1. Introduction
Tuberculosis (TB), primarily caused by
Mycobacterium tuberculosis (
M. tb) and
Mycobacterium bovis (
M. bovis), is a leading cause of death from a single infectious agent worldwide [
1], remaining a global health threat, and
M. bovis contributes to approximately 10% of human TB cases. This situation is exacerbated by the limitations of early detection, the limited efficacy of the
Bacille Calmette–Guerin (BCG) vaccine, derived from an attenuated strain of
M. bovis, and the emergence of drug-resistant strains [
2,
3,
4]. These issues often arise from an incomplete understanding of TB pathogenesis.
As a class of obligate intracellular bacteria, both M. tb and M. bovis has developed complicated strategies to evade host immune responses, utilizing different host factors to establish and sustain infection. Thus, understanding these interactions is vital for unraveling the complexities of TB pathogenesis.
Nitric oxide (NO) is an essential signaling molecule that plays a crucial role in the immune response against TB [
5,
6]. It activates the immune response of the host to eliminate intracellular
M. tb. NO production is an essential aspect of the innate immune response against TB, promoting the destruction of pathogens by generating reactive nitrogen species (RNS) that directly damage bacterial components and inhibit their replication. However, maintaining an appropriate level of NO is critical, as excessive production can lead to tissue damage and inflammation, complicating the disease outcome [
7,
8,
9,
10,
11,
12,
13].
Historically, NmrA-like proteins have been discovered in some fungi and bacteria, including PadA from
Dictyostelium discoideum, PcNMRAL1 from
Phytophthora capsica, and QOR2 from
Escherichia coli [
14,
15,
16]. In mammals, an NmrA-like protein was reported in humans, designated as HSCARG, while, in animals such as mice, rats, and bovines, it was known as NMRAL1 [
17]. NMRAL1 negatively regulates innate immunity by suppressing the activities of the NF-κB and RIG-I-like receptor (RLR) pathway [
18]. It serves as a crucial link connecting the cellular redox status with other signaling pathways. It inhibits NO production through interacting with argininosuccinate synthetase (AS) [
18]. While the regulation of NO by NMRA-like proteins is well-established in other contexts, their specific roles in TB infection remain unexplored.
Previously, we noticed another unannotated gene ENSBTAG00000011305, which contains a NmrA domain beside the NMRAL1 gene (ENSBTAG00000010015), and designated it as the NMRAL2_Bovine (hereafter NMRAL2). This study aimed to demonstrate the function of the NMRAL2 in the host defense against M. bovis infection related to the NO production and inflammatory response. As a result, NMRAL2 knockout and knockdown led to a marked increase in NO production, and a reduction in cellular death induced by M. bovis and number of intracellular M. bovis. These findings contribute to a more comprehensive understanding of TB pathogenesis and the development of novel control measures for both human and animal TB.
2. Materials and Methods
2.1. Cell and Bacterial Culture
The embryonic bovine lung (EBL) cell lines were generously provided by M. Heller from the Friedrich-Loeffler-Institute in Jena, Germany. These cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, Melbourne, Australia), which was supplemented with non-essential amino acids (NEAA, Gibco, Melbourne, Australia) and 10% heat-inactivated fetal bovine serum (FBS, Gibco, Melbourne, Australia). The HEK293FT cell line was procured from the Cell Bank of the Chinese Academy of Sciences in Shanghai, China, and cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin, under conditions of 37 °C and 5% CO2.
M. bovis was obtained from Professor Junyan Liu from Wuhan University. Professor Luiz Bermudez from Oregon State University supplied BCG-Pasteur strain. BCG-Pasteur strain expressing red fluorescent protein DsRed (BCG-red) was provided by Professor Gang Cao from Huazhong Agriculture University. Bacterial strains were cultured in Middlebrook 7H9 broth with 0.5% glycerol, 10% oleic acid-albumin-dextrosecatalase (OADC; BD PharMingen, San Diego, CA, USA), and 0.05% Tween 80, or on Middlebrook 7H11 agar plates containing 0.5% glycerol and 10% OADC.
Prior to infection, the optical densities of bacterial cultures were standardized at 600 nm (OD600) to achieve the desired multiplicity of infection (MOI). The cultures underwent centrifugation at 5000× g for 10 min, after which the bacterial pellet was resuspended in a medium and dispersed using an insulin syringe. Subsequently, a 100 μL aliquot of the 10-fold serially diluted bacterial suspension was plated on Middlebrook 7H11 agar to quantify viable bacteria as colony-forming units (CFUs).
2.2. Construction of Gene Knockout Cell Line
To generate the NMRAL2 knockout cell line (NMRAL2-KO), NMRAL2 sgRNA sequence (NMRAL2_sgRNA-F: CACCGGTATCTCGGTTGCTGATAT; NMRAL2_sgRNA-R: AAACATATCAGCAACCGAGATACC) were inserted into the lentiCRISPR-V2 vector. This construct was subsequently co-transfected with the pMD2.G and psPAX2 plasmids into HEK293FT cells using JetPRIME reagent (Polyplus, Illkirch, France), following the manufacturer’s protocol. After 60 h post-transfection, the supernatants of the cell cultures were collected, filtered using 0.45 μm low protein binding membrane (Millipore Corporation, Billerica, MA, USA), and centrifuged at 150,000× g for 2.5 h at 4 °C. Lentivirus pellets with NMRAL2 sgRNA were resuspended in PBS (pH 7.4), aliquoted, and stored at −80°C. Following infection with the lentivirus, EBL cells underwent selection for successful infection using puromycin (Invivo Gen, San Diego, CA, USA) at a concentration of 5 μg/mL. The knockout of the NMRAL2 gene was confirmed using Sanger sequencing and RT-qPCR analysis. The specific forward primers (5ʹ-AGCGACTCAGTGTTACATGGTG-3ʹ) and reverse primers (5ʹ-TTCACCCCAACTCTGCTGTG-3ʹ) were utilized for polymerase chain reaction (PCR) amplification, followed by Sanger sequencing (Tsingke, Beijing, China).
2.3. RNAi
siRNA of NMRAL2 and NMRAL1_Bovine (Si-NMRAL1: CAGUGCAUUUCGGUCAGACCUGAUU; Si-NMRAL2: UGGCUUCUGCUAAGCUACCUCUGAG) gene was synthesized by Tsingke (Tsingke, Beijing, China). EBL cells were cultured in 12-well plates and transfected with 80 pmol of siRNA utilizing Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) when they reached approximately 60% confluence, in accordance with the manufacturer’s instructions. A negative control siRNA (NC siRNA) was employed as a control. Subsequent to a 48 h post-transfection incubation period, the cells were subjected to further functional analyses.
2.4. RNA-Seq
Total RNA extraction was performed utilizing the Trizol Reagent (Invitrogen, Carlsbad, CA, USA). The concentration, quality, and integrity of the extracted RNA were evaluated using a NanoDrop spectrophotometer (Thermo Scientific, Waltham, MA, USA). Subsequent sequencing libraries were prepared following established protocols. mRNA was selectively isolated from the total RNA using poly-T oligonucleotide-attached magnetic beads. Fragmentation of the mRNA was achieved through the application of divalent cations at elevated temperatures within an Illumina proprietary fragmentation buffer. The synthesis of first-strand complementary DNA (cDNA) was performed utilizing random oligonucleotides in conjunction with SuperScript II reverse transcriptase. This was followed by the synthesis of the second-strand cDNA, which was achieved using DNA Polymerase I and RNase H. To convert any remaining overhangs into blunt ends, exonuclease and polymerase activities were employed, after which the enzymes were removed. Subsequently, Illumina paired-end (PE) adapted oligonucleotides were ligated to the DNA fragments, following adenylation of their 3′ ends, to facilitate hybridization. To preferentially select cDNA fragments ranging from 400 to 500 base pairs in length, library fragments were purified utilizing the AMPure XP system (Beckman Coulter, Beverly, CA, USA). DNA fragments with adaptor molecules ligated at both ends were selectively enriched through a 15-cycle PCR reaction employing the Illumina PCR Primer Cocktail. The resulting products were then purified again and quantified. The sequencing library was subsequently processed using the NovaSeq 6000 platform (Illumina, San Diego, CA, USA) by Shanghai Personal Biotechnology Co., Ltd (Shanghai, China). The RNA sequencing services were facilitated by Personal Biotechnology Co., Ltd., located in Shanghai, China. Data analysis was conducted utilizing the online platform Personalbio GenesCloud (
https://www.genescloud.cn, accessed on 11 August 2022). Furthermore, genes associated with the inflammatory response were randomly selected for validation. The primer sequences used for RT-qPCR are detailed in
Table 1.
2.5. RT-qPCR
Total RNA was extracted from the cells utilizing the TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). cDNA synthesis was performed by reverse transcription of the isolated RNA using the HiScript III RT SuperMix with gDNA Eraser (Vazyme, Nanjing, China) in a reaction volume of 20 μL. Amplification of target genes was achieved using a reaction mixture comprising 5 μL of SYBR Green Mix, 0.5 μL of gene-specific primers, and 3 μL of double-distilled H2O, culminating in a total volume of 10 μL. The PCR protocol was conducted using a Bio-Rad IQ5 instrument (Bio-Rad, Hercules, CA, USA) and comprised an initial denaturation phase of 5 min at 95 °C, followed by 40 cycles consisting of 10 s at 95 °C and 30 s at 60 °C.
2.6. EdU Fluorescence and CCK-8 Assays
Cell viability and proliferation were assessed utilizing the CCK-8 and EdU fluorescence assays. NMRAL2-KO and WT EBL cells were initially seeded into 96-well plates. Each well was subsequently treated with CCK-8 solution (Dojindo, Kumamoto, Japan), and absorbance was recorded at 450 nm. For the EdU fluorescence assay, NMRAL2-KO and WT EBL cells were cultured in 12-well plates. The EdU cell proliferation assay was then performed using the BeyoClick EdU kit (Beyotime Biotechnology, Shanghai, China) with Alexa Fluor 488, following the manufacturer’s instructions. Fluorescence microscopy was used to visualize treated cells, and ImageJ software (ImageJ software, 1.52a; National Institutes of Health, Bethesda, MD, USA) calculated the proportion of EdU-positive cells. Images were taken from a random field of view in each of three independent wells.
2.7. Western Blotting and Antibodies
Cells were lysed at 4 °C for 30 min using a cell lysis buffer supplemented with a 1× complete protease inhibitor cocktail (Roche, Mannheim, Germany). Cellular debris was eliminated by centrifugation at 12,000× g for 15 min at 4 °C. The supernatant derived from the lysed cells was subsequently subjected to heating at 95 °C for a duration of 5 min in the presence of loading buffer. Proteins were then denatured, resolved via SDS-PAGE, and transferred onto a polyvinylidene fluoride (PVDF; Millipore Corporation, Bedford, MA, USA) membrane. The PVDF membranes underwent a blocking procedure for a minimum duration of three hours utilizing Tris-buffered saline with Tween-20 (TBST) supplemented with 5% skim milk. Subsequently, the membranes were incubated with primary antibodies in TBST, a process conducted either overnight at 4 °C. For Western blot analysis, primary antibodies from Cell Signaling Technology (Cell Signaling Technology, Boston,MA, USA) were utilized, including rabbit monoclonal antibodies for phospho-p65 (Ser536) (93H1) (#3033), p38 (#9212), phospho-p38 (Thr180/Tyr182) (#9211), p44/42 (Erk1/2) (137F5) (#4695), phospho-p44/42 (Erk1/2) (Thr202/Tyr204) (D13.14.4E) XP® (#4370), JNK (56G8) (#9258), and phospho-SAPK/JNK (Thr183/Tyr185) (98F2) (#4671). Mouse monoclonal antibodies for NF-κB subunit p65 (L8F6) (#6956) was also obtained from the same supplier. For loading controls, Anti-β-actin antibody was obtained from Proteintech (Proteintech, Wuhan, China). Secondary antibodies specific to mouse and rabbit (Abbkine, Wuhan, China) were utilized in TBST for a duration of one hour at room temperature. Protein detection was subsequently performed using ECL Prime Western blot detection reagents (Bio-Rad, Hercules, CA, USA).
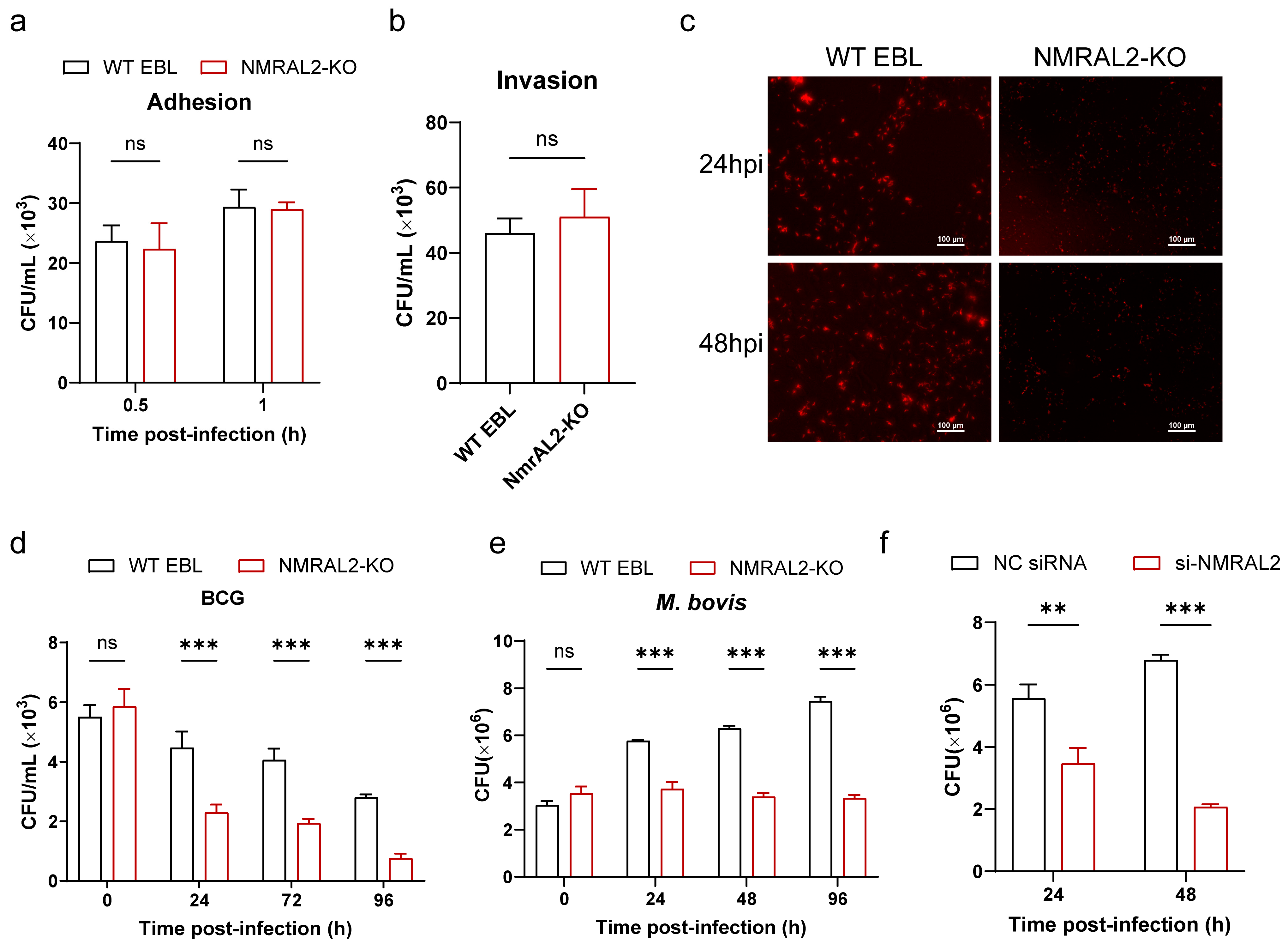
2.8. Invasion and Adhesion Assays
In the invasion and adhesion assay, both NMRAL2-KO and wild-type (WT) EBL cells were exposed to M. bovis at an MOI of 10:1. For the invasion assay, cells were incubated at 37 °C for 1 h, whereas, for the adhesion assay, incubation was conducted at 4 °C for 30 min. Following incubation, the cells underwent three washes with 1× phosphate-buffered saline (PBS; Gibco, Melbourne, Australia) and were subsequently lysed using 0.025% Tween-20 for a duration of 10 min. The resulting cell lysate was subjected to a 10-fold serial dilution prior to being plated on 7H11 agar. Bacterial colony-forming units (CFUs) were quantified after an incubation period of three weeks.
2.9. Intracellular Survival Assay
NMRAL2-KO and WT EBL cells were inoculated with M. bovis at an MOI of 10. Following infection, the cells underwent three washes with PBS and were subsequently incubated in fresh DMEM supplemented with 100 μg/mL gentamicin to eradicate extracellular bacteria. The cells were then washed and lysed using 0.025% Tween-20 for 10 min at intervals of 0, 24, 48, and 72 h post-infection. The resulting lysates were subjected to serial ten-fold dilutions and plated on 7H11 agar. Colony formation was assessed after an incubation period of 3 to 4 weeks at 37 °C.
2.10. Analysis of Intracellular NO Production
The release of intracellular NO was evaluated utilizing a fluorescent microscope in conjunction with the NO-sensitive fluorescence probe DAF-FM DA (Beyotime Biotechnology, Shanghai, China). Both NMRAL2-KO and WT cells were exposed to M. bovis for a duration of 24 h, followed by treatment with 1 μM DAF-FM DA for 20 min. Post-treatment, the medium was aspirated, and the cells underwent three washes with phosphate-buffered saline (PBS). Confocal microscopy (Nikon, Tokyo, Japan) was employed to capture fluorescence images, which were subsequently analyzed to determine cellular fluorescence intensity. Additionally, quantitative measurement of NO concentration was performed using the Griess reagent kit assay (Beyotime Biotechnology, Shanghai, China). The concentration of NO in cell culture supernatants was quantified utilizing a Griess-reaction-based NO assay kit. Both NMRAL2-KO and WT cells were infected with M. bovis for durations of 0, 24, and 48 h, adhering to the manufacturer’s protocol for sample preparation.
2.11. Statistical Analysis
All assays and experiments were conducted in triplicate, with data reported as means ± standard errors of the mean (SEMs) derived from these triplicates. Statistical analyses were performed utilizing GraphPad Prism software (GraphPad Prism, version 9.0, Boston, MA, USA). The Student’s t-test was applied for comparisons between two groups, whereas analysis of variance (ANOVA) was utilized for comparisons involving more than two groups. Statistical significance was categorized as non-significant (ns, p > 0.05), * p < 0.05, ** p < 0.01, and *** p < 0.001.
4. Discussion
In this study, we elucidate the pivotal role of NMRAL2 in modulating the response of EBL cells to M. bovis infection. Our results demonstrate that the knockout of NMRAL2 significantly reduces cell death induced by M. bovis, revealing a previously unrecognized mechanism that enhances host cell survival against M. bovis infection. This discovery underscores the importance of NMRAL2 in NO production, a key factor in activating anti-tuberculosis innate immune pathways. The regulation of NO synthesis and the immune response by NMRAL2 provides novel insights into the dynamics of host–pathogen interactions. Consequently, this study offers a new perspective regarding the interplay between cellular defense mechanisms and M. bovis infection.
The modulation of NO signaling involves not only redox molecule dynamics but also interactions with regulatory proteins like NMRAL1 [
18]. NMRAL1 directly affects NO synthesis by reducing argininosuccinate synthetase activity [
18]. Our findings align with this, as the presence of specific conserved motifs in NMRAL2 indicates potential functional similarities with NMRAL1. However, the distinct outcomes observed when knocking down
NMRAL1 versus
NMRAL2 highlight the functional divergence within this protein family. The broader NmrA protein family, including PadA, QOR2, and PcNMRAL1, is known for binding to coenzymes NAD(H) or NADP(H) without dehydrogenase activity [
17], characterized by a short-chain dehydrogenase/reductase-like structure. This family features an N-terminal Rossmann fold essential for coenzyme binding [
19]. Despite the topological resemblance to NmrA, NMRAL1 shows a distinct affinity for reduced NADPH due to structural variations, such as the absence of NmrA-like Thr-276 in its C-terminal region [
17]. We also found that the 276th amino acid of NMRAL2 is His, while the 276th amino acid of NMRAL1 is Leu. Such variations underscore the functional divergence within the NmrA protein family, particularly in their respective roles in NO regulation and redox sensing. Our results suggest that minor structural changes can lead to functional differences in the NmrA protein family.
Our comparative analyses across species highlight the high conservation of NMRAL1, suggesting its ubiquitous role in biological functions, particularly in NO production and cellular survival mechanisms. The structural similarity of NMRAL2 to bovine-derived NMRAL11 implies a potential functional overlap. However, our results show that knocking down both NMRAL1 and NMRAL2 in EBL cells yields distinct outcomes. Specifically, NMRAL1 knockdown led to more cell death compared to both NMRAL2 knockdown and WT cells. Conversely, knocking out or knocking down NMRAL2 enhanced cell survival following M. bovis infection. This discrepancy may stem from structural differences between NMRAL1 and NMRAL2. Notably, while NMRAL2 shares a structure and motifs with NMRAL1, it lacks Motifs 7–9 and has an additional Motif 10 at its C-terminus. The absence of certain motifs in NMRAL2_Bovine, combined with an additional motif at its C-terminus, may account for the functional distinctions between the two homologs proteins, NMRAL2 and NMRAL1.
NMRAL1 is known to regulate NO production by sensing cellular redox states inhibiting excessive oxidative stress through a negative feedback mechanism, preventing cellular damage [
20]. NMRAL1 is an important regulator of NO playing a dual role, and its absence may disrupt the cellular redox balance, leading to cytotoxicity, with excessive levels causing cellular damage [
21,
22,
23]. Therefore, we hypothesized that the knockout of
NMRAL2 may lead to a moderate increase in NO levels, effectively controlling infection without inducing excessive inflammatory damage. Additionally, NMRAL2 may function as an auxiliary pathway for NO regulation, potentially increasing NO production upon deletion, thereby enhancing
M. bovis clearance and promoting cell survival. However, these hypotheses warrant further investigation to elucidate the specific molecular interactions and pathway analyses. Furthermore, the evolutionary aspects of the NmrA-like protein family, particularly the differences and similarities between species, remain an open area for exploration. Gene family analyses indicates that
NMRAL2 may have evolved through duplication and mutation events within the
NMRAL1 gene family, a hypothesis that requires validation through evolutionary biology studies.
NMRAL1 acts a negative regulator of innate immune responses, inhibiting cytokine-induced NF-κB activation [
24,
25,
26]. In resting cells, NF-κB is sequestered in the cytoplasm by IκB proteins, which suppress the transcriptional activity of the p50 and p65 subunits [
27]. In the classical NF-κB signaling pathway, cytokines like TNF-α or IL-1β trigger the ubiquitination and degradation of IκB, allowing p50 and p65 to translocate to the nucleus and regulate transcription [
28,
29]. NMRAL1 interacts with IKKβ and NEMO, influencing post-translational modifications such as IKKβ dephosphorylation by PP2A and NEMO deubiquitination by USP7 [
24,
25,
26]. Given the structural similarities between NMRAL2 and NMRAL1, it is plausible that NMRAL2 exert similar functions within the NF-κB pathway. Our research indicates that the knockout of
NMRAL2 leads to the significant activation of innate immune pathways, marked by an increased phosphorylation of the p65 signaling molecule, suggesting its involvement in the NF-κB signaling cascade. However, whether NMRAL2 modulates this pathway through the same strategies requires further investigation to fully elucidate its role in innate immune response regulation.
Our research on bovine NMRAL2 offers insights relevant to human TB, due to the shared pathogenic mechanisms between
M. bovis and
M. tb. Although these mycobacterial strains differ in host specificity, they utilize analogous strategies to circumvent host immune responses [
30,
31], indicating conserved roles in pathogenesis. The NmrA-like protein family, encompassing NMRAL1 and NMRAL2, is highly conserved across species and plays crucial roles in immune modulation. In humans, NMRAL1 (HSCARG), a homolog of NMRAL2, has been shown to modulate immune responses, particularly by influencing NO production and cytokine expression [
17]. In this study, we observed that the knockdown of
NMRAL2 in bovine cells facilitated the clearance of
M. bovis and enhanced host cell survival by augmenting NO levels and pro-inflammatory cytokine production. Conversely, the knockdown of
NMRAL1 in bovine cells resulted in increased cell death. These findings highlight the functional diversity within the NmrA protein family and suggest a conserved role for NMRAL1 in the pathogenesis of TB. We hypothesize that the moderate inhibition of NMRAL1 in humans could enhance NO levels, thereby facilitating pathogen clearance while minimizing cytotoxicity and tissue damage. Further research is required to confirm the role of NMRAL1 in human TB, which may uncover novel strategies for host-directed therapies targeting redox-sensitive pathways.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}