



A Novel Protozoa Parasite-Derived Protein Adjuvant Is Effective in Immunization with Cancer Cells to Activate the Cancer-Specific Protective Immunity and Inhibit the Cancer Growth in a Murine Model of Colorectal Cancer
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Tumor Cell Lines
2.3. Immunization with Non-Replicable MC38 CRC Cells
2.4. Challenge Implantation of MC38 CRC and EL4 Lymphoma Cells
2.5. Immunohistochemical Staining for CD4+ and CD8+ T Cells That Infiltrated into MC38 Tumors
2.6. Counting Numbers of CD4+ and CD8+ T Cells That Had Infiltrated into the MC38 Tumors
2.7. Purification of CD4+ and CD8+ T Cells and Their Cultures with MC38 CRC Cells
2.8. Statistical Analysis
3. Results
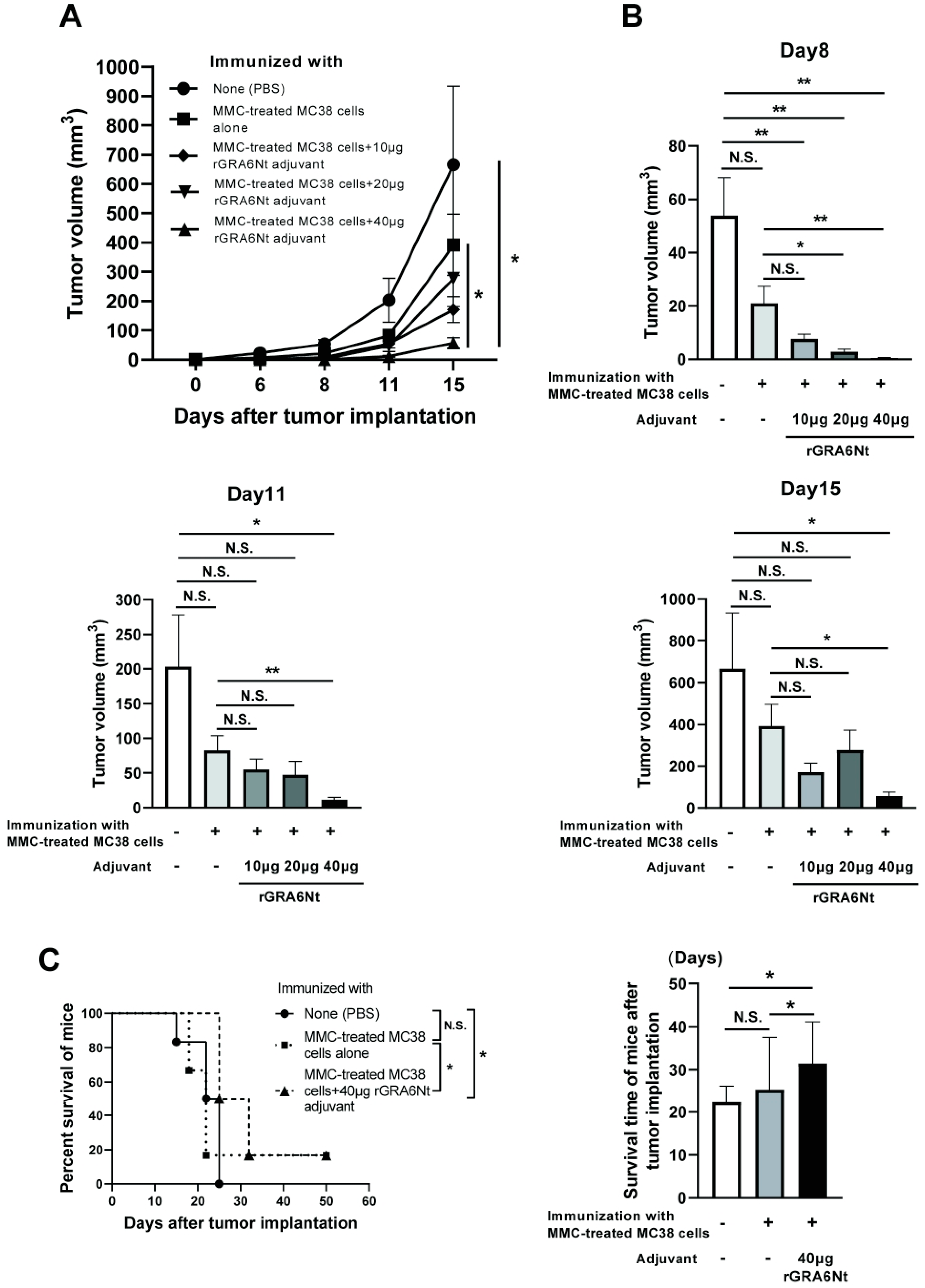
3.1. Immunization with MMC-Treated MC38 CRC Cells with rGRA6Nt Adjuvant Confers Potent Protections against a Challenge Implantation of This Cancer Cells in a Dose-Dependent Manner of the Adjuvant
3.2. Immunization with Irradiated MC38 CRC Cells with rGRA6Nt Adjuvant Confers Potent Protections against a Challenge Implantation of This Cancer Cells in a Dose-Dependent Manner of the Adjuvant
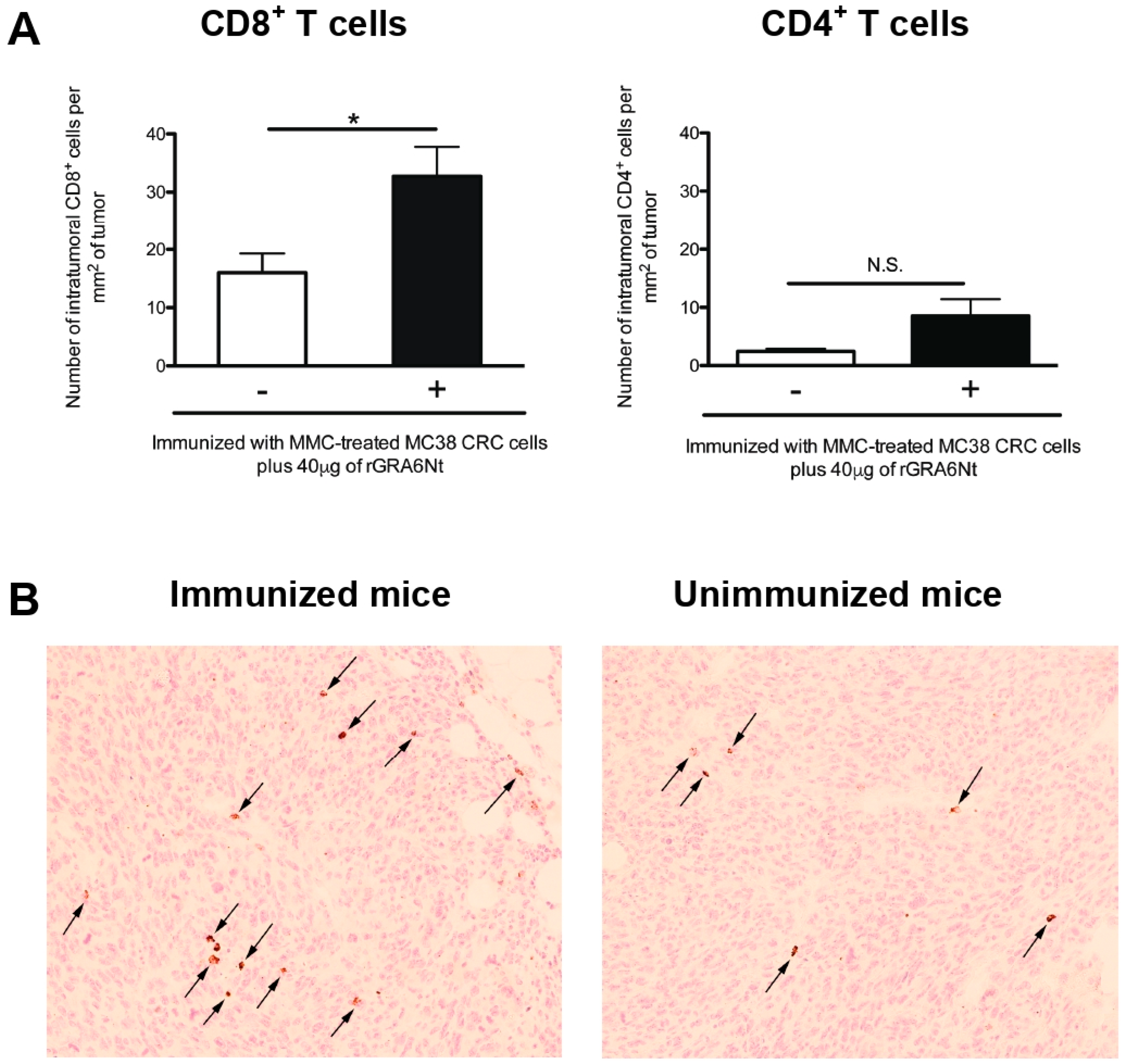
3.3. Immunizations with Non-Replicable MC38 CRC Cells with rGRA6Nt Adjuvant Induces Increased Infiltration of CD8+ T Cells into MC38 Tumors Implanted into These Mice
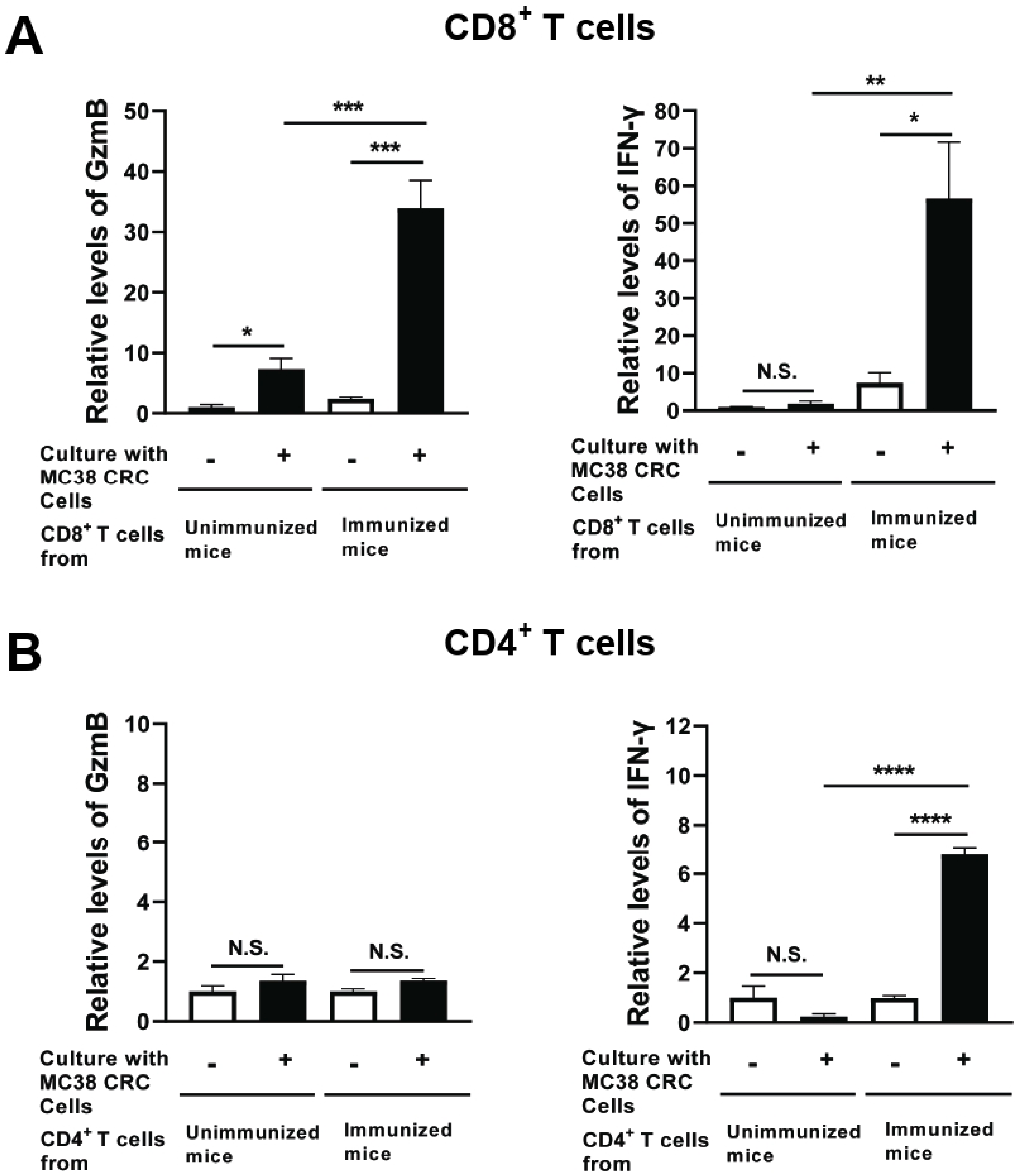
3.4. Immunization with Non-Replicable MC38 CRC Cells with rGRA6Nt Adjuvant Activates CD8+ Cytotoxic and IFN-γ-Producing T Cells against the Tumor Cells
3.5. The Protective Effects of Immunization with Non-Replicable MC38 CRC Cells with rGRA6Nt Adjuvant Are Specific to MC38 CRC
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Passardi, A.; Canale, M.; Valgiusti, M.; Ulivi, P. Immune checkpoints as a target for colorectal cancer treatment. Int. J. Mol. Sci. 2017, 18, 1324. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.P.; Mahalingam, D. Immunotherapy in colorectal cancer: For the select few or all? J. Gastrointest. Oncol. 2018, 9, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Hao, J.; Wang, H.; Fang, Y.; Tan, L. Efficacy and safety of immune checkpoint inhibitors in non-small cell lung cancer. Oncoimmunology 2018, 7, e1457600. [Google Scholar] [CrossRef]
- Aguiar, P.N., Jr.; De Mello, R.A.; Barreto, C.M.N.; Perry, L.A.; Penny-Dimri, J.; Tadokoro, H.; Lopes, G.L., Jr. Immune checkpoint inhibitors for advanced non-small cell lung cancer: Emerging sequencing for new treatment targets. ESMO Open 2017, 2, e000200. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Salem, J.E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal Toxic effects associated with immune checkpoint inhibitors: A systematic review and meta-analysis. JAMA 2018, 4, 1721–1728. [Google Scholar] [CrossRef]
- Jackson, H.J.; Rafiq, S.; Brentjens, R.J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 2016, 13, 370–383. [Google Scholar] [CrossRef]
- Fuge, O.; Vasdev, N.; Allchorne, P.; Green, J.S. Immunotherapy for bladder cancer. Res. Rep. Urol. 2015, 7, 65–79. [Google Scholar]
- Kidner, T.B.; Morton, D.L.; Lee, D.J.; Hoban, M.; Foshag, L.J.; Turner, R.R.; Faries, M.B. Combined intralesional Bacille Calmette-Guerin (BCG) and topical imiquimod for in-transit melanoma. J. Immunother. 2012, 35, 716–720. [Google Scholar] [CrossRef]
- Kim, K.D.; Lee, H.G.; Kim, J.K.; Park, S.N.; Choe, I.S.; Choe, Y.K.; Kim, S.J.; Lee, E.; Lim, J.S. Enhanced antigen-presenting activity and tumour necrosis factor-alpha-independent activation of dendritic cells following treatment with Mycobacterium bovis bacillus Calmette-Guerin. Immunology 1999, 97, 626–633. [Google Scholar] [CrossRef]
- Thurnher, M.; Ramoner, R.; Gastl, G.; Radmayr, C.; Bock, G.; Herold, M.; Klocker, H.; Bartsch, G. Bacillus Calmette-Guerin mycobacteria stimulate human blood dendritic cells. Int. J. Cancer 1997, 70, 128–134. [Google Scholar] [CrossRef]
- Antonelli, A.C.; Binyamin, A.; Hohl, T.M.; Glickman, M.S.; Redelman-Sidi, G. Bacterial immunotherapy for cancer induces CD4-dependent tumor-specific immunity through tumor-intrinsic interferon-gamma signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 18627–18637. [Google Scholar] [CrossRef] [PubMed]
- Reis e Sousa, C.; Hieny, S.; Scharton-Kersten, T.; Jankovic, D.; Charest, H.; Germain, R.N.; Sher, A. In Vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J. Exp. Med. 1997, 186, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Sher, A.; Hieny, S.; Charest, H.; Scharton-Kersten, T.; Collazo, C.; Germain, R.N.; Reis e Sousa, C. The role of dendritic cells in the initiation of host resistance to Toxoplasma gondii. Adv. Exp. Med. Biol. 1998, 452, 103–110. [Google Scholar] [PubMed]
- Suzuki, Y.; Muto, M.; Kobayashi, A. Antitumor effect of formalin-fixed Toxoplasma gondii organisms on EL4 lymphoma in Toxoplasma-infected mice. J. Biol. Response Modif. 1986, 5, 288–293. [Google Scholar]
- Suzuki, Y.; Kobayashi, A. Antitumor effect of intralesional injection with formalin-fixed Toxoplasma gondii organisms on Lewis lung carcinoma in Toxoplasma-infected mice. Cancer Lett. 1985, 25, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.R.; Byrne, K.T.; Lizotte, P.H.; Toraya-Brown, S.; Scarlett, U.K.; Alexander, M.P.; Sheen, M.R.; Fox, B.A.; Bzik, D.J.; Bosenberg, M.; et al. Immune-mediated regression of established B16F10 melanoma by intratumoral injection of attenuated Toxoplasma gondii protects against rechallenge. J. Immunol. 2013, 190, 469–478. [Google Scholar] [CrossRef]
- Baird, J.R.; Fox, B.A.; Sanders, K.L.; Lizotte, P.H.; Cubillos-Ruiz, J.R.; Scarlett, U.K.; Rutkowski, J.R.; Conejo-Garcia, J.R.; Fiering, S.; Bzik, D.J. Avirulent Toxoplasma gondii generates therapeutic antitumor immunity by reversing immunosuppression in the ovarian cancer microenvironment. Cancer Res. 2013, 73, 3842–3851. [Google Scholar] [CrossRef]
- Fox, B.A.; Sanders, K.L.; Bzik, D.J. Non-replicating Toxoplasma gondii reverses tumor-associated immunosuppression. Oncoimmunology 2013, 2, e26296. [Google Scholar] [CrossRef]
- Sanders, K.L.; Fox, B.A.; Bzik, D.J. Attenuated Toxoplasma gondii therapy of disseminated pancreatic cancer generates long-lasting immunity to pancreatic cancer. Oncoimmunology 2016, 5, e1104447. [Google Scholar] [CrossRef]
- Zhu, Y.C.; Elsheikha, H.M.; Wang, J.H.; Fang, S.; He, J.J.; Zhu, X.Q.; Chen, J. Synergy between Toxoplasma gondii type I DeltaGRA17 immunotherapy and PD-L1 checkpoint inhibition triggers the regression of targeted and distal tumors. J. Immunother. Cancer 2021, 9, e002970. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.; Mercier, C.; Delauw, M.F.; Suzuki, Y. Immunization with the amino-terminus region of dense granule protein 6 (GRA6) of Toxoplasma gondii activates CD8+ cytotoxic T cells capable of removing tissue cysts of the parasite through antigen presentation by human HLA-A2.1. Microbes Infect. 2023, 25, 105182. [Google Scholar] [CrossRef] [PubMed]
- Sa, Q.; Mercier, C.; Cesbron-Delauw, M.F.; Suzuki, Y. The amino-terminal region of dense granule protein 6 of Toxoplasma gondii stimulates IFN-gamma production by microglia. Microbes Infect. 2020, 22, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Roff, S.R.; Noon-Song, E.N.; Yamamoto, J.K. The Significance of Interferon-gamma in HIV-1 Pathogenesis, Therapy, and Prophylaxis. Front. Immunol. 2014, 4, 498. [Google Scholar] [CrossRef]
- Whitmire, J.K.; Tan, J.T.; Whitton, J.L. Interferon-gamma acts directly on CD8+ T cells to increase their abundance during virus infection. J. Exp. Med. 2005, 201, 1053–1059. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2015, 102, 18538–18543. [Google Scholar] [CrossRef]
- Chow, Z.; Johnson, J.; Chauhan, A.; Izumi, T.; Cavnar, M.; Weiss, H.; Townsend, C.M., Jr.; Anthony, L.; Wasilchenko, C.; Melton, M.L.; et al. PI3K/mTOR Dual Inhibitor PF-04691502 Is a Schedule-Dependent Radiosensitizer for Gastroenteropancreatic Neuroendocrine Tumors. Cells 2021, 10, 1261. [Google Scholar] [CrossRef]
- Johnson, J.; Chow, Z.; Lee, E.; Weiss, H.L.; Evers, B.M.; Rychahou, P. Role of AMPK and Akt in triple negative breast cancer lung colonization. Neoplasia 2021, 23, 429–438. [Google Scholar] [CrossRef]
- Tiwari, A.; Hannah, R.; Lutshumba, J.; Ochiai, E.; Weiss, L.M.; Suzuki, Y. Penetration of CD8+ cytotoxic T cells into large target, tissue cysts of Toxoplasma gondii, leads to Its elimination. Am. J. Pathol. 2019, 189, 1594–1607. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Jiga, L.P.; Bauer, T.M.; Chuang, J.J.; Opelz, G.; Terness, P. Generation of tolerogenic dendritic cells by treatment with mitomycin C: Inhibition of allogeneic T-cell response is mediated by downregulation of ICAM-1, CD80, and CD86. Transplantation 2004, 77, 1761–1764. [Google Scholar] [CrossRef] [PubMed]
- Terness, P.; Oelert, T.; Ehser, S.; Chuang, J.J.; Lahdou, I.; Kleist, C.; Velten, F.; Hammerling, G.J.; Arnold, B.; Opelz, G. Mitomycin C-treated dendritic cells inactivate autoreactive T cells: Toward the development of a tolerogenic vaccine in autoimmune diseases. Proc. Natl. Acad. Sci. USA 2008, 105, 18442–18447. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [PubMed]
- Hoves, S.; Ooi, C.H.; Wolter, C.; Sade, H.; Bissinger, S.; Schmittnaegel, M.; Ast, O.; Giusti, A.M.; Wartha, K.; Runza, V.; et al. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J. Exp. Med. 2018, 215, 859–876. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Cui, W.; Chi, Z.; Xiao, Q.; Hu, T.; Ye, Q.; Zhu, K.; Yu, W.; Wang, Z.; Yu, C.; et al. Tumor-associated macrophages are shaped by intratumoral high potassium via Kir2.1. Cell Metab. 2022, 34, 1843–1859. [Google Scholar] [CrossRef] [PubMed]
- Qiao, T.; Yang, W.; He, X.; Song, P.; Chen, X.; Liu, R.; Xiao, J.; Yang, X.; Li, M.; Gao, Y.; et al. Dynamic differentiation of F4/80+ tumor-associated macrophage and its role in tumor vascularization in a syngeneic mouse model of colorectal liver metastasis. Cell Death Dis. 2023, 14, 117. [Google Scholar] [CrossRef]
- Cianciaruso, C.; Beltraminelli, T.; Duval, F.; Nassiri, S.; Hamelin, R.; Mozes, A.; Gallert-Ayala, H.; Torres, G.C.; Torchia, B.; Ries, C.H.; et al. Molecular Profiling and Functional Analysis of Macrophage-Derived Tumor Extracellular Vesicles. Cell Rep. 2019, 27, 3062–3080. [Google Scholar] [CrossRef]
- Krieg, A.M. Development of TLR9 agonists for cancer therapy. J. Clin. Investig. 2007, 117, 1184–1194. [Google Scholar] [CrossRef]
- Carpentier, A.; Laigle-Donadey, F.; Zohar, S.; Capelle, L.; Behin, A.; Tibi, A.; Martin-Duverneuil, N.; Sanson, M.; Lacomblez, L.; Taillibert, S.; et al. Phase 1 trial of a CpG oligodeoxynucleotide for patients with recurrent glioblastoma. Neuro-Oncol. 2006, 8, 60–66. [Google Scholar] [CrossRef]
- Muraoka, D.; Kato, T.; Wang, L.; Maeda, Y.; Noguchi, T.; Harada, N.; Takeda, K.; Yagita, H.; Guillaume, P.; Luescher, I.; et al. Peptide vaccine induces enhanced tumor growth associated with apoptosis induction in CD8+ T cells. J. Immunol. 2010, 185, 3768–3776. [Google Scholar] [CrossRef] [PubMed]
- Sultan, H.; Wu, J.; Fesenkova, V.I.; Fan, A.E.; Addis, D.; Salazar, A.M.; Celis, E. Poly-IC enhances the effectiveness of cancer immunotherapy by promoting T cell tumor infiltration. J. Immunother. Cancer 2020, 8, e001224. [Google Scholar] [CrossRef] [PubMed]
- De Waele, J.; Verhezen, T.; van der Heijden, S.; Berneman, Z.N.; Peeters, M.; Lardon, F.; Wouters, A.; Smits, E. A systematic review on poly(I:C) and poly-ICLC in glioblastoma: Adjuvants coordinating the unlocking of immunotherapy. J. Exp. Clin. Cancer Res. 2021, 40, 213. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mani, R.; Martin, C.G.; Balu, K.E.; Wang, Q.; Rychahou, P.; Izumi, T.; Evers, B.M.; Suzuki, Y. A Novel Protozoa Parasite-Derived Protein Adjuvant Is Effective in Immunization with Cancer Cells to Activate the Cancer-Specific Protective Immunity and Inhibit the Cancer Growth in a Murine Model of Colorectal Cancer. Cells 2024, 13, 111. https://doi.org/10.3390/cells13020111
Mani R, Martin CG, Balu KE, Wang Q, Rychahou P, Izumi T, Evers BM, Suzuki Y. A Novel Protozoa Parasite-Derived Protein Adjuvant Is Effective in Immunization with Cancer Cells to Activate the Cancer-Specific Protective Immunity and Inhibit the Cancer Growth in a Murine Model of Colorectal Cancer. Cells. 2024; 13(2):111. https://doi.org/10.3390/cells13020111
Chicago/Turabian StyleMani, Rajesh, Chloe G. Martin, Kanal E. Balu, Qingding Wang, Piotr Rychahou, Tadahide Izumi, B. Mark Evers, and Yasuhiro Suzuki. 2024. "A Novel Protozoa Parasite-Derived Protein Adjuvant Is Effective in Immunization with Cancer Cells to Activate the Cancer-Specific Protective Immunity and Inhibit the Cancer Growth in a Murine Model of Colorectal Cancer" Cells 13, no. 2: 111. https://doi.org/10.3390/cells13020111
APA StyleMani, R., Martin, C. G., Balu, K. E., Wang, Q., Rychahou, P., Izumi, T., Evers, B. M., & Suzuki, Y. (2024). A Novel Protozoa Parasite-Derived Protein Adjuvant Is Effective in Immunization with Cancer Cells to Activate the Cancer-Specific Protective Immunity and Inhibit the Cancer Growth in a Murine Model of Colorectal Cancer. Cells, 13(2), 111. https://doi.org/10.3390/cells13020111