mice [259]. The finding that osteoclasts derived from mice with a specific deletion of either β1, β2 or β3 integrins displayed a milder phenotype than kindlin-3−/− osteoclasts suggested that only combined loss of kindlin-3-dependent integrin activity resulted in the osteopetrosis phenotype. Similar to LAD-1, the only available therapy is allogeneic hematopoietic stem cell transplantation, accompanied by erythrocyte/platelet transfusions [260].
3.5. Role of β2 Integrins in Autoimmunity and Tumorigenesis and Receptor-Specific Therapeutic Approaches
As reflected by the impaired immune state of LAD-1 patients, immune cells critically depend on β2 integrins to eradicate pathogens but also to maintain immune homeostasis to prevent autoimmunity. Further, tumor-induced immunoregulatory cell types support tumorigenesis in a β2 integrin-dependent manner. Consequently, the suitability of β2 integrin agonists and antagonists for therapeutic applications has been assessed in a number of pre-clinical and clinical studies, as recently reviewed by Pang et al. [11].
3.5.1. Autoimmunity
Autoimmune reactions are often characterized by a reduced number of functionally active Tregs that are critical to maintaining self-tolerance [261]. For example, reduced levels of Treg have been described in psoriatic arthritis [262], systemic SLE [263,264], and Kawasaki disease [261,265]. Further, we and others showed that LFA-1 was required for TH1/TH2 polarization [73,100,206]. An imbalance between TH1 and TH2 has been associated with several autoimmune diseases like MS, RA, and type-1 diabetes [266]. In this regard, LFA-1 signaling in combination with TCR stimulation triggered glycogen synthase kinase 3β-dependent Notch1 activation by γ-secretase proteolytic cleavage [267]. This upregulated T-bet expression and thereby favored a shift to TH1-associated cytokine production, specifically of IL-2 and IFN-γ. Notch constitutes a critical differentiation factor of T cell effector cells [268]. According to the so-called ‘second touch hypothesis’ [269], Notch1 signaling induced by LFA-1/ICAM interaction enhances T-cell effector priming and migration to sites of inflammation [73].
Furthermore, elevated expression of LFA-1 on T cells has been shown to correlate with systemic sclerosis and SLE, as well as RA and autoimmune thrombocytopenia [270]. mAb interfering with LFA-1/ICAM-1 interactions has been extensively analyzed in numerous pre-clinical studies [271]. Often, LFA-1 signaling interruption has been combined with additional therapies like the blockade of either ICAM-1 or CD40L. For example, in a preclinical study, combined treatment with anti-LFA-1 and -CD40L mAb after islet transplantation in vivo resulted in a tolerance that was defined as ‘dominant’, which was not achieved in the case of monotherapy with either murine mAb [272].
CD2 is known to upregulate LFA-1 avidity and, therefore, cell adhesion [273]. In a clinical phase II study, anti-LFA-1 and -CD2 mAb therapy in combination with T cell depletion prevented the rejection of transplanted bone marrow for the treatment of acute lymphoblastic leukemia in children [274]. However, this effect was not observed in adult patients [275].
Efalizumab, a humanized IgG1 anti-LFA-1 antibody, blocks LFA-1/ICAM-1 interactions in order to suppress T cell priming and effector functions of immune cells, which diminishes inflammatory cell recruitment and the reversal of keratinocyte hyperplasia at psoriatic lesions [276,277,278]. Despite this beneficial effect on skin lesions, efalizumab was not effective in psoriatic arthritis [279] and was less effective in psoriasis than other established therapies like cyclosporine and ultraviolet radiation-based therapies [280]. In 2009, efalizumab was withdrawn from the market because of a high risk of John Cunningham polyomavirus reactivation and the development of progressive multifocal leukoencephalopathy in patients under long-term administration [281,282,283]. In general, LFA-1-targeted therapies that aim to modulate the activation state of T cells carry the inherent problem that β2 integrins exert their function in a cell-specific manner and often have divergent functions on different cell types. For example, they limit T cell activation when expressed on DC [284], but they activate T cell functions when expressed on T cells [3].
MAC-1 was shown to suppress the differentiation of TH17 cells, which are associated with autoimmunity, and CD11b deficiency in mice was reported to cause elevated IL-6 production by APC, which in turn promoted TH17 differentiation [166]. In a murine collagen-induced arthritis model, CD11b−/− mice presented with an earlier onset, higher incidence, and increased severity of arthritis compared to wild-type animals. This was due to markedly increased IL6 production by CD11b−/− DC and elevated TH17 differentiation. In agreement, arthritis severity could be treated by the administration of IL-6-specific mAb treatments and the adoptive transfer of wild-type DCs [285]. Tang et al. investigated the role of MAC-1-FcγR interactions in immune complex-mediated injuries and compared the course of Fc-dependent anti-glomerular basement membrane nephritis in CD11b−/− and wild-type mice. They found that initial glomerular PMN accumulation was comparable in both strains, but whereas it further increased in wild-type mice, PMN numbers decreased in CD11b-deficient mice. Furthermore, CD11b−/− mice showed no proteinuria in contrast to wild-type controls [286]. In the context of the autoimmune disease bullous pemphigoid (BP), MAC-1 was shown to be important for disease development since CD11b−/− mice were resistant to experimental BP, which was associated with less PMN infiltration into the skin [287]. Further, MAC-1 has been implicated in EAE induction, as CD11b-deficient mice displayed a significantly delayed onset and attenuated course of EAE [288]. We observed a similar outcome for mice with a DC-specific knockdown of β2 integrins [162]. β2 integrins on macrophages also play a significant role in the pathogenesis of osteoarthritis, and the current state of knowledge has recently been reviewed [289].
Several nonsynonymous single nucleotide polymorphisms in the ITGAM gene that encodes CD11b have been associated with an increased risk of developing SLE [290,291,292]. Cd11b agonists have been found to be promising candidates for the treatment of SLE [293]. Aside from SLE, these gene variants do not seem to be associated with an increased risk for other autoimmune diseases such as MS, type-1 diabetes, or Sjögren’s syndrome [294,295].
3.5.2. Tumorigenesis
Tumors exploit several immune evasion mechanisms, including the induction and expansion of regulatory immune cell types comprising Tregs [296], tumor-associated macrophages (TAMs) [297], and myeloid-derived suppressor cells (MDSCs) [298]. The latter can be broadly defined as Ly-6C+CD11b+ monocytic MDSC and Ly-6G+CD11b+ granulocytic (g)MDSC that originate from monocyte and PMN progenitors, respectively [299]. PMN and gMDSC are scarcely distinguishable on the phenotypic level. Further, tumor-associated neutrophils (TANs) have been reported to exert tumoricidal (N1) and tumor-promoting (N2) properties within the tumor microenvironment (TME), respectively [300]. N1-TANs were reported to kill tumor cells via antibody-dependent cytotoxic effects (ADCC), like Fc receptor-dependent phagocytosis [301], which was shown to require MAC-1 [302,303]. Besides classical ADCC, Matlung and colleagues reported that N1-TAN engaged tumor cells opsonized by therapeutic antibodies via Fc receptors and MAC-1 [304]. Within the cytotoxic synapse, this resulted in disruption and internalization of part of the tumor cell membrane by N1-TAN, thereby inducing cell death by trogoptosis. Aside from ADCC, N1 TAN may kill Fas-expressing tumor cells via the Fas ligand [305], ROS [306], and nitric oxide [307]. Of note, MAC-1-dependent signaling plays an important role in both ROS [308,309] and nitric oxide [310] production.
TME-derived soluble mediators like G-CSF [311], IL-8 [312], and high mobility group box 1 [313] were demonstrated to promote NETosis. Whereas Schedel and coworkers reported that NET exerted anti-tumor effects by inducing tumor cell necrosis and inhibiting tumor cell motility [314], numerous other studies have reported on the tumor-promoting role of NETs via several mechanisms [315]. For example, Munir and coworkers demonstrated that cancer-associated fibroblasts (CAFs) released amyloid β peptide, which, presumably via MAC-mediated uptake, activated PMN to undergo NETosis [316]. NETs supported tumor metastasis by activating CAF [317], which suggests a positive feedback loop comprising CAF and PMN/gMDSC-derived NETs and promotes the binding of circulating tumor cells to liver sinusoids [318]. This in turn activated CAF, thereby promoting tumor growth.
N2-TANs have been reported to bind disseminated tumor cells via MAC-1/ICAM-1 and to facilitate their endothelial transmigration, thus favoring tumor metastasis [319]. Sprouse and coworkers demonstrated that interaction of N2-TAN/gMDSC with circulating tumor cells enhanced Notch1 receptor expression in the latter via ROS production [320]. This enhanced the engagement of JAGGED on N2-TAN/gMDSC, known to favor tumor cell proliferation [321], and elevated NODAL production and release by tumor cells [320]. NODAL, in a paracrine manner, activated N2-TAN/gMDSC by binding to the Cripto receptor.
Besides direct stimulation of tumor growth and metastasis, immunoregulatory cell types comprising N2-TAN, MDSC, and Treg exert immune-inhibitory effects by overlapping and distinctly soluble and receptor-dependent mechanisms both in the TME and in the periphery [322]. Aarts and coworkers reported that human MDSC required MAC-1-dependent cell–cell contact to suppress T cell proliferation, depending on ROS, degranulation, and trogoptosis [323]. As discussed above, all of these mechanisms were reported to rely in part on MAC-1 activity [126,132,304]. Likewise, as outlined above, Tregs require LFA-1 at least in part to exert their inhibitory effects on APC [226,228,229] and T cells [212,324]. Furthermore, MDSCs and Tregs have been shown to interact via soluble mediators and receptors, resulting in mutual activation, which may depend on β2 integrins as well [322].
CD11a−/− mice were shown to display attenuated tumor growth, which was associated with lower Treg numbers [325]. In agreement, the treatment of wild-type mice bearing a subcutaneous tumor with a pharmacological LFA-1 antagonist (BIRT377) yielded similar results. Soloviev and coworkers reported that CD11b−/− mice presented with attenuated melanoma growth due to impaired infiltration of the tumor by monocytes and PMN and reduced vascular endothelial growth factor production by the latter [326]. On the contrary, Schmid and coworkers showed that CD11b was necessary for the polarization of M1 macrophages, exerting tumoricidal activity by inducing the expression of miRNA Let7a, thereby attenuating tumor growth [142]. However, several studies showed that the application of CD11b-blocking antibodies attenuated tumor growth, especially when combined with radiation and immunostimulation by co-applied immune checkpoint inhibitors, respectively [327]. Pharmacological activation of MAC-1 using GB1275 (Leukadherin-1), which binds MAC-1 and activates this β2 integrin by outside-in signaling [328], yielded pronounced anti-tumor effects in numerous studies [142,329,330]. This outcome has been attributed largely to reduced tumor infiltration by myeloid cells and repolarization of MDSC toward an immunostimulatory phenotype, which was accompanied by elevated anti-tumor CD4+ and CD8+ T cell responses. In addition, Liu and coworkers demonstrated that CD11b agonists reprogramed murine and human TAM by inhibiting NF-κB activity and inducing stimulators of IFN genes and STAT signaling, resulting in IFN I production [331]. However, the oral MAC-1 agonist GB1275 failed to show therapeutic benefit in human patients when applied as monotherapy or in combination with immune checkpoint inhibition for the therapy of metastatic pancreatic adenocarcinoma in a clinical phase 1/2 trial (NCT04060342).
Altogether, these studies underscore the overall relevance of β2 integrins for tumorigeneses. However, data on their effect on tumor growth and modulation of the tumor microenvironment (TME) are still controversial, most likely due to the cell-type-specific, activation-dependent, and multi-facetted role of β2 integrins and especially MAC-1.
4. Conclusions
αβ integrins, and among these, β2 integrins, are highly relevant for immune functions by controlling intercellular communication, cell differentiation, activation, migration, and various effector mechanisms required to maintain self-tolerance and for pathogen killing. In line with this, patients lacking β2 integrin activity suffer from severe infections, and at the same time, they are prone to developing autoimmune diseases. All leukocytes express β2 integrins in a cell-type-specific pattern that is dynamically regulated in terms of expression intensity and ligand affinity in response to stimulatory events. So far, most studies on the cellular and molecular role of β2 integrins have been performed using mice with a constitutive knockout of either α (CD11a–d) or the common β (CD18) subunit. Therefore, it has been challenging to delineate the cell type-specific role of β2 integrins, especially in vivo, since functional alterations of immune cells may be due to intrinsic or extrinsic effects of integrin receptor deficiencies. To dissect our observations, we generated a mouse with a floxed CD18 gene, which enabled the establishment of sublines with a cell-type-specific knockdown of β2 integrins.
The modulation of integrin receptor activity by antibodies and pharmacological compounds has been shown to yield therapeutic effects in inflammatory diseases and for tumor therapy in preclinical studies, but in clinical trials, it quite often yields no major beneficial effects and is even associated with significant side effects. These outcomes are most likely a consequence of the systemic modulation of integrin activity. Hence, cell-type-directed delivery of integrin receptor-modulating agents by cell-type-targeting nanoformulations may be necessary to achieve a major breakthrough.
Author Contributions
Conceptualization, T.K., M.B. and S.G.; original draft writing, T.K., C.H. and M.B.; supervision, S.G. All authors have read and agreed to the published version of the manuscript.
Funding
S.G. is funded by the Deutsche Forschungsgemeinschaft (TR156); grant number: B11.
Acknowledgments
We thank the Research Center for Immunotherapy (FZI; University Medical Center, Mainz, Germany) for their continuous support. All graphs were generated using BioRender.com (assessed on 18 September 2023).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| ABC | Age-associated B cell |
| ADCC | Antibody-dependent cytotoxic effects |
| ANA | Antinuclear antibody |
| APC | Antigen-presenting cell |
| BM | Bone marrow |
| BP | Bullous pemphigoid |
| CAF | Cancer-associated fibroblast |
| cAMP | Cyclic adenosine monophosphate |
| CCL | Chemokines like chemokine ligand |
| CLL | Chronic lymphocytic leukemia |
| CMV | Cytomegalovirus |
| CNS | Central nervous system |
| CR3 | Complement receptor 3 |
| c-SMAC | Central supramolecular activation cluster |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein |
| CYTIP | Cytohesin-1-interacting protein |
| DC | Dendritic cell |
| EAE | Experimental autoimmune encephalomyelitits |
| ECM | Extracellular matrix |
| ESM | Endothelial cell-specific molecule |
| FAK | Focal adhesion kinase |
| FDC | Follicular dendritic cells |
| Foxp3 | Forkhead box p3 |
| gMDSC | Granulocytic myeloid-derived suppressor cell |
| HSC | Hematopoietic stem cell |
| IBD | Inflammatory bowel disease |
| IC | Immune complex |
| ICAM | Intercellular adhesion molecule |
| IFN | Interferon |
| Ig | Immune globulin |
| IL | Interleukin |
| IS | Immunolocial synapse |
| iTreg | Induced Treg |
| JAM | Junctional adhesion molecule |
| KO | Knockout |
| LAD-1 | Leukocyte adhesion deficiency 1 |
| LDV | Leu-Asp-Val |
| LFA-1 | Leukocyte function-associated antigen 1 |
| LPS | Lipopolysaccharide |
| mAb | Monoclonal antibody |
| MAC-1 | Macrophage-1 antigen |
| MAPK | Mitogen-activated protein kinase |
| MCB | Memory B cell |
| MDSC | Myeloid-derived suppressor cell |
| MHC | Major histocompatibility complex |
| MIDAS | Metal ion-dependent adhesion site |
| MIP | Macrophage inflammatory protein |
| MS | Multiple sclerosis |
| NET | Neutrophil extracellular trap |
| NF-κB | Nuclear factor k-light-chain-enhancer of activated B cells |
| NK cells | Natural killer cells |
| NOX2 | NADPH oxidase 2 |
| PMN | Polymorphonuclear neutrophil |
| p-SMAC | Peripheral supramolecular activation cluster |
| pTreg | Peripheral Treg |
| RA | Rheumatoid arthritis |
| RGD | Arginylglycylaspartic acid |
| RIAM | Rap-1-GTP interacting adaptor molecule |
| ROS | Reactive oxygen species |
| SKI | Sloan-Kettering Institute proto-oncogene |
| SLE | Systemic lupus erythematodes |
| sLex | Sialyl Lewis x |
| SMAC | Supramolecular activation cluster |
| SMAD7 | Mothers against decapentaplegic family member 7 |
| SMURF2 | SMAD Ubiquitination Regulatory Factor 2 |
| SOCS | Suppressor of cytokine signaling |
| STAT | Signal transducer and activator of transcription |
| TAM | Tumor-associated macrophage |
| TCR | T cell receptor |
| Teff | Effector T cell |
| TET | T cell extracellular trap |
| TGF-ß | Transforming growth factor |
| TH | T helper cell |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| Tr1 | Type 1 pTreg |
| Treg | Regulatory T cell |
| tTreg | Thymus-derived Treg |
| VCAM | Vascular cell adhesion protein |
| WT | Wild-type |
References
- Johnson, M.S.; Lu, N.; Denessiouk, K.; Heino, J.; Gullberg, D. Integrins during evolution: Evolutionary trees and model organisms. Biochim. Biophys. Acta 2009, 1788, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Mitroulis, I.; Alexaki, V.I.; Kourtzelis, I.; Ziogas, A.; Hajishengallis, G.; Chavakis, T. Leukocyte integrins: Role in leukocyte recruitment and as therapeutic targets in inflammatory disease. Pharmacol. Ther. 2015, 147, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Fagerholm, S.C.; Guenther, C.; Llort Asens, M.; Savinko, T.; Uotila, L.M. β2-Integrins and Interacting Proteins in Leukocyte Trafficking, Immune Suppression, and Immunodeficiency Disease. Front. Immunol. 2019, 10, 254. [Google Scholar] [CrossRef] [PubMed]
- Blythe, E.N.; Weaver, L.C.; Brown, A.; Dekaban, G.A. β2 Integrin CD11d/CD18: From Expression to an Emerging Role in Staged Leukocyte Migration. Front. Immunol. 2021, 12, 775447. [Google Scholar] [CrossRef] [PubMed]
- Mezu-Ndubuisi, O.J.; Maheshwari, A. The role of integrins in inflammation and angiogenesis. Pediatr. Res. 2021, 89, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef]
- Bednarczyk, M.; Stege, H.; Grabbe, S.; Bros, M. β2 Integrins-Multi-Functional Leukocyte Receptors in Health and Disease. Int. J. Mol. Sci. 2020, 21, 1402. [Google Scholar] [CrossRef]
- LaFoya, B.; Munroe, J.A.; Miyamoto, A.; Detweiler, M.A.; Crow, J.J.; Gazdik, T.; Albig, A.R. Beyond the Matrix: The Many Non-ECM Ligands for Integrins. Int. J. Mol. Sci. 2018, 19, 449. [Google Scholar] [CrossRef]
- Clements, J.M.; Newham, P.; Shepherd, M.; Gilbert, R.; Dudgeon, T.J.; Needham, L.A.; Edwards, R.M.; Berry, L.; Brass, A.; Humphries, M.J. Identification of a key integrin-binding sequence in VCAM-1 homologous to the LDV active site in fibronectin. J. Cell Sci. 1994, 107 Pt 8, 2127–2135. [Google Scholar] [CrossRef]
- Pang, X.; He, X.; Qiu, Z.; Zhang, H.; Xie, R.; Liu, Z.; Gu, Y.; Zhao, N.; Xiang, Q.; Cui, Y. Targeting integrin pathways: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wu, Q.; Dong, Z.; Liu, K. Integrins in cancer: Emerging mechanisms and therapeutic opportunities. Pharmacol. Ther. 2023, 247, 108458. [Google Scholar] [CrossRef]
- Plow, E.F.; Haas, T.A.; Zhang, L.; Loftus, J.; Smith, J.W. Ligand binding to integrins. J. Biol. Chem. 2000, 275, 21785–21788. [Google Scholar] [CrossRef] [PubMed]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tissue Res. 2010, 339, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, B.S.; Kessler, H.; Kossatz, S.; Reuning, U. RGD-Binding Integrins Revisited: How Recently Discovered Functions and Novel Synthetic Ligands (Re-)Shape an Ever-Evolving Field. Cancers 2021, 13, 1711. [Google Scholar] [CrossRef] [PubMed]
- Nader, D.; Curley, G.F.; Kerrigan, S.W. A new perspective in sepsis treatment: Could RGD-dependent integrins be novel targets? Drug Discov. Today 2020, 25, 2317–2325. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, C.F. Sepsis heterogeneity. World J. Pediatr. 2023, 19, 919–927. [Google Scholar] [CrossRef]
- Conroy, K.P.; Kitto, L.J.; Henderson, N.C. αv integrins: Key regulators of tissue fibrosis. Cell Tissue Res. 2016, 365, 511–519. [Google Scholar] [CrossRef]
- Altrock, E.; Sens, C.; Wuerfel, C.; Vasel, M.; Kawelke, N.; Dooley, S.; Sottile, J.; Nakchbandi, I.A. Inhibition of fibronectin deposition improves experimental liver fibrosis. J. Hepatol. 2015, 62, 625–633. [Google Scholar] [CrossRef]
- Khurana, A.; Sayed, N.; Allawadhi, P.; Weiskirchen, R. It’s all about the spaces between cells: Role of extracellular matrix in liver fibrosis. Ann. Transl. Med. 2021, 9, 728. [Google Scholar] [CrossRef]
- Hussein, H.A.; Walker, L.R.; Abdel-Raouf, U.M.; Desouky, S.A.; Montasser, A.K.; Akula, S.M. Beyond RGD: Virus interactions with integrins. Arch. Virol. 2015, 160, 2669–2681. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Nabors, L.B.; Fink, K.L.; Mikkelsen, T.; Grujicic, D.; Tarnawski, R.; Nam, D.H.; Mazurkiewicz, M.; Salacz, M.; Ashby, L.; Zagonel, V.; et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: Results of the open-label, controlled, randomized phase II CORE study. Neuro Oncol. 2015, 17, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.R.; Hart, I.R.; Watson, A.R.; Welti, J.C.; Silva, R.G.; Robinson, S.D.; Da Violante, G.; Gourlaouen, M.; Salih, M.; Jones, M.C.; et al. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat. Med. 2009, 15, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Stipp, C.S. Laminin-binding integrins and their tetraspanin partners as potential antimetastatic targets. Expert. Rev. Mol. Med. 2010, 12, e3. [Google Scholar] [CrossRef]
- Tsuruta, D.; Kobayashi, H.; Imanishi, H.; Sugawara, K.; Ishii, M.; Jones, J.C. Laminin-332-integrin interaction: A target for cancer therapy? Curr. Med. Chem. 2008, 15, 1968–1975. [Google Scholar] [CrossRef] [PubMed]
- Hemler, M.E.; Jacobson, J.G.; Brenner, M.B.; Mann, D.; Strominger, J.L. VLA-1: A T cell surface antigen which defines a novel late stage of human T cell activation. Eur. J. Immunol. 1985, 15, 502–508. [Google Scholar] [CrossRef]
- Conrad, C.; Boyman, O.; Tonel, G.; Tun-Kyi, A.; Laggner, U.; de Fougerolles, A.; Kotelianski, V.; Gardner, H.; Nestle, F.O. α1β1 integrin is crucial for accumulation of epidermal T cells and the development of psoriasis. Nat. Med. 2007, 13, 836–842. [Google Scholar] [CrossRef]
- Breuer, J.; Schneider-Hohendorf, T.; Ostkamp, P.; Herich, S.; Rakhade, S.; Antonijevic, I.; Klotz, L.; Wiendl, H.; Schwab, N. VLA-2 blockade in vivo by vatelizumab induces CD4+FoxP3+ regulatory T cells. Int. Immunol. 2019, 31, 407–412. [Google Scholar] [CrossRef]
- Sheremata, W.A.; Minagar, A.; Alexander, J.S.; Vollmer, T. The role of alpha-4 integrin in the aetiology of multiple sclerosis: Current knowledge and therapeutic implications. CNS Drugs 2005, 19, 909–922. [Google Scholar] [CrossRef]
- Weinstock-Guttman, B. An update on new and emerging therapies for relapsing-remitting multiple sclerosis. Am. J. Manag. Care 2013, 19, s343–s354. [Google Scholar]
- Bamias, G.; Clark, D.J.; Rivera-Nieves, J. Leukocyte traffic blockade as a therapeutic strategy in inflammatory bowel disease. Curr. Drug Targets 2013, 14, 1490–1500. [Google Scholar] [CrossRef]
- Lin, K.K.; Mahadevan, U. Etrolizumab: Anti-β7—A novel therapy for ulcerative colitis. Gastroenterology 2014, 146, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, S.; O’Byrne, S.; Keir, M.; Williams, M.; Lu, T.T.; Mansfield, J.C.; Lamb, C.A.; Feagan, B.G.; Panes, J.; Salas, A.; et al. Etrolizumab as induction therapy for ulcerative colitis: A randomised, controlled, phase 2 trial. Lancet 2014, 384, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, E.J.; Chomas, J.E.; Ley, K. Role of primary and secondary capture for leukocyte accumulation in vivo. Circ. Res. 1998, 82, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Picarella, D.; Hurlbut, P.; Rottman, J.; Shi, X.; Butcher, E.; Ringler, D.J. Monoclonal antibodies specific for β7 integrin and mucosal addressin cell adhesion molecule-1 (MAdCAM-1) reduce inflammation in the colon of scid mice reconstituted with CD45RBhigh CD4+ T cells. J. Immunol. 1997, 158, 2099–2106. [Google Scholar] [CrossRef] [PubMed]
- Rettig, M.P.; Ansstas, G.; DiPersio, J.F. Mobilization of hematopoietic stem and progenitor cells using inhibitors of CXCR4 and VLA-4. Leukemia 2012, 26, 34–53. [Google Scholar] [CrossRef]
- Ghobadi, A.; Rettig, M.P.; Cooper, M.L.; Holt, M.S.; Ritchey, J.K.; Eissenberg, L.; DiPersio, J.F. Bortezomib is a rapid mobilizer of hematopoietic stem cells in mice via modulation of the VCAM-1/VLA-4 axis. Blood 2014, 124, 2752–2754. [Google Scholar] [CrossRef]
- Arnaout, M.A. Biology and structure of leukocyte β2 integrins and their role in inflammation. F1000Res 2016, 5, 2433. [Google Scholar] [CrossRef]
- Guenther, C. β2-Integrins—Regulatory and Executive Bridges in the Signaling Network Controlling Leukocyte Trafficking and Migration. Front. Immunol. 2022, 13, 809590. [Google Scholar] [CrossRef]
- McCaffery, P.J.; Berridge, M.V. Expression of the leukocyte functional molecule (LFA-1) on mouse platelets. Blood 1986, 67, 1757–1764. [Google Scholar] [CrossRef]
- Kummer, D.; Ebnet, K. Junctional Adhesion Molecules (JAMs): The JAM-Integrin Connection. Cells 2018, 7, 25. [Google Scholar] [CrossRef]
- Fraemohs, L.; Koenen, R.R.; Ostermann, G.; Heinemann, B.; Weber, C. The functional interaction of the β2 integrin lymphocyte function-associated antigen-1 with junctional adhesion molecule-A is mediated by the I domain. J. Immunol. 2004, 173, 6259–6264. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Hyun, Y.M.; Lambert-Emo, K.; Topham, D.J.; Kim, M. Visualization of integrin Mac-1 in vivo. J. Immunol. Methods 2015, 426, 120–127. [Google Scholar] [CrossRef]
- Li, N.; Yang, H.; Wang, M.; Lu, S.; Zhang, Y.; Long, M. Ligand-specific binding forces of LFA-1 and Mac-1 in neutrophil adhesion and crawling. Mol. Biol. Cell 2018, 29, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.I. Opening the field of integrin biology to “biased agonism”. Circ. Res. 2011, 109, 1199–1201. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C.; Agbanyo, F.R.; Plescia, J.; Ginsberg, M.H.; Edgington, T.S.; Plow, E.F. A unique recognition site mediates the interaction of fibrinogen with the leukocyte integrin Mac-1 (CD11b/CD18). J. Biol. Chem. 1990, 265, 12119–12122. [Google Scholar] [CrossRef]
- Kanse, S.M.; Matz, R.L.; Preissner, K.T.; Peter, K. Promotion of leukocyte adhesion by a novel interaction between vitronectin and the β2 integrin Mac-1 (αMβ2, CD11b/CD18). Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2251–2256. [Google Scholar] [CrossRef]
- Peter, K.; Schwarz, M.; Conradt, C.; Nordt, T.; Moser, M.; Kubler, W.; Bode, C. Heparin inhibits ligand binding to the leukocyte integrin Mac-1 (CD11b/CD18). Circulation 1999, 100, 1533–1539. [Google Scholar] [CrossRef] [PubMed]
- Chavakis, T.; Bierhaus, A.; Al-Fakhri, N.; Schneider, D.; Witte, S.; Linn, T.; Nagashima, M.; Morser, J.; Arnold, B.; Preissner, K.T.; et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: A novel pathway for inflammatory cell recruitment. J. Exp. Med. 2003, 198, 1507–1515. [Google Scholar] [CrossRef]
- Wolf, D.; Anto-Michel, N.; Blankenbach, H.; Wiedemann, A.; Buscher, K.; Hohmann, J.D.; Lim, B.; Bauml, M.; Marki, A.; Mauler, M.; et al. A ligand-specific blockade of the integrin Mac-1 selectively targets pathologic inflammation while maintaining protective host-defense. Nat. Commun. 2018, 9, 525. [Google Scholar] [CrossRef] [PubMed]
- Lishko, V.K.; Podolnikova, N.P.; Yakubenko, V.P.; Yakovlev, S.; Medved, L.; Yadav, S.P.; Ugarova, T.P. Multiple binding sites in fibrinogen for integrin αMβ2 (Mac-1). J. Biol. Chem. 2004, 279, 44897–44906. [Google Scholar] [CrossRef] [PubMed]
- Walzog, B.; Schuppan, D.; Heimpel, C.; Hafezi-Moghadam, A.; Gaehtgens, P.; Ley, K. The leukocyte integrin Mac-1 (CD11b/CD18) contributes to binding of human granulocytes to collagen. Exp. Cell Res. 1995, 218, 28–38. [Google Scholar] [CrossRef]
- Bajic, G.; Yatime, L.; Sim, R.B.; Vorup-Jensen, T.; Andersen, G.R. Structural insight on the recognition of surface-bound opsonins by the integrin I domain of complement receptor 3. Proc. Natl. Acad. Sci. USA 2013, 110, 16426–16431. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.; Pluss, C.J.; Ricklin, D. The Promiscuous Profile of Complement Receptor 3 in Ligand Binding, Immune Modulation, and Pathophysiology. Front. Immunol. 2021, 12, 662164. [Google Scholar] [CrossRef] [PubMed]
- Sandor, N.; Lukacsi, S.; Ungai-Salanki, R.; Orgovan, N.; Szabo, B.; Horvath, R.; Erdei, A.; Bajtay, Z. CD11c/CD18 Dominates Adhesion of Human Monocytes, Macrophages and Dendritic Cells over CD11b/CD18. PLoS ONE 2016, 11, e0163120. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Garcia-Aguilar, J.; Bickford, J.K.; Corbi, A.L.; Springer, T.A. The I domain is a major recognition site on the leukocyte integrin Mac-1 (CD11b/CD18) for four distinct adhesion ligands. J. Cell Biol. 1993, 120, 1031–1043. [Google Scholar] [CrossRef]
- Blackford, J.; Reid, H.W.; Pappin, D.J.; Bowers, F.S.; Wilkinson, J.M. A monoclonal antibody, 3/22, to rabbit CD11c which induces homotypic T cell aggregation: Evidence that ICAM-1 is a ligand for CD11c/CD18. Eur. J. Immunol. 1996, 26, 525–531. [Google Scholar] [CrossRef]
- Ihanus, E.; Uotila, L.M.; Toivanen, A.; Varis, M.; Gahmberg, C.G. Red-cell ICAM-4 is a ligand for the monocyte/macrophage integrin CD11c/CD18: Characterization of the binding sites on ICAM-4. Blood 2007, 109, 802–810. [Google Scholar] [CrossRef]
- Sadhu, C.; Ting, H.J.; Lipsky, B.; Hensley, K.; Garcia-Martinez, L.F.; Simon, S.I.; Staunton, D.E. CD11c/CD18: Novel ligands and a role in delayed-type hypersensitivity. J. Leukoc. Biol. 2007, 81, 1395–1403. [Google Scholar] [CrossRef]
- Ingalls, R.R.; Golenbock, D.T. CD11c/CD18, a transmembrane signaling receptor for lipopolysaccharide. J. Exp. Med. 1995, 181, 1473–1479. [Google Scholar] [CrossRef] [PubMed]
- Loike, J.D.; Sodeik, B.; Cao, L.; Leucona, S.; Weitz, J.I.; Detmers, P.A.; Wright, S.D.; Silverstein, S.C. CD11c/CD18 on neutrophils recognizes a domain at the N terminus of the A alpha chain of fibrinogen. Proc. Natl. Acad. Sci. USA 1991, 88, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Garnotel, R.; Rittie, L.; Poitevin, S.; Monboisse, J.C.; Nguyen, P.; Potron, G.; Maquart, F.X.; Randoux, A.; Gillery, P. Human blood monocytes interact with type I collagen through αxβ2 integrin (CD11c-CD18, gp150-95). J. Immunol. 2000, 164, 5928–5934. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.H.; Cui, K.; Das, M.; Brown, K.E.; Ardell, C.L.; Febbraio, M.; Pluskota, E.; Han, J.; Wu, H.; Ballantyne, C.M.; et al. The Upregulation of Integrin αDβ2 (CD11d/CD18) on Inflammatory Macrophages Promotes Macrophage Retention in Vascular Lesions and Development of Atherosclerosis. J. Immunol. 2017, 198, 4855–4867. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.J.; Scornik, J.C. Serum antibody reactivity of broadly sensitized patients with HLA-matched peripheral blood T lymphocytes. Transplantation 1986, 41, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Brandon, L.; Walling, M.K. LFA-1 in T Cell Migration and Differentiation. Front. Immunol. 2018, 9, 952. [Google Scholar] [CrossRef]
- Kondo, N.; Ueda, Y.; Kinashi, T. LFA1 Activation: Insights from a Single-Molecule Approach. Cells 2022, 11, 1751. [Google Scholar] [CrossRef]
- Luo, B.H.; Carman, C.V.; Springer, T.A. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007, 25, 619–647. [Google Scholar] [CrossRef]
- Shimaoka, M.; Xiao, T.; Liu, J.H.; Yang, Y.; Dong, Y.; Jun, C.D.; McCormack, A.; Zhang, R.; Joachimiak, A.; Takagi, J.; et al. Structures of the αL I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell 2003, 112, 99–111. [Google Scholar] [CrossRef]
- Li, J.; Su, Y.; Xia, W.; Qin, Y.; Humphries, M.J.; Vestweber, D.; Cabanas, C.; Lu, C.; Springer, T.A. Conformational equilibria and intrinsic affinities define integrin activation. EMBO J. 2017, 36, 629–645. [Google Scholar] [CrossRef]
- Li, J.; Springer, T.A. Energy landscape differences among integrins establish the framework for understanding activation. J. Cell Biol. 2018, 217, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, K.; Maeda, A.; Shimonaka, M.; Kinashi, T. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nat. Immunol. 2003, 4, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.K.; Kelleher, D. Not Just an Adhesion Molecule: LFA-1 Contact Tunes the T Lymphocyte Program. J. Immunol. 2017, 199, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Witte, A.; Meineke, B.; Sticht, J.; Philipsen, L.; Kuropka, B.; Muller, A.J.; Freund, C.; Schraven, B.; Kliche, S. D120 and K152 within the PH Domain of T Cell Adapter SKAP55 Regulate Plasma Membrane Targeting of SKAP55 and LFA-1 Affinity Modulation in Human T Lymphocytes. Mol. Cell Biol. 2017, 37, e00509-16. [Google Scholar] [CrossRef] [PubMed]
- Gerard, A.; Cope, A.P.; Kemper, C.; Alon, R.; Kochl, R. LFA-1 in T cell priming, differentiation, and effector functions. Trends Immunol. 2021, 42, 706–722. [Google Scholar] [CrossRef] [PubMed]
- Pflugfelder, S.C.; Stern, M.; Zhang, S.; Shojaei, A. LFA-1/ICAM-1 Interaction as a Therapeutic Target in Dry Eye Disease. J. Ocul. Pharmacol. Ther. 2017, 33, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Schmidt, T.; Cho, E.G.; Ye, F.; Ulmer, T.S.; Ginsberg, M.H. Basic amino-acid side chains regulate transmembrane integrin signalling. Nature 2011, 481, 209–213. [Google Scholar] [CrossRef]
- Verma, N.K.; Kelleher, D. Adaptor regulation of LFA-1 signaling in T lymphocyte migration: Potential druggable targets for immunotherapies? Eur. J. Immunol. 2014, 44, 3484–3499. [Google Scholar] [CrossRef]
- Hogg, N.; Patzak, I.; Willenbrock, F. The insider’s guide to leukocyte integrin signalling and function. Nat. Rev. Immunol. 2011, 11, 416–426. [Google Scholar] [CrossRef]
- Swaim, C.D.; Scott, A.F.; Canadeo, L.A.; Huibregtse, J.M. Extracellular ISG15 Signals Cytokine Secretion through the LFA-1 Integrin Receptor. Mol. Cell 2017, 68, 581–590.e585. [Google Scholar] [CrossRef]
- Sharma, A.; Lawry, S.M.; Klein, B.S.; Wang, X.; Sherer, N.M.; Zumwalde, N.A.; Gumperz, J.E. LFA-1 Ligation by High-Density ICAM-1 Is Sufficient to Activate IFN-γ Release by Innate T Lymphocytes. J. Immunol. 2018, 201, 2452–2461. [Google Scholar] [CrossRef] [PubMed]
- Legate, K.R.; Wickstrom, S.A.; Fassler, R. Genetic and cell biological analysis of integrin outside-in signaling. Genes. Dev. 2009, 23, 397–418. [Google Scholar] [CrossRef] [PubMed]
- Hieber, C.; Grabbe, S.; Bros, M. Counteracting Immunosenescence-Which Therapeutic Strategies Are Promising? Biomolecules 2023, 13, 1085. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, A.; Horwitz, A.R. Integrins in cell migration. Cold Spring Harb. Perspect. Biol. 2011, 3, a005074. [Google Scholar] [CrossRef]
- Simon, S.I.; Hu, Y.; Vestweber, D.; Smith, C.W. Neutrophil tethering on E-selectin activates β2 integrin binding to ICAM-1 through a mitogen-activated protein kinase signal transduction pathway. J. Immunol. 2000, 164, 4348–4358. [Google Scholar] [CrossRef] [PubMed]
- Schubert, K.; Polte, T.; Bönisch, U.; Schader, S.; Holtappels, R.; Hildebrandt, G.; Lehmann, J.; Simon, J.C.; Anderegg, U.; Saalbach, A. Thy-1 (CD90) regulates the extravasation of leukocytes during inflammation. Eur. J. Immunol. 2011, 41, 645–656. [Google Scholar] [CrossRef]
- Phillipson, M.; Heit, B.; Colarusso, P.; Liu, L.; Ballantyne, C.M.; Kubes, P. Intraluminal crawling of neutrophils to emigration sites: A molecularly distinct process from adhesion in the recruitment cascade. J. Exp. Med. 2006, 203, 2569–2575. [Google Scholar] [CrossRef]
- Hyun, Y.M.; Choe, Y.H.; Park, S.A.; Kim, M. LFA-1 (CD11a/CD18) and Mac-1 (CD11b/CD18) distinctly regulate neutrophil extravasation through hotspots I and II. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Sun, H.; Zhi, K.; Hu, L.; Fan, Z. The Activation and Regulation of β2 Integrins in Phagocytes and Phagocytosis. Front. Immunol. 2021, 12, 633639. [Google Scholar] [CrossRef]
- Aderem, A.; Underhill, D.M. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 1999, 17, 593–623. [Google Scholar] [CrossRef] [PubMed]
- Kourtzelis, I.; Hajishengallis, G.; Chavakis, T. Phagocytosis of Apoptotic Cells in Resolution of Inflammation. Front. Immunol. 2020, 11, 553. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, A.G.; Caron, E. Integrin-dependent phagocytosis: Spreading from microadhesion to new concepts. J. Cell Sci. 2008, 121, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Rosales, C.; Uribe-Querol, E. Phagocytosis: A Fundamental Process in Immunity. Biomed. Res. Int. 2017, 2017, 9042851. [Google Scholar] [CrossRef] [PubMed]
- Lukacsi, S.; Nagy-Balo, Z.; Erdei, A.; Sandor, N.; Bajtay, Z. The role of CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in complement-mediated phagocytosis and podosome formation by human phagocytes. Immunol. Lett. 2017, 189, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Skoberne, M.; Somersan, S.; Almodovar, W.; Truong, T.; Petrova, K.; Henson, P.M.; Bhardwaj, N. The apoptotic-cell receptor CR3, but not αvβ5, is a regulator of human dendritic-cell immunostimulatory function. Blood 2006, 108, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Amarilyo, G.; Verbovetski, I.; Atallah, M.; Grau, A.; Wiser, G.; Gil, O.; Ben-Neriah, Y.; Mevorach, D. iC3b-opsonized apoptotic cells mediate a distinct anti-inflammatory response and transcriptional NF-κB-dependent blockade. Eur. J. Immunol. 2010, 40, 699–709. [Google Scholar] [CrossRef]
- Shi, H.; Shao, B. LFA-1 Activation in T-Cell Migration and Immunological Synapse Formation. Cells 2023, 12, 1136. [Google Scholar] [CrossRef]
- Wurzer, H.; Hoffmann, C.; Al Absi, A.; Thomas, C. Actin Cytoskeleton Straddling the Immunological Synapse between Cytotoxic Lymphocytes and Cancer Cells. Cells 2019, 8, 463. [Google Scholar] [CrossRef]
- Varga, G.; Nippe, N.; Balkow, S.; Peters, T.; Wild, M.K.; Seeliger, S.; Beissert, S.; Krummen, M.; Roth, J.; Sunderkotter, C.; et al. LFA-1 contributes to signal I of T-cell activation and to the production of Th1 cytokines. J. Investig. Dermatol. 2010, 130, 1005–1012. [Google Scholar] [CrossRef]
- Balkow, S.; Heinz, S.; Schmidbauer, P.; Kolanus, W.; Holzmann, B.; Grabbe, S.; Laschinger, M. LFA-1 activity state on dendritic cells regulates contact duration with T cells and promotes T-cell priming. Blood 2010, 116, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Varga, G.; Balkow, S.; Wild, M.K.; Stadtbaeumer, A.; Krummen, M.; Rothoeft, T.; Higuchi, T.; Beissert, S.; Wethmar, K.; Scharffetter-Kochanek, K.; et al. Active MAC-1 (CD11b/CD18) on DCs inhibits full T-cell activation. Blood 2007, 109, 661–669. [Google Scholar] [CrossRef]
- Futosi, K.; Fodor, S.; Mócsai, A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int. Immunopharmacol. 2013, 17, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Hellberg, C.; Molony, L.; Zheng, L.; Andersson, T. Ca2+ signalling mechanisms of the β2 integrin on neutrophils: Involvement of phospholipase Cγ2 and Ins(1,4,5)P3. Biochem. J. 1996, 317 Pt 2, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Giagulli, C.; Ottoboni, L.; Caveggion, E.; Rossi, B.; Lowell, C.; Constantin, G.; Laudanna, C.; Berton, G. The Src family kinases Hck and Fgr are dispensable for inside-out, chemoattractant-induced signaling regulating β2 integrin affinity and valency in neutrophils, but are required for β2 integrin-mediated outside-in signaling involved in sustained adhesion. J. Immunol. 2006, 177, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Han, C.; Jin, J.; Qin, K.; Zhang, H.; Li, T.; Li, N.; Cao, X. Integrin CD11b attenuates colitis by strengthening Src-Akt pathway to polarize anti-inflammatory IL-10 expression. Sci. Rep. 2016, 6, 26252. [Google Scholar] [CrossRef] [PubMed]
- Margraf, A.; Germena, G.; Drexler, H.C.A.; Rossaint, J.; Ludwig, N.; Prystaj, B.; Mersmann, S.; Thomas, K.; Block, H.; Gottschlich, W.; et al. The integrin-linked kinase is required for chemokine-triggered high-affinity conformation of the neutrophil β2-integrin LFA-1. Blood 2020, 136, 2200–2205. [Google Scholar] [CrossRef]
- Raab, M.; Lu, Y.; Kohler, K.; Smith, X.; Strebhardt, K.; Rudd, C.E. LFA-1 activates focal adhesion kinases FAK1/PYK2 to generate LAT-GRB2-SKAP1 complexes that terminate T-cell conjugate formation. Nat. Commun. 2017, 8, 16001. [Google Scholar] [CrossRef]
- Sun, X.; Huang, B.; Pan, Y.; Fang, J.; Wang, H.; Ji, Y.; Ling, Y.; Guo, P.; Lin, J.; Li, Q.; et al. Spatiotemporal characteristics of P-selectin-induced β2 integrin activation of human neutrophils under flow. Front. Immunol. 2022, 13, 1023865. [Google Scholar] [CrossRef]
- Bros, M.; Haas, K.; Moll, L.; Grabbe, S. RhoA as a Key Regulator of Innate and Adaptive Immunity. Cells 2019, 8, 733. [Google Scholar] [CrossRef]
- Rezzonico, R.; Imbert, V.; Chicheportiche, R.; Dayer, J.M. Ligation of CD11b and CD11c β2 integrins by antibodies or soluble CD23 induces macrophage inflammatory protein 1α (MIP-1α) and MIP-1β production in primary human monocytes through a pathway dependent on nuclear factor–κB. Blood 2001, 97, 2932–2940. [Google Scholar] [CrossRef]
- Conley, H.E.; Sheats, M.K. Targeting Neutrophil β2-Integrins: A Review of Relevant Resources, Tools, and Methods. Biomolecules 2023, 13, 892. [Google Scholar] [CrossRef] [PubMed]
- Heit, B.; Colarusso, P.; Kubes, P. Fundamentally different roles for LFA-1, Mac-1 and α4-integrin in neutrophil chemotaxis. J. Cell Sci. 2005, 118, 5205–5220. [Google Scholar] [CrossRef]
- Basoni, C.; Nobles, M.; Grimshaw, A.; Desgranges, C.; Davies, D.; Perretti, M.; Kramer, I.M.; Genot, E. Inhibitory control of TGF-β1 on the activation of Rap1, CD11b, and transendothelial migration of leukocytes. FASEB J. 2005, 19, 822–824. [Google Scholar] [CrossRef] [PubMed]
- Aleisa, F.A.; Sakashita, K.; Lee, J.M.; AbuSamra, D.B.; Al Alwan, B.; Nozue, S.; Tehseen, M.; Hamdan, S.M.; Habuchi, S.; Kusakabe, T.; et al. Functional binding of E-selectin to its ligands is enhanced by structural features beyond its lectin domain. J. Biol. Chem. 2020, 295, 3719–3733. [Google Scholar] [CrossRef] [PubMed]
- Zen, K.; Cui, L.B.; Zhang, C.Y.; Liu, Y. Critical role of mac-1 sialyl lewis x moieties in regulating neutrophil degranulation and transmigration. J. Mol. Biol. 2007, 374, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Haist, M.; Ries, F.; Gunzer, M.; Bednarczyk, M.; Siegel, E.; Kuske, M.; Grabbe, S.; Radsak, M.; Bros, M.; Teschner, D. Neutrophil-Specific Knockdown of β2 Integrins Impairs Antifungal Effector Functions and Aggravates the Course of Invasive Pulmonal Aspergillosis. Front. Immunol. 2022, 13, 823121. [Google Scholar] [CrossRef] [PubMed]
- Teschner, D.; Cholaszczyńska, A.; Ries, F.; Beckert, H.; Theobald, M.; Grabbe, S.; Radsak, M.; Bros, M. CD11b Regulates Fungal Outgrowth but Not Neutrophil Recruitment in a Mouse Model of Invasive Pulmonary Aspergillosis. Front. Immunol. 2019, 10, 123. [Google Scholar] [CrossRef]
- Tasaka, S.; Richer, S.E.; Mizgerd, J.P.; Doerschuk, C.M. Very late antigen-4 in CD18-independent neutrophil emigration during acute bacterial pneumonia in mice. Am. J. Respir. Crit. Care Med. 2002, 166, 53–60. [Google Scholar] [CrossRef]
- Mobberley-Schuman, P.S.; Weiss, A.A. Influence of CR3 (CD11b/CD18) expression on phagocytosis of Bordetella pertussis by human neutrophils. Infect. Immun. 2005, 73, 7317–7323. [Google Scholar] [CrossRef]
- Demirdjian, S.; Hopkins, D.; Cumbal, N.; Lefort, C.T.; Berwin, B. Distinct Contributions of CD18 Integrins for Binding and Phagocytic Internalization of Pseudomonas aeruginosa. Infect. Immun. 2020, 88, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Saggu, G.; Okubo, K.; Chen, Y.; Vattepu, R.; Tsuboi, N.; Rosetti, F.; Cullere, X.; Washburn, N.; Tahir, S.; Rosado, A.M.; et al. Cis interaction between sialylated FcγRIIA and the αI-domain of Mac-1 limits antibody-mediated neutrophil recruitment. Nat. Commun. 2018, 9, 5058. [Google Scholar] [CrossRef] [PubMed]
- Thakur, N.; Sodani, R.; Chandra, J.; Singh, V. Leukocyte adhesion defect type 1 presenting with recurrent pyoderma gangrenosum. Indian. J. Dermatol. 2013, 58, 158. [Google Scholar] [CrossRef] [PubMed]
- Gresham, H.D.; Graham, I.L.; Anderson, D.C.; Brown, E.J. Leukocyte adhesion-deficient neutrophils fail to amplify phagocytic function in response to stimulation. Evidence for CD11b/CD18-dependent and -independent mechanisms of phagocytosis. J. Clin. Investig. 1991, 88, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Gierlikowska, B.; Stachura, A.; Gierlikowski, W.; Demkow, U. Phagocytosis, Degranulation and Extracellular Traps Release by Neutrophils-The Current Knowledge, Pharmacological Modulation and Future Prospects. Front. Pharmacol. 2021, 12, 666732. [Google Scholar] [CrossRef] [PubMed]
- Horie, S.; Kita, H. CD11b/CD18 (Mac-1) is required for degranulation of human eosinophils induced by human recombinant granulocyte-macrophage colony-stimulating factor and platelet-activating factor. J. Immunol. 1994, 152, 5457–5467. [Google Scholar] [CrossRef] [PubMed]
- Vorobjeva, N.V.; Chernyak, B.V. NETosis: Molecular Mechanisms, Role in Physiology and Pathology. Biochemistry 2020, 85, 1178–1190. [Google Scholar] [CrossRef]
- Cahilog, Z.; Zhao, H.; Wu, L.; Alam, A.; Eguchi, S.; Weng, H.; Ma, D. The Role of Neutrophil NETosis in Organ Injury: Novel Inflammatory Cell Death Mechanisms. Inflammation 2020, 43, 2021–2032. [Google Scholar] [CrossRef]
- Behnen, M.; Leschczyk, C.; Möller, S.; Batel, T.; Klinger, M.; Solbach, W.; Laskay, T. Immobilized immune complexes induce neutrophil extracellular trap release by human neutrophil granulocytes via FcγRIIIB and Mac-1. J. Immunol. 2014, 193, 1954–1965. [Google Scholar] [CrossRef]
- Papp, K.; Végh, P.; Hóbor, R.; Szittner, Z.; Vokó, Z.; Podani, J.; Czirják, L.; Prechl, J. Immune complex signatures of patients with active and inactive SLE revealed by multiplex protein binding analysis on antigen microarrays. PLoS ONE 2012, 7, e44824. [Google Scholar] [CrossRef]
- McDonald, B.; Urrutia, R.; Yipp, B.G.; Jenne, C.N.; Kubes, P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012, 12, 324–333. [Google Scholar] [CrossRef]
- Silva, J.C.; Rodrigues, N.C.; Thompson-Souza, G.A.; Muniz, V.S.; Neves, J.S.; Figueiredo, R.T. Mac-1 triggers neutrophil DNA extracellular trap formation to Aspergillus fumigatus independently of PAD4 histone citrullination. J. Leukoc. Biol. 2020, 107, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Moutsopoulos, N.M.; Konkel, J.; Sarmadi, M.; Eskan, M.A.; Wild, T.; Dutzan, N.; Abusleme, L.; Zenobia, C.; Hosur, K.B.; Abe, T.; et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci. Transl. Med. 2014, 6, 229ra240. [Google Scholar] [CrossRef] [PubMed]
- Uzel, G.; Kleiner, D.E.; Kuhns, D.B.; Holland, S.M. Dysfunctional LAD-1 neutrophils and colitis. Gastroenterology 2001, 121, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Yuki, K.; Hou, L. Role of β2 Integrins in Neutrophils and Sepsis. Infect. Immun. 2020, 88, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Schittenhelm, L.; Hilkens, C.M.; Morrison, V.L. β2 Integrins as Regulators of Dendritic Cell, Monocyte, and Macrophage Function. Front. Immunol. 2017, 8, 1866. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.K.; McIntyre, L.L.; Marsh, S.E.; Schneider, C.A.; Hoover, E.M.; Walsh, C.M.; Lodoen, M.B.; Blurton-Jones, M.; Inlay, M.A. CD11a expression distinguishes infiltrating myeloid cells from plaque-associated microglia in Alzheimer’s disease. Glia 2019, 67, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Torr, E.E.; Gardner, D.H.; Thomas, L.; Goodall, D.M.; Bielemeier, A.; Willetts, R.; Griffiths, H.R.; Marshall, L.J.; Devitt, A. Apoptotic cell-derived ICAM-3 promotes both macrophage chemoattraction to and tethering of apoptotic cells. Cell Death Differ. 2012, 19, 671–679. [Google Scholar] [CrossRef]
- Kristóf, E.; Zahuczky, G.; Katona, K.; Doró, Z.; Nagy, É.; Fésüs, L. Novel role of ICAM3 and LFA-1 in the clearance of apoptotic neutrophils by human macrophages. Apoptosis 2013, 18, 1235–1251. [Google Scholar] [CrossRef]
- Tang, Z.; Davidson, D.; Li, R.; Zhong, M.C.; Qian, J.; Chen, J.; Veillette, A. Inflammatory macrophages exploit unconventional pro-phagocytic integrins for phagocytosis and anti-tumor immunity. Cell Rep. 2021, 37, 110111. [Google Scholar] [CrossRef]
- Yuan, D.; Zhao, Y.; Banks, W.A.; Bullock, K.M.; Haney, M.; Batrakova, E.; Kabanov, A.V. Macrophage exosomes as natural nanocarriers for protein delivery to inflamed brain. Biomaterials 2017, 142, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.C.; Khan, S.Q.; Kaneda, M.M.; Pathria, P.; Shepard, R.; Louis, T.L.; Anand, S.; Woo, G.; Leem, C.; Faridi, M.H.; et al. Integrin CD11b activation drives anti-tumor innate immunity. Nat. Commun. 2018, 9, 5379. [Google Scholar] [CrossRef] [PubMed]
- Khalaji, S.; Zondler, L.; KleinJan, F.; Nolte, U.; Mulaw, M.A.; Danzer, K.M.; Weishaupt, J.H.; Gottschalk, K.E. Age Increases Monocyte Adhesion on Collagen. Sci. Rep. 2017, 7, 46532. [Google Scholar] [CrossRef] [PubMed]
- Droho, S.; Rajesh, A.; Cuda, C.M.; Perlman, H.; Lavine, J.A. CD11c+ macrophages are proangiogenic and necessary for experimental choroidal neovascularization. JCI Insight 2023, 8, e168142. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Ardell, C.L.; Podolnikova, N.P.; Yakubenko, V.P. Distinct Migratory Properties of M1, M2, and Resident Macrophages Are Regulated by αDβ2 and αMβ2 Integrin-Mediated Adhesion. Front. Immunol. 2018, 9, 2650. [Google Scholar] [CrossRef] [PubMed]
- Van der Vieren, M.; Crowe, D.T.; Hoekstra, D.; Vazeux, R.; Hoffman, P.A.; Grayson, M.H.; Bochner, B.S.; Gallatin, W.M.; Staunton, D.E. The leukocyte integrin αDβ2 binds VCAM-1: Evidence for a binding interface between I domain and VCAM-1. J. Immunol. 1999, 163, 1984–1990. [Google Scholar] [CrossRef] [PubMed]
- Yakubenko, V.P.; Belevych, N.; Mishchuk, D.; Schurin, A.; Lam, S.C.; Ugarova, T.P. The role of integrin αDβ2 (CD11d/CD18) in monocyte/macrophage migration. Exp. Cell Res. 2008, 314, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Zhang, X.; Chen, Z.; Robinson, M.K.; Simon, D.I. Leukocyte integrin Mac-1 recruits toll/interleukin-1 receptor superfamily signaling intermediates to modulate NF-κB activity. Circ. Res. 2001, 89, 859–865. [Google Scholar] [CrossRef]
- Han, C.; Jin, J.; Xu, S.; Liu, H.; Li, N.; Cao, X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat. Immunol. 2010, 11, 734–742. [Google Scholar] [CrossRef]
- Wang, L.; Gordon, R.A.; Huynh, L.; Su, X.; Park Min, K.H.; Han, J.; Arthur, J.S.; Kalliolias, G.D.; Ivashkiv, L.B. Indirect inhibition of Toll-like receptor and type I interferon responses by ITAM-coupled receptors and integrins. Immunity 2010, 32, 518–530. [Google Scholar] [CrossRef]
- Lagarrigue, F.; Kim, C.; Ginsberg, M.H. The Rap1-RIAM-talin axis of integrin activation and blood cell function. Blood 2016, 128, 479–487. [Google Scholar] [CrossRef]
- Querrey, M.; Chiu, S.; Lecuona, E.; Wu, Q.; Sun, H.; Anderson, M.; Kelly, M.; Ravi, S.; Misharin, A.V.; Kreisel, D.; et al. CD11b suppresses TLR activation of nonclassical monocytes to reduce primary graft dysfunction after lung transplantation. J. Clin. Invest. 2022, 132, e157262. [Google Scholar] [CrossRef] [PubMed]
- Yee, N.K.; Hamerman, J.A. β2 integrins inhibit TLR responses by regulating NF-κB pathway and p38 MAPK activation. Eur. J. Immunol. 2013, 43, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Singh-Jasuja, H.; Thiolat, A.; Ribon, M.; Boissier, M.C.; Bessis, N.; Rammensee, H.G.; Decker, P. The mouse dendritic cell marker CD11c is down-regulated upon cell activation through Toll-like receptor triggering. Immunobiology 2013, 218, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Flores-Langarica, A.; Cook, C.; Müller Luda, K.; Persson, E.K.; Marshall, J.L.; Beristain-Covarrubias, N.; Yam-Puc, J.C.; Dahlgren, M.; Persson, J.J.; Uematsu, S.; et al. Intestinal CD103+CD11b+ cDC2 Conventional Dendritic Cells Are Required for Primary CD4+ T and B Cell Responses to Soluble Flagellin. Front. Immunol. 2018, 9, 2409. [Google Scholar] [CrossRef] [PubMed]
- Ling, G.S.; Bennett, J.; Woollard, K.J.; Szajna, M.; Fossati-Jimack, L.; Taylor, P.R.; Scott, D.; Franzoso, G.; Cook, H.T.; Botto, M. Integrin CD11b positively regulates TLR4-induced signalling pathways in dendritic cells but not in macrophages. Nat. Commun. 2014, 5, 3039. [Google Scholar] [CrossRef] [PubMed]
- Stadtmann, A.; Zarbock, A. The role of kindlin in neutrophil recruitment to inflammatory sites. Curr. Opin. Hematol. 2017, 24, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Quast, T.; Tappertzhofen, B.; Schild, C.; Grell, J.; Czeloth, N.; Forster, R.; Alon, R.; Fraemohs, L.; Dreck, K.; Weber, C.; et al. Cytohesin-1 controls the activation of RhoA and modulates integrin-dependent adhesion and migration of dendritic cells. Blood 2009, 113, 5801–5810. [Google Scholar] [CrossRef] [PubMed]
- Heufler, C.; Ortner, D.; Hofer, S. Cybr, CYTIP or CASP: An attempt to pinpoint a molecule’s functions and names. Immunobiology 2008, 213, 729–732. [Google Scholar] [CrossRef]
- Theodoridis, A.A.; Eich, C.; Figdor, C.G.; Steinkasserer, A. Infection of dendritic cells with herpes simplex virus type 1 induces rapid degradation of CYTIP, thereby modulating adhesion and migration. Blood 2011, 118, 107–115. [Google Scholar] [CrossRef]
- Grosche, L.; Drassner, C.; Muhl-Zurbes, P.; Kamm, L.; Le-Trilling, V.T.K.; Trilling, M.; Steinkasserer, A.; Heilingloh, C.S. Human Cytomegalovirus-Induced Degradation of CYTIP Modulates Dendritic Cell Adhesion and Migration. Front. Immunol. 2017, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Bednarczyk, M.; Bolduan, V.; Haist, M.; Stege, H.; Hieber, C.; Johann, L.; Schelmbauer, C.; Blanfeld, M.; Karram, K.; Schunke, J.; et al. β2 Integrins on Dendritic Cells Modulate Cytokine Signaling and Inflammation-Associated Gene Expression, and Are Required for Induction of Autoimmune Encephalomyelitis. Cells 2022, 11, 2188. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.H.; Zhang, Y.; Jiang, L.Q.; Wang, S.; Lei, C.Q.; Sun, M.S.; Shu, H.B.; Liu, Y. WDFY1 mediates TLR3/4 signaling by recruiting TRIF. EMBO Rep. 2015, 16, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Bartuzi, P.; Hofker, M.H.; van de Sluis, B. Tuning NF-κB activity: A touch of COMMD proteins. Biochim. Biophys. Acta 2013, 1832, 2315–2321. [Google Scholar] [CrossRef] [PubMed]
- Podgrabinska, S.; Kamalu, O.; Mayer, L.; Shimaoka, M.; Snoeck, H.; Randolph, G.J.; Skobe, M. Inflamed lymphatic endothelium suppresses dendritic cell maturation and function via Mac-1/ICAM-1-dependent mechanism. J. Immunol. 2009, 183, 1767–1779. [Google Scholar] [CrossRef] [PubMed]
- Ehirchiou, D.; Xiong, Y.; Xu, G.; Chen, W.; Shi, Y.; Zhang, L. CD11b facilitates the development of peripheral tolerance by suppressing Th17 differentiation. J. Exp. Med. 2007, 204, 1519–1524. [Google Scholar] [CrossRef]
- Nording, H.; Sauter, M.; Lin, C.; Steubing, R.; Geisler, S.; Sun, Y.; Niethammer, J.; Emschermann, F.; Wang, Y.; Zieger, B.; et al. Activated Platelets Upregulate β2 Integrin Mac-1 (CD11b/CD18) on Dendritic Cells, Which Mediates Heterotypic Cell-Cell Interaction. J. Immunol. 2022, 208, 1729–1741. [Google Scholar] [CrossRef]
- Zhao, Z.B.; Lu, F.T.; Ma, H.D.; Wang, Y.H.; Yang, W.; Long, J.; Miao, Q.; Zhang, W.; Tian, Z.; Ridgway, W.M.; et al. Liver-resident NK cells suppress autoimmune cholangitis and limit the proliferation of CD4+ T cells. Cell Mol. Immunol. 2020, 17, 178–189. [Google Scholar] [CrossRef]
- Mahapatra, S.; Shearer, W.T.; Minard, C.G.; Mace, E.; Paul, M.; Orange, J.S. NK cells in treated HIV-infected children display altered phenotype and function. J. Allergy Clin. Immunol. 2019, 144, 294–303.e213. [Google Scholar] [CrossRef]
- Crozat, K.; Eidenschenk, C.; Jaeger, B.N.; Krebs, P.; Guia, S.; Beutler, B.; Vivier, E.; Ugolini, S. Impact of β2 integrin deficiency on mouse natural killer cell development and function. Blood 2011, 117, 2874–2882. [Google Scholar] [CrossRef]
- Siegers, G.M.; Barreira, C.R.; Postovit, L.M.; Dekaban, G.A. CD11d β2 integrin expression on human NK, B, and γδ T cells. J. Leukoc. Biol. 2017, 101, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Urlaub, D.; Höfer, K.; Müller, M.L.; Watzl, C. LFA-1 Activation in NK Cells and Their Subsets: Influence of Receptors, Maturation, and Cytokine Stimulation. J. Immunol. 2017, 198, 1944–1951. [Google Scholar] [CrossRef] [PubMed]
- Nandagopal, S.; Li, C.G.; Xu, Y.; Sodji, Q.H.; Graves, E.E.; Giaccia, A.J. C3aR Signaling Inhibits NK-cell Infiltration into the Tumor Microenvironment in Mouse Models. Cancer Immunol. Res. 2022, 10, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.C.; Cohnen, A.; Ludwig, T.; Watzl, C. 2B4 engagement mediates rapid LFA-1 and actin-dependent NK cell adhesion to tumor cells as measured by single cell force spectroscopy. J. Immunol. 2011, 186, 2757–2764. [Google Scholar] [CrossRef] [PubMed]
- Enqvist, M.; Ask, E.H.; Forslund, E.; Carlsten, M.; Abrahamsen, G.; Béziat, V.; Andersson, S.; Schaffer, M.; Spurkland, A.; Bryceson, Y.; et al. Coordinated expression of DNAM-1 and LFA-1 in educated NK cells. J. Immunol. 2015, 194, 4518–4527. [Google Scholar] [CrossRef] [PubMed]
- Pariani, A.P.; Almada, E.; Hidalgo, F.; Borini-Etichetti, C.; Vena, R.; Marín, L.; Favre, C.; Goldenring, J.R.; Cecilia Larocca, M. Identification of a novel mechanism for LFA-1 organization during NK cytolytic response. J. Cell Physiol. 2023, 238, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Perez, O.D.; Mitchell, D.; Jager, G.C.; Nolan, G.P. LFA-1 signaling through p44/42 is coupled to perforin degranulation in CD56+CD8+ natural killer cells. Blood 2004, 104, 1083–1093. [Google Scholar] [CrossRef]
- March, M.E.; Long, E.O. β2 integrin induces TCRζ-Syk-phospholipase C-γ phosphorylation and paxillin-dependent granule polarization in human NK cells. J. Immunol. 2011, 186, 2998–3005. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Smyth, M.J. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J. Immunol. 2006, 176, 1517–1524. [Google Scholar] [CrossRef]
- Thomas, L.M.; Peterson, M.E.; Long, E.O. Cutting edge: NK cell licensing modulates adhesion to target cells. J. Immunol. 2013, 191, 3981–3985. [Google Scholar] [CrossRef]
- Milićević, N.M.; Milićević, Z.; Westermann, J. Lymphocyte function-associated antigen-1 and intercellular adhesion molecule-1 expression on B-cell subsets and the effects of splenectomy-experimental studies. Leuk. Lymphoma 2002, 43, 2071–2074. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Tsuno, N.H.; Matsuhashi, M.; Kitayama, J.; Osada, T.; Yamada, J.; Tsuchiya, T.; Yoneyama, S.; Watanabe, T.; Takahashi, K.; et al. CD11b-mediated migratory property of peripheral blood B cells. J. Allergy Clin. Immunol. 2005, 116, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Golinski, M.L.; Demeules, M.; Derambure, C.; Riou, G.; Maho-Vaillant, M.; Boyer, O.; Joly, P.; Calbo, S. CD11c+ B Cells Are Mainly Memory Cells, Precursors of Antibody Secreting Cells in Healthy Donors. Front. Immunol. 2020, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Makgoba, M.W.; Sanders, M.E.; Ginther Luce, G.E.; Dustin, M.L.; Springer, T.A.; Clark, E.A.; Mannoni, P.; Shaw, S. ICAM-1 a ligand for LFA-1-dependent adhesion of B, T and myeloid cells. Nature 1988, 331, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Camponeschi, A.; Gerasimcik, N.; Wang, Y.; Fredriksson, T.; Chen, D.; Farroni, C.; Thorarinsdottir, K.; Sjökvist Ottsjö, L.; Aranburu, A.; Cardell, S.; et al. Dissecting Integrin Expression and Function on Memory B Cells in Mice and Humans in Autoimmunity. Front. Immunol. 2019, 10, 534. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Ma, Y.; Chen, X.; Liu, M.; Cai, Y.; Hu, X.; Xiang, D.; Nath, S.; Zhang, H.G.; Ye, H.; et al. Integrin CD11b negatively regulates BCR signalling to maintain autoreactive B cell tolerance. Nat. Commun. 2013, 4, 2813. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, M. The ontogeny of murine B-1a cells. Int. J. Hematol. 2020, 111, 622–627. [Google Scholar] [CrossRef]
- Griffin, D.O.; Rothstein, T.L. A small CD11b+ human B1 cell subpopulation stimulates T cells and is expanded in lupus. J. Exp. Med. 2011, 208, 2591–2598. [Google Scholar] [CrossRef]
- Rubtsov, A.V.; Rubtsova, K.; Fischer, A.; Meehan, R.T.; Gillis, J.Z.; Kappler, J.W.; Marrack, P. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c⁺ B-cell population is important for the development of autoimmunity. Blood 2011, 118, 1305–1315. [Google Scholar] [CrossRef]
- Erdei, A.; Kovács, K.G.; Nagy-Baló, Z.; Lukácsi, S.; Mácsik-Valent, B.; Kurucz, I.; Bajtay, Z. New aspects in the regulation of human B cell functions by complement receptors CR1, CR2, CR3 and CR4. Immunol. Lett. 2021, 237, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Nagy-Balo, Z.; Kiss, R.; Menge, A.; Bodor, C.; Bajtay, Z.; Erdei, A. Activated Human Memory B Lymphocytes Use CR4 (CD11c/CD18) for Adhesion, Migration, and Proliferation. Front. Immunol. 2020, 11, 565458. [Google Scholar] [CrossRef] [PubMed]
- Van der Poel, C.E.; Bajic, G.; Macaulay, C.W.; van den Broek, T.; Ellson, C.D.; Bouma, G.; Victora, G.D.; Degn, S.E.; Carroll, M.C. Follicular Dendritic Cells Modulate Germinal Center B Cell Diversity through FcγRIIB. Cell Rep. 2019, 29, 2745–2755.e2744. [Google Scholar] [CrossRef] [PubMed]
- Rincon-Arevalo, H.; Wiedemann, A.; Stefanski, A.L.; Lettau, M.; Szelinski, F.; Fuchs, S.; Frei, A.P.; Steinberg, M.; Kam-Thong, T.; Hatje, K.; et al. Deep Phenotyping of CD11c+ B Cells in Systemic Autoimmunity and Controls. Front. Immunol. 2021, 12, 635615. [Google Scholar] [CrossRef] [PubMed]
- Lúcio, P.J.; Faria, M.T.; Pinto, A.M.; da Silva, M.R.; Correia Júnior, M.E.; da Costa, R.J.; Parreira, A.B. Expression of adhesion molecules in chronic B-cell lymphoproliferative disorders. Haematologica 1998, 83, 104–111. [Google Scholar] [PubMed]
- Uzonyi, B.; Mácsik-Valent, B.; Lukácsi, S.; Kiss, R.; Török, K.; Kremlitzka, M.; Bajtay, Z.; Demeter, J.; Bödör, C.; Erdei, A. Functional studies of chronic lymphocytic leukemia B cells expressing β2-integrin type complement receptors CR3 and CR4. Immunol. Lett. 2017, 189, 73–81. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Song, W.; Antao, O.Q.; Condiff, E.; Sanchez, G.M.; Chernova, I.; Zembrzuski, K.; Steach, H.; Rubtsova, K.; Angeletti, D.; Lemenze, A.; et al. Development of Tbet- and CD11c-expressing B cells in a viral infection requires T follicular helper cells outside of germinal centers. Immunity 2022, 55, 290–307.e295. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L. Cell adhesion molecules and actin cytoskeleton at immune synapses and kinapses. Curr. Opin. Cell Biol. 2007, 19, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Ueda, Y.; Kita, T.; Ozawa, M.; Tomiyama, T.; Yasuda, K.; Lim, D.S.; Kinashi, T. NDR1-Dependent Regulation of Kindlin-3 Controls High-Affinity LFA-1 Binding and Immune Synapse Organization. Mol. Cell Biol. 2017, 37, e00424-16. [Google Scholar] [CrossRef]
- Alarcon, B.; Mestre, D.; Martinez-Martin, N. The immunological synapse: A cause or consequence of T-cell receptor triggering? Immunology 2011, 133, 420–425. [Google Scholar] [CrossRef]
- Comrie, W.A.; Burkhardt, J.K. Action and Traction: Cytoskeletal Control of Receptor Triggering at the Immunological Synapse. Front. Immunol. 2016, 7, 68. [Google Scholar] [CrossRef]
- Le Floc’h, A.; Huse, M. Molecular mechanisms and functional implications of polarized actin remodeling at the T cell immunological synapse. Cell Mol. Life Sci. 2015, 72, 537–556. [Google Scholar] [CrossRef]
- Xia, S.; Chen, Q.; Niu, B. CD28: A New Drug Target for Immune Disease. Curr. Drug Targets 2020, 21, 589–598. [Google Scholar] [CrossRef]
- Dustin, M.L. Hunter to gatherer and back: Immunological synapses and kinapses as variations on the theme of amoeboid locomotion. Annu. Rev. Cell Dev. Biol. 2008, 24, 577–596. [Google Scholar] [CrossRef] [PubMed]
- Shimaoka, M.; Takagi, J.; Springer, T.A. Conformational regulation of integrin structure and function. Annu. Rev. Biophys. Biomol. Struct. 2002, 31, 485–516. [Google Scholar] [CrossRef]
- Bleijs, D.A.; Binnerts, M.E.; van Vliet, S.J.; Figdor, C.G.; van Kooyk, Y. Low-affinity LFA-1/ICAM-3 interactions augment LFA-1/ICAM-1-mediated T cell adhesion and signaling by redistribution of LFA-1. J. Cell Sci. 2000, 113 Pt 3, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.K.; Dempsey, E.; Long, A.; Davies, A.; Barry, S.P.; Fallon, P.G.; Volkov, Y.; Kelleher, D. Leukocyte function-associated antigen-1/intercellular adhesion molecule-1 interaction induces a novel genetic signature resulting in T-cells refractory to transforming growth factor-beta signaling. J. Biol. Chem. 2012, 287, 27204–27216. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.K.; Dourlat, J.; Davies, A.M.; Long, A.; Liu, W.Q.; Garbay, C.; Kelleher, D.; Volkov, Y. STAT3-stathmin interactions control microtubule dynamics in migrating T-cells. J. Biol. Chem. 2009, 284, 12349–12362. [Google Scholar] [CrossRef] [PubMed]
- Shevyrev, D.; Tereshchenko, V. Treg Heterogeneity, Function, and Homeostasis. Front. Immunol. 2019, 10, 3100. [Google Scholar] [CrossRef]
- Rocamora-Reverte, L.; Melzer, F.L.; Wurzner, R.; Weinberger, B. The Complex Role of Regulatory T Cells in Immunity and Aging. Front. Immunol. 2020, 11, 616949. [Google Scholar] [CrossRef]
- Hogquist, K.A.; Bevan, M.J. The nature of the peptide/MHC ligand involved in positive selection. Semin. Immunol. 1996, 8, 63–68. [Google Scholar] [CrossRef]
- Ness, S.; Lin, S.; Gordon, J.R. Regulatory Dendritic Cells, T Cell Tolerance, and Dendritic Cell Therapy for Immunologic Disease. Front. Immunol. 2021, 12, 633436. [Google Scholar] [CrossRef] [PubMed]
- Marski, M.; Kandula, S.; Turner, J.R.; Abraham, C. CD18 is required for optimal development and function of CD4+CD25+ T regulatory cells. J. Immunol. 2005, 175, 7889–7897. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Gatzka, M.; Peters, T.; Borkner, L.; Hainzl, A.; Wang, H.; Sindrilaru, A.; Scharffetter-Kochanek, K. Reduced CD18 levels drive regulatory T cell conversion into Th17 cells in the CD18hypo PL/J mouse model of psoriasis. J. Immunol. 2013, 190, 2544–2553. [Google Scholar] [CrossRef] [PubMed]
- Verhagen, J.; Wraith, D.C. Blockade of LFA-1 augments in vitro differentiation of antigen-induced Foxp3⁺ Treg cells. J. Immunol. Methods 2014, 414, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Powrie, F.; Carlino, J.; Leach, M.W.; Mauze, S.; Coffman, R.L. A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J. Exp. Med. 1996, 183, 2669–2674. [Google Scholar] [CrossRef] [PubMed]
- Goswami, T.K.; Singh, M.; Dhawan, M.; Mitra, S.; Emran, T.B.; Rabaan, A.A.; Mutair, A.A.; Alawi, Z.A.; Alhumaid, S.; Dhama, K. Regulatory T cells (Tregs) and their therapeutic potential against autoimmune disorders—Advances and challenges. Hum. Vaccin. Immunother. 2022, 18, 2035117. [Google Scholar] [CrossRef]
- Gondek, D.C.; Lu, L.F.; Quezada, S.A.; Sakaguchi, S.; Noelle, R.J. Cutting edge: Contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J. Immunol. 2005, 174, 1783–1786. [Google Scholar] [CrossRef]
- Grossman, W.J.; Verbsky, J.W.; Tollefsen, B.L.; Kemper, C.; Atkinson, J.P.; Ley, T.J. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood 2004, 104, 2840–2848. [Google Scholar] [CrossRef]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef]
- Fletcher, J.M.; Lonergan, R.; Costelloe, L.; Kinsella, K.; Moran, B.; O’Farrelly, C.; Tubridy, N.; Mills, K.H. CD39+Foxp3+ regulatory T Cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J. Immunol. 2009, 183, 7602–7610. [Google Scholar] [CrossRef] [PubMed]
- Safinia, N.; Scotta, C.; Vaikunthanathan, T.; Lechler, R.I.; Lombardi, G. Regulatory T Cells: Serious Contenders in the Promise for Immunological Tolerance in Transplantation. Front. Immunol. 2015, 6, 438. [Google Scholar] [CrossRef] [PubMed]
- Read, S.; Malmstrom, V.; Powrie, F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J. Exp. Med. 2000, 192, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Onishi, Y.; Fehervari, Z.; Yamaguchi, T.; Sakaguchi, S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc. Natl. Acad. Sci. USA 2008, 105, 10113–10118. [Google Scholar] [CrossRef] [PubMed]
- Tran, D. Dysfunctional FOXP3+ Regulatory T cells in Leukocyte Adhesion Deficiency Type 1 (LAD-1) Patients with Reversion Mutations and Inflammatory Bowel Disease. J. Allergy Clin. Immunol. 2009, 123, S15. [Google Scholar] [CrossRef]
- Chen, J.; Ganguly, A.; Mucsi, A.D.; Meng, J.; Yan, J.; Detampel, P.; Munro, F.; Zhang, Z.; Wu, M.; Hari, A.; et al. Strong adhesion by regulatory T cells induces dendritic cell cytoskeletal polarization and contact-dependent lethargy. J. Exp. Med. 2017, 214, 327–338. [Google Scholar] [CrossRef]
- Klaus, T.; Wilson, A.S.; Vicari, E.; Hadaschik, E.; Klein, M.; Helbich, S.S.C.; Kamenjarin, N.; Hodapp, K.; Schunke, J.; Haist, M.; et al. Impaired Treg-DC interactions contribute to autoimmunity in leukocyte adhesion deficiency type 1. JCI Insight 2022, 7, e162580. [Google Scholar] [CrossRef]
- Wang, M.S.; Hu, Y.; Sanchez, E.E.; Xie, X.; Roy, N.H.; de Jesus, M.; Winer, B.Y.; Zale, E.A.; Jin, W.; Sachar, C.; et al. Mechanically active integrins target lytic secretion at the immune synapse to facilitate cellular cytotoxicity. Nat. Commun. 2022, 13, 3222. [Google Scholar] [CrossRef]
- Basu, R.; Whitlock, B.M.; Husson, J.; Le Floc’h, A.; Jin, W.; Oyler-Yaniv, A.; Dotiwala, F.; Giannone, G.; Hivroz, C.; Biais, N.; et al. Cytotoxic T Cells Use Mechanical Force to Potentiate Target Cell Killing. Cell 2016, 165, 100–110. [Google Scholar] [CrossRef]
- Ouyang, K.; Oparaugo, N.; Nelson, A.M.; Agak, G.W. T Cell Extracellular Traps: Tipping the Balance Between Skin Health and Disease. Front. Immunol. 2022, 13, 900634. [Google Scholar] [CrossRef]
- Koh, C.C.; Wardini, A.B.; Vieira, M.; Passos, L.S.A.; Martinelli, P.M.; Neves, E.G.A.; Antonelli, L.; Barbosa, D.F.; Velikkakam, T.; Gutseit, E.; et al. Human CD8+ T Cells Release Extracellular Traps Co-Localized with Cytotoxic Vesicles That Are Associated with Lesion Progression and Severity in Human Leishmaniasis. Front. Immunol. 2020, 11, 594581. [Google Scholar] [CrossRef] [PubMed]
- Roos, D.; van Leeuwen, K.; Madkaikar, M.; Kambli, P.M.; Gupta, M.; Mathews, V.; Rawat, A.; Kuhns, D.B.; Holland, S.M.; de Boer, M.; et al. Hematologically important mutations: Leukocyte adhesion deficiency (second update). Blood Cells Mol. Dis. 2023, 99, 102726. [Google Scholar] [CrossRef] [PubMed]
- Fekadu, J.; Modlich, U.; Bader, P.; Bakhtiar, S. Understanding the Role of LFA-1 in Leukocyte Adhesion Deficiency Type I (LAD I): Moving towards Inflammation? Int. J. Mol. Sci. 2022, 23, 3578. [Google Scholar] [CrossRef]
- Cox, D.P.; Weathers, D.R. Leukocyte adhesion deficiency type 1: An important consideration in the clinical differential diagnosis of prepubertal periodontitis. A case report and review of the literature. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. Endod. 2008, 105, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, T.W.; Van Lier, R.A.; Hamann, D.; de Boer, M.; Thung, L.Y.; Weening, R.S.; Verhoeven, A.J.; Roos, D. Leukocyte adhesion deficiency type 1 (LAD-1)/variant. A novel immunodeficiency syndrome characterized by dysfunctional β2 integrins. J. Clin. Investig. 1997, 100, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, T.W.; van Bruggen, R.; Kamerbeek, N.; Tool, A.T.; Hicsonmez, G.; Gurgey, A.; Karow, A.; Verhoeven, A.J.; Seeger, K.; Sanal, O.; et al. Natural history and early diagnosis of LAD-1/variant syndrome. Blood 2007, 109, 3529–3537. [Google Scholar] [CrossRef]
- Kambli, P.M.; Bargir, U.A.; Yadav, R.M.; Gupta, M.R.; Dalvi, A.D.; Hule, G.; Kelkar, M.; Sawant-Desai, S.; Setia, P.; Jodhawat, N.; et al. Clinical and Genetic Spectrum of a Large Cohort of Patients with Leukocyte Adhesion Deficiency Type 1 and 3: A Multicentric Study from India. Front. Immunol. 2020, 11, 612703. [Google Scholar] [CrossRef]
- Almarza Novoa, E.; Kasbekar, S.; Thrasher, A.J.; Kohn, D.B.; Sevilla, J.; Nguyen, T.; Schwartz, J.D.; Bueren, J.A. Leukocyte adhesion deficiency-I: A comprehensive review of all published cases. J. Allergy Clin. Immunol. Pract. 2018, 6, 1418–1420.e1410. [Google Scholar] [CrossRef]
- Cabanillas, D.; Regairaz, L.; Deswarte, C.; Garcia, M.; Richard, M.E.; Casanova, J.L.; Bustamante, J.; Perez, L. Leukocyte Adhesion Deficiency Type 1 (LAD1) with Expressed but Nonfunctional CD11/CD18. J. Clin. Immunol. 2016, 36, 627–630. [Google Scholar] [CrossRef]
- De Rose, D.U.; Giliani, S.; Notarangelo, L.D.; Lougaris, V.; Lanfranchi, A.; Moratto, D.; Martire, B.; Specchia, F.; Tommasini, A.; Plebani, A.; et al. Long term outcome of eight patients with type 1 Leukocyte Adhesion Deficiency (LAD-1): Not only infections, but high risk of autoimmune complications. Clin. Immunol. 2018, 191, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Hanna, S.; Etzioni, A. Leukocyte adhesion deficiencies. Ann. N. Y. Acad. Sci. 2012, 1250, 50–55. [Google Scholar] [CrossRef]
- Justiz, V. Leukocyte Adhesion Deficiency; SatPearls: St. Petersburg, FL, USA, 2022. [Google Scholar]
- Tran, D.Q.; Glass, D.D.; Uzel, G.; Darnell, D.A.; Spalding, C.; Holland, S.M.; Shevach, E.M. Analysis of adhesion molecules, target cells, and role of IL-2 in human FOXP3+ regulatory T cell suppressor function. J. Immunol. 2009, 182, 2929–2938. [Google Scholar] [CrossRef] [PubMed]
- Uzel, G.; Tng, E.; Rosenzweig, S.D.; Hsu, A.P.; Shaw, J.M.; Horwitz, M.E.; Linton, G.F.; Anderson, S.M.; Kirby, M.R.; Oliveira, J.B.; et al. Reversion mutations in patients with leukocyte adhesion deficiency type-1 (LAD-1). Blood 2008, 111, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Qasim, W.; Cavazzana-Calvo, M.; Davies, E.G.; Davis, J.; Duval, M.; Eames, G.; Farinha, N.; Filopovich, A.; Fischer, A.; Friedrich, W.; et al. Allogeneic hematopoietic stem-cell transplantation for leukocyte adhesion deficiency. Pediatrics 2009, 123, 836–840. [Google Scholar] [CrossRef] [PubMed]
- Skurska, E.; Szulc, B.; Maszczak-Seneczko, D.; Wiktor, M.; Wiertelak, W.; Makowiecka, A.; Olczak, M. Incorporation of fucose into glycans independent of the GDP-fucose transporter SLC35C1 preferentially utilizes salvaged over de novo GDP-fucose. J. Biol. Chem. 2022, 298, 102206. [Google Scholar] [CrossRef]
- Jin, F.; Wang, F. The physiological and pathological roles and applications of sialyl Lewis x, a common carbohydrate ligand of the three selectins. Glycoconj. J. 2020, 37, 277–291. [Google Scholar] [CrossRef]
- Tahata, S.; Raymond, K.; Quade, M.; Barnes, S.; Boyer, S.; League, S.; Kumanovics, A.; Abraham, R.; Jacob, E.; Menon, P.; et al. Defining the mild variant of leukocyte adhesion deficiency type II (SLC35C1-congenital disorder of glycosylation) and response to l-fucose therapy: Insights from two new families and review of the literature. Am. J. Med. Genet. A 2022, 188, 2005–2018. [Google Scholar] [CrossRef]
- Lühn, K.; Marquardt, T.; Harms, E.; Vestweber, D. Discontinuation of fucose therapy in LADII causes rapid loss of selectin ligands and rise of leukocyte counts. Blood 2001, 97, 330–332. [Google Scholar] [CrossRef]
- Harris, E.S.; Smith, T.L.; Springett, G.M.; Weyrich, A.S.; Zimmerman, G.A. Leukocyte adhesion deficiency-I variant syndrome (LAD-Iv, LAD-III): Molecular characterization of the defect in an index family. Am. J. Hematol. 2012, 87, 311–313. [Google Scholar] [CrossRef]
- Fagerholm, S.C.; Lek, H.S.; Morrison, V.L. Kindlin-3 in the immune system. Am. J. Clin. Exp. Immunol. 2014, 3, 37–42. [Google Scholar]
- Svensson, L.; Howarth, K.; McDowall, A.; Patzak, I.; Evans, R.; Ussar, S.; Moser, M.; Metin, A.; Fried, M.; Tomlinson, I.; et al. Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat. Med. 2009, 15, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Manevich-Mendelson, E.; Feigelson, S.W.; Pasvolsky, R.; Aker, M.; Grabovsky, V.; Shulman, Z.; Kilic, S.S.; Rosenthal-Allieri, M.A.; Ben-Dor, S.; Mory, A.; et al. Loss of Kindlin-3 in LAD-III eliminates LFA-1 but not VLA-4 adhesiveness developed under shear flow conditions. Blood 2009, 114, 2344–2353. [Google Scholar] [CrossRef]
- Kilic, S.S.; Etzioni, A. The clinical spectrum of leukocyte adhesion deficiency (LAD) III due to defective CalDAG-GEF1. J. Clin. Immunol. 2009, 29, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Bledzka, K.; Smyth, S.S.; Plow, E.F. Integrin αIIbβ3: From discovery to efficacious therapeutic target. Circ. Res. 2013, 112, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Crazzolara, R.; Maurer, K.; Schulze, H.; Zieger, B.; Zustin, J.; Schulz, A.S. A new mutation in the KINDLIN-3 gene ablates integrin-dependent leukocyte, platelet, and osteoclast function in a patient with leukocyte adhesion deficiency-III. Pediatr. Blood Cancer 2015, 62, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Nakchbandi, I.; Ruppert, R.; Kawelke, N.; Hess, M.W.; Pfaller, K.; Jurdic, P.; Fässler, R.; Moser, M. Kindlin-3-mediated signaling from multiple integrin classes is required for osteoclast-mediated bone resorption. J. Cell Biol. 2011, 192, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Saultier, P.; Szepetowski, S.; Canault, M.; Falaise, C.; Poggi, M.; Suchon, P.; Barlogis, V.; Michel, G.; Loyau, S.; Jandrot-Perrus, M.; et al. Long-term management of leukocyte adhesion deficiency type III without hematopoietic stem cell transplantation. Haematologica 2018, 103, e264–e267. [Google Scholar] [CrossRef] [PubMed]
- Dejaco, C.; Duftner, C.; Grubeck-Loebenstein, B.; Schirmer, M. Imbalance of regulatory T cells in human autoimmune diseases. Immunology 2006, 117, 289–300. [Google Scholar] [CrossRef]
- Cao, D.; van Vollenhoven, R.; Klareskog, L.; Trollmo, C.; Malmstrom, V. CD25brightCD4+ regulatory T cells are enriched in inflamed joints of patients with chronic rheumatic disease. Arthritis Res. Ther. 2004, 6, R335–R346. [Google Scholar] [CrossRef]
- Liu, M.F.; Wang, C.R.; Fung, L.L.; Wu, C.R. Decreased CD4+CD25+ T cells in peripheral blood of patients with systemic lupus erythematosus. Scand. J. Immunol. 2004, 59, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Crispin, J.C.; Martinez, A.; Alcocer-Varela, J. Quantification of regulatory T cells in patients with systemic lupus erythematosus. J. Autoimmun. 2003, 21, 273–276. [Google Scholar] [CrossRef]
- Furuno, K.; Yuge, T.; Kusuhara, K.; Takada, H.; Nishio, H.; Khajoee, V.; Ohno, T.; Hara, T. CD25+CD4+ regulatory T cells in patients with Kawasaki disease. J. Pediatr. 2004, 145, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.P.; Hansda, A.K.; Goswami, R. Rheumatoid arthritis: ‘melting pot’ of T helper subsets. Int. Rev. Immunol. 2019, 38, 212–231. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.K.; Fazil, M.H.; Ong, S.T.; Chalasani, M.L.; Low, J.H.; Kottaiswamy, A.; Praseetha, P.; Kizhakeyil, A.; Kumar, S.; Panda, A.K.; et al. LFA-1/ICAM-1 Ligation in Human T Cells Promotes Th1 Polarization through a GSK3β Signaling-Dependent Notch Pathway. J. Immunol. 2016, 197, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Brandstadter, J.D.; Maillard, I. Notch signalling in T cell homeostasis and differentiation. Open Biol. 2019, 9, 190187. [Google Scholar] [CrossRef] [PubMed]
- Ley, K. The second touch hypothesis: T cell activation, homing and polarization. F1000Res 2014, 3, 37. [Google Scholar] [CrossRef] [PubMed]
- Gensterblum, E.; Renauer, P.; Coit, P.; Strickland, F.M.; Kilian, N.C.; Miller, S.; Ognenovski, M.; Wren, J.D.; Tsou, P.S.; Lewis, E.E.; et al. CD4+CD28+KIR+CD11ahi T cells correlate with disease activity and are characterized by a pro-inflammatory epigenetic and transcriptional profile in lupus patients. J. Autoimmun. 2018, 86, 19–28. [Google Scholar] [CrossRef]
- Nicolls, M.R.; Gill, R.G. LFA-1 (CD11a) as a therapeutic target. Am. J. Transplant. 2006, 6, 27–36. [Google Scholar] [CrossRef]
- Nicolls, M.R.; Coulombe, M.; Beilke, J.; Gelhaus, H.C.; Gill, R.G. CD4-dependent generation of dominant transplantation tolerance induced by simultaneous perturbation of CD154 and LFA-1 pathways. J. Immunol. 2002, 169, 4831–4839. [Google Scholar] [CrossRef]
- Springer, T.A. Adhesion receptors of the immune system. Nature 1990, 346, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Bordigoni, P.; Michel, G.; Esperou, H.; Souillet, G.; Leblanc, T.; Stephan, J.L.; Vannier, J.P.; Mechinaud, F.; Reiffers, J.; et al. A phase II trial of partially incompatible bone marrow transplantation for high-risk acute lymphoblastic leukaemia in children: Prevention of graft rejection with anti-LFA-1 and anti-CD2 antibodies. Br. J. Haematol. 1996, 93, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Maraninchi, D.; Mawas, C.; Stoppa, A.M.; Gaspard, M.H.; Marit, G.; Van Ekthoven, A.; Reiffers, J.; Olive, D.; Hirn, M.; Delaage, M.; et al. Anti LFA1 monoclonal antibody for the prevention of graft rejection after T cell-depleted HLA-matched bone marrow transplantation for leukemia in adults. Bone Marrow Transplant. 1989, 4, 147–150. [Google Scholar] [PubMed]
- Guttman-Yassky, E.; Vugmeyster, Y.; Lowes, M.A.; Chamian, F.; Kikuchi, T.; Kagen, M.; Gilleaudeau, P.; Lee, E.; Hunte, B.; Howell, K.; et al. Blockade of CD11a by efalizumab in psoriasis patients induces a unique state of T-cell hyporesponsiveness. J. Invest. Dermatol. 2008, 128, 1182–1191. [Google Scholar] [CrossRef]
- Dedrick, R.L.; Walicke, P.; Garovoy, M. Anti-adhesion antibodies efalizumab, a humanized anti-CD11a monoclonal antibody. Transpl. Immunol. 2002, 9, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.; Krueger, J.G.; Bright, R.; Ling, M.; Lebwohl, M.; Kang, S.; Feldman, S.; Spellman, M.; Wittkowski, K.; Ochs, H.D.; et al. Effects of administration of a single dose of a humanized monoclonal antibody to CD11a on the immunobiology and clinical activity of psoriasis. J. Am. Acad. Dermatol. 2000, 42, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Caro, I.; Leung, H.M.; Garovoy, M.; Mease, P.J. Efalizumab for the treatment of psoriatic arthritis. J. Cutan. Med. Surg. 2007, 11, 57–66. [Google Scholar] [CrossRef]
- Stern, R.S. A promising step forward in psoriasis therapy. JAMA 2003, 290, 3133–3135. [Google Scholar] [CrossRef]
- Schwab, N.; Ulzheimer, J.C.; Fox, R.J.; Schneider-Hohendorf, T.; Kieseier, B.C.; Monoranu, C.M.; Staugaitis, S.M.; Welch, W.; Jilek, S.; Du Pasquier, R.A.; et al. Fatal PML associated with efalizumab therapy: Insights into integrin αLβ2 in JC virus control. Neurology 2012, 78, 458–467. [Google Scholar] [CrossRef]
- Sterry, W.; Bagot, M.; Ferrandiz, C.; Kragballe, K.; Papp, K.; Stingl, G. Immunosuppressive therapy in dermatology and PML. J. Dtsch. Dermatol. Ges. 2009, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Weger, W. Current status and new developments in the treatment of psoriasis and psoriatic arthritis with biological agents. Br. J. Pharmacol. 2010, 160, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Morrison, V.L.; James, M.J.; Grzes, K.; Cook, P.; Glass, D.G.; Savinko, T.; Lek, H.S.; Gawden-Bone, C.; Watts, C.; Millington, O.R.; et al. Loss of β2-integrin-mediated cytoskeletal linkage reprogrammes dendritic cells to a mature migratory phenotype. Nat. Commun. 2014, 5, 5359. [Google Scholar] [CrossRef] [PubMed]
- Stevanin, M.; Busso, N.; Chobaz, V.; Pigni, M.; Ghassem-Zadeh, S.; Zhang, L.; Acha-Orbea, H.; Ehirchiou, D. CD11b regulates the Treg/Th17 balance in murine arthritis via IL-6. Eur. J. Immunol. 2017, 47, 637–645. [Google Scholar] [CrossRef]
- Tang, T.; Rosenkranz, A.; Assmann, K.J.; Goodman, M.J.; Gutierrez-Ramos, J.C.; Carroll, M.C.; Cotran, R.S.; Mayadas, T.N. A role for Mac-1 (CDIIb/CD18) in immune complex-stimulated neutrophil function in vivo: Mac-1 deficiency abrogates sustained Fcγ receptor-dependent neutrophil adhesion and complement-dependent proteinuria in acute glomerulonephritis. J. Exp. Med. 1997, 186, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhao, M.; Li, N.; Diaz, L.A.; Mayadas, T.N. Differential roles for β2 integrins in experimental autoimmune bullous pemphigoid. Blood 2006, 107, 1063–1069. [Google Scholar] [CrossRef]
- Bullard, D.C.; Hu, X.; Schoeb, T.R.; Axtell, R.C.; Raman, C.; Barnum, S.R. Critical requirement of CD11b (Mac-1) on T cells and accessory cells for development of experimental autoimmune encephalomyelitis. J. Immunol. 2005, 175, 6327–6333. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Zhang, Z.; Deng, C.; Ma, X.; Liu, X. Effects of β2 Integrins on Osteoclasts, Macrophages, Chondrocytes, and Synovial Fibroblasts in Osteoarthritis. Biomolecules 2022, 12, 1653. [Google Scholar] [CrossRef]
- Tsokos, G.C. Systemic lupus erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef]
- Nath, S.K.; Han, S.; Kim-Howard, X.; Kelly, J.A.; Viswanathan, P.; Gilkeson, G.S.; Chen, W.; Zhu, C.; McEver, R.P.; Kimberly, R.P.; et al. A nonsynonymous functional variant in integrin-αM (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat. Genet. 2008, 40, 152–154. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, L.; Luo, C.; Tang, W.; Dai, R.; An, Y.; Tang, X. Clinical characteristics of early-onset paediatric systemic lupus erythematosus in a single centre in China. Rheumatology 2023, 62, 3373–3381. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, V.; Li, X.; Jimenez, V.; Faridi, H.M.; Gupta, V. CD11b agonists offer a novel approach for treating lupus nephritis. Transl. Res. 2022, 245, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Anaya, J.M.; Kim-Howard, X.; Prahalad, S.; Cherñavsky, A.; Cañas, C.; Rojas-Villarraga, A.; Bohnsack, J.; Jonsson, R.; Bolstad, A.I.; Brun, J.G.; et al. Evaluation of genetic association between an ITGAM non-synonymous SNP (rs1143679) and multiple autoimmune diseases. Autoimmun. Rev. 2012, 11, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Rosetti, F.; Mayadas, T.N. The many faces of Mac-1 in autoimmune disease. Immunol. Rev. 2016, 269, 175–193. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Contribution of regulatory T cells to cancer: A review. J. Cell Physiol. 2019, 234, 7983–7993. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. A timeline of tumour-associated macrophage biology. Nat. Rev. Cancer 2023, 23, 238–257. [Google Scholar] [CrossRef]
- Dysthe, M.; Parihar, R. Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1224, 117–140. [Google Scholar] [CrossRef]
- Hegde, S.; Leader, A.M.; Merad, M. MDSC: Markers, development, states, and unaddressed complexity. Immunity 2021, 54, 875–884. [Google Scholar] [CrossRef]
- Antuamwine, B.B.; Bosnjakovic, R.; Hofmann-Vega, F.; Wang, X.; Theodosiou, T.; Iliopoulos, I.; Brandau, S. N1 versus N2 and PMN-MDSC: A critical appraisal of current concepts on tumor-associated neutrophils and new directions for human oncology. Immunol. Rev. 2023, 314, 250–279. [Google Scholar] [CrossRef]
- Brandsma, A.M.; Bondza, S.; Evers, M.; Koutstaal, R.; Nederend, M.; Jansen, J.H.M.; Rösner, T.; Valerius, T.; Leusen, J.H.W.; Ten Broeke, T. Potent Fc Receptor Signaling by IgA Leads to Superior Killing of Cancer Cells by Neutrophils Compared to IgG. Front. Immunol. 2019, 10, 704. [Google Scholar] [CrossRef]
- Kushner, B.H.; Cheung, N.K. Absolute requirement of CD11/CD18 adhesion molecules, FcRII and the phosphatidylinositol-linked FcRIII for monoclonal antibody-mediated neutrophil antihuman tumor cytotoxicity. Blood 1992, 79, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Van Spriel, A.B.; van Ojik, H.H.; Bakker, A.; Jansen, M.J.; van de Winkel, J.G. Mac-1 (CD11b/CD18) is crucial for effective Fc receptor-mediated immunity to melanoma. Blood 2003, 101, 253–258. [Google Scholar] [CrossRef]
- Matlung, H.L.; Babes, L.; Zhao, X.W.; van Houdt, M.; Treffers, L.W.; van Rees, D.J.; Franke, K.; Schornagel, K.; Verkuijlen, P.; Janssen, H.; et al. Neutrophils Kill Antibody-Opsonized Cancer Cells by Trogoptosis. Cell Rep. 2018, 23, 3946–3959.e3946. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Qin, W.; Song, M.; Liu, L.; Yu, Y.; Qi, X.; Sun, H. Neutrophil Suppresses Tumor Cell Proliferation via Fas/Fas Ligand Pathway Mediated Cell Cycle Arrested. Int. J. Biol. Sci. 2018, 14, 2103–2113. [Google Scholar] [CrossRef] [PubMed]
- Kalafati, L.; Kourtzelis, I.; Schulte-Schrepping, J.; Li, X.; Hatzioannou, A.; Grinenko, T.; Hagag, E.; Sinha, A.; Has, C.; Dietz, S.; et al. Innate Immune Training of Granulopoiesis Promotes Anti-tumor Activity. Cell 2020, 183, 771–785.e712. [Google Scholar] [CrossRef]
- Hirschhorn, D.; Budhu, S.; Kraehenbuehl, L.; Gigoux, M.; Schröder, D.; Chow, A.; Ricca, J.M.; Gasmi, B.; De Henau, O.; Mangarin, L.M.B.; et al. T cell immunotherapies engage neutrophils to eliminate tumor antigen escape variants. Cell 2023, 186, 1432–1447.e1417. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef] [PubMed]
- Canino, J.; Guidetti, G.F.; Galgano, L.; Vismara, M.; Minetti, G.; Torti, M.; Canobbio, I. The proline-rich tyrosine kinase Pyk2 modulates integrin-mediated neutrophil adhesion and reactive oxygen species generation. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118799. [Google Scholar] [CrossRef] [PubMed]
- Pendino, K.J.; Gardner, C.R.; Quinones, S.; Laskin, D.L. Stimulation of nitric oxide production in rat lung lavage cells by anti-Mac-1beta antibody: Effects of ozone inhalation. Am. J. Respir. Cell Mol. Biol. 1996, 14, 327–333. [Google Scholar] [CrossRef]
- He, K.; Liu, X.; Hoffman, R.D.; Shi, R.Z.; Lv, G.Y.; Gao, J.L. G-CSF/GM-CSF-induced hematopoietic dysregulation in the progression of solid tumors. FEBS Open Bio 2022, 12, 1268–1285. [Google Scholar] [CrossRef]
- Fousek, K.; Horn, L.A.; Palena, C. Interleukin-8: A chemokine at the intersection of cancer plasticity, angiogenesis, and immune suppression. Pharmacol. Ther. 2021, 219, 107692. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Espinosa, S.; Morales, X.; Senent, Y.; Alignani, D.; Tavira, B.; Macaya, I.; Ruiz, B.; Moreno, H.; Remírez, A.; Sainz, C.; et al. Complement C5a induces the formation of neutrophil extracellular traps by myeloid-derived suppressor cells to promote metastasis. Cancer Lett. 2022, 529, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Schedel, F.; Mayer-Hain, S.; Pappelbaum, K.I.; Metze, D.; Stock, M.; Goerge, T.; Loser, K.; Sunderkötter, C.; Luger, T.A.; Weishaupt, C. Evidence and impact of neutrophil extracellular traps in malignant melanoma. Pigment. Cell Melanoma Res. 2020, 33, 63–73. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Del Vecchio, S.; Carriero, M.V. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front. Immunol. 2020, 11, 1749. [Google Scholar] [CrossRef] [PubMed]
- Munir, H.; Jones, J.O.; Janowitz, T.; Hoffmann, M.; Euler, M.; Martins, C.P.; Welsh, S.J.; Shields, J.D. Stromal-driven and Amyloid β-dependent induction of neutrophil extracellular traps modulates tumor growth. Nat. Commun. 2021, 12, 683. [Google Scholar] [CrossRef]
- Takesue, S.; Ohuchida, K.; Shinkawa, T.; Otsubo, Y.; Matsumoto, S.; Sagara, A.; Yonenaga, A.; Ando, Y.; Kibe, S.; Nakayama, H.; et al. Neutrophil extracellular traps promote liver micrometastasis in pancreatic ductal adenocarcinoma via the activation of cancer-associated fibroblasts. Int. J. Oncol. 2020, 56, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Rayes, R.F.; Mouhanna, J.G.; Nicolau, I.; Bourdeau, F.; Giannias, B.; Rousseau, S.; Quail, D.; Walsh, L.; Sangwan, V.; Bertos, N.; et al. Primary tumors induce neutrophil extracellular traps with targetable metastasis promoting effects. JCI Insight 2019, 5, e128008. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.D.; Wang, J.H.; Condron, C.; Bouchier-Hayes, D.; Redmond, H.P. Human neutrophils facilitate tumor cell transendothelial migration. Am. J. Physiol. Cell Physiol. 2001, 280, C814–C822. [Google Scholar] [CrossRef]
- Sprouse, M.L.; Welte, T.; Boral, D.; Liu, H.N.; Yin, W.; Vishnoi, M.; Goswami-Sewell, D.; Li, L.; Pei, G.; Jia, P.; et al. PMN-MDSCs Enhance CTC Metastatic Properties through Reciprocal Interactions via ROS/Notch/Nodal Signaling. Int. J. Mol. Sci. 2019, 20, 1916. [Google Scholar] [CrossRef]
- BeLow, M.; Osipo, C. Notch Signaling in Breast Cancer: A Role in Drug Resistance. Cells 2020, 9, 2204. [Google Scholar] [CrossRef]
- Haist, M.; Stege, H.; Grabbe, S.; Bros, M. The Functional Crosstalk between Myeloid-Derived Suppressor Cells and Regulatory T Cells within the Immunosuppressive Tumor Microenvironment. Cancers 2021, 13, 210. [Google Scholar] [CrossRef] [PubMed]
- Aarts, C.E.M.; Hiemstra, I.H.; Béguin, E.P.; Hoogendijk, A.J.; Bouchmal, S.; van Houdt, M.; Tool, A.T.J.; Mul, E.; Jansen, M.H.; Janssen, H.; et al. Activated neutrophils exert myeloid-derived suppressor cell activity damaging T cells beyond repair. Blood Adv. 2019, 3, 3562–3574. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Peters, T.; Sindrilaru, A.; Kess, D.; Oreshkova, T.; Yu, X.Z.; Seier, A.M.; Schreiber, H.; Wlaschek, M.; Blakytny, R.; et al. TGF-beta-dependent suppressive function of Tregs requires wild-type levels of CD18 in a mouse model of psoriasis. J. Clin. Investig. 2008, 118, 2629–2639. [Google Scholar] [CrossRef] [PubMed]
- Niu, T.; Li, Z.; Huang, Y.; Ye, Y.; Liu, Y.; Ye, Z.; Jiang, L.; He, X.; Wang, L.; Li, J. LFA-1 knockout inhibited the tumor growth and is correlated with treg cells. Cell Commun. Signal 2023, 21, 233. [Google Scholar] [CrossRef]
- Soloviev, D.A.; Hazen, S.L.; Szpak, D.; Bledzka, K.M.; Ballantyne, C.M.; Plow, E.F.; Pluskota, E. Dual role of the leukocyte integrin αMβ2 in angiogenesis. J. Immunol. 2014, 193, 4712–4721. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.N.; Kolomeyevskaya, N.; Singel, K.L.; Grimm, M.J.; Moysich, K.B.; Daudi, S.; Grzankowski, K.S.; Lele, S.; Ylagan, L.; Webster, G.A.; et al. Targeting myeloid cells in the tumor microenvironment enhances vaccine efficacy in murine epithelial ovarian cancer. Oncotarget 2015, 6, 11310–11326. [Google Scholar] [CrossRef] [PubMed]
- Maiguel, D.; Faridi, M.H.; Wei, C.; Kuwano, Y.; Balla, K.M.; Hernandez, D.; Barth, C.J.; Lugo, G.; Donnelly, M.; Nayer, A.; et al. Small molecule-mediated activation of the integrin CD11b/CD18 reduces inflammatory disease. Sci. Signal 2011, 4, ra57. [Google Scholar] [CrossRef]
- Panni, R.Z.; Herndon, J.M.; Zuo, C.; Hegde, S.; Hogg, G.D.; Knolhoff, B.L.; Breden, M.A.; Li, X.; Krisnawan, V.E.; Khan, S.Q.; et al. Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Galkin, A.; Dupont, J.; Zhou, L.; Bendell, J. GB1275, a first-in-class CD11b modulator: Rationale for immunotherapeutic combinations in solid tumors. J. Immunother. Cancer 2021, 9, e003005. [Google Scholar] [CrossRef]
- Liu, X.; Hogg, G.D.; Zuo, C.; Borcherding, N.C.; Baer, J.M.; Lander, V.E.; Kang, L.I.; Knolhoff, B.L.; Ahmad, F.; Osterhout, R.E.; et al. Context-dependent activation of STING-interferon signaling by CD11b agonists enhances anti-tumor immunity. Cancer Cell 2023, 41, 1073–1090.e1012. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).