
Pharmacological Blockade of the Adenosine A2B Receptor Is Protective of Proteinuria in Diabetic Rats, through Affecting Focal Adhesion Kinase Activation and the Adhesion Dynamics of Podocytes

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. The A2BAR Antagonist Attenuates Diabetes-Induced FPE In Vivo
2.2. A2BAR Affects Human Immortalized Podocyte Adhesion Dynamics In Vitro
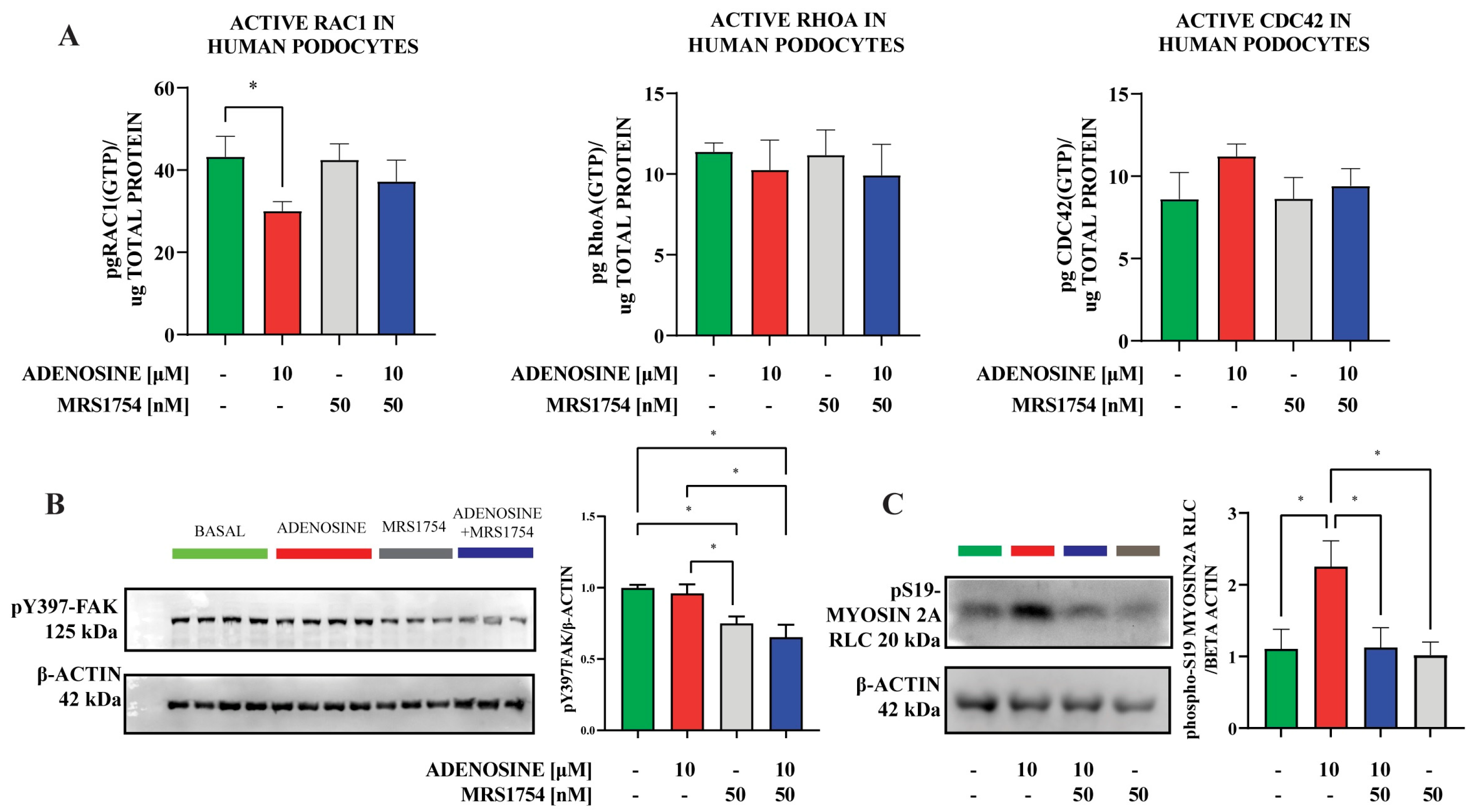
2.3. A2BAR Stimulation Increases the Abundance of S19-Phosphorylated Myosin2A
3. Discussion
4. Materials and Methods
4.1. Experimental Diabetes
4.2. Urine Analyses
4.3. Transmission Electron Microscopy
4.4. Cell Culture
4.5. Time-Lapse Video Microscopy
4.6. Immunofluorescence and Histofluorescence
4.7. Cell Adhesion Assays
4.8. G-LISA
4.9. Adhesion to Stiffness-Controlled Matrices
4.10. Western Blots Analysis
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| A2BAR | Adenosine A2B receptor subtype |
| CKD | Chronic kidney disease |
| DN | Diabetic nephropathy |
| FPs | Podocytes foot processes |
| FPE | Foot processes effacement |
| GBM | Glomerular basement membrane |
| GFB | Glomerular filtration barrier |
| STZ | Streptozotocin |
| FAK | Focal adhesion kinase |
References
- Mills, K.T.; Xu, Y.; Zhang, W.; Bundy, J.D.; Chen, C.S.; Kelly, T.N.; Chen, J.; He, J. A systematic analysis of worldwide population-based data on the global burden of chronic kidney disease in 2010. Kidney Int. 2015, 88, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lerman, L.O. The metabolic syndrome and chronic kidney disease. Transl. Res. 2017, 183, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, C.; Borrelli, S.; Minutolo, R.; Chiodini, P.; De Nicola, L.; Conte, G. A systematic review and meta-analysis suggests obesity predicts onset of chronic kidney disease in the general population. Kidney Int. 2017, 91, 1224–1235. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Afkarian, M.; Sachs, M.C.; Kestenbaum, B.; Hirsch, I.B.; Tuttle, K.R.; Himmelfarb, J.; de Boer, I.H. Kidney disease and increased mortality risk in type 2 diabetes. J. Am. Soc. Nephrol. 2013, 24, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.M.; Su, Y.; Li, J.; Wang, L.F. Recent advances in understanding the biochemical and molecular mechanism of diabetic nephropathy. Biochem. Biophys. Res. Commun. 2013, 433, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Kakio, Y.; Uchida, H.A.; Takeuchi, H.; Okuyama, Y.; Okuyama, M.; Umebayashi, R.; Wada, K.; Sugiyama, H.; Sugimoto, K.; Rakugi, H.; et al. Diabetic nephropathy is associated with frailty in patients with chronic hemodialysis. Geriatr. Gerontol. Int. 2018, 18, 1597–1602. [Google Scholar] [CrossRef]
- Pavenstädt, H.; Kriz, W.; Kretzler, M. Cell biology of the glomerular podocyte. Physiol. Rev. 2003, 83, 253–307. [Google Scholar] [CrossRef]
- Turing, A.M. The chemical basis of morphogenesis. Bull Math Biol. 1990, 52, 153–197. [Google Scholar] [CrossRef]
- Faul, C.; Asanuma, K.; Yanagida-Asanuma, E.; Kim, K.; Mundel, P. Actin up: Regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007, 17, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Lemley, K.V. Mechanical challenges to the glomerular filtration barrier: Adaptations and pathway to sclerosis. Pediatr. Nephrol. 2017, 32, 405–417. [Google Scholar] [CrossRef]
- Kriz, W.; Shirato, I.; Nagata, M.; LeHir, M.; Lemley, K.V. The podocyte’s response to stress: The enigma of foot process effacement. Am. J. Physiol. Renal Physiol. 2013, 304, F333–F347. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Jiang, S.; Hou, Q.; Qiu, D.; Shi, J.; Wang, L.; Chen, Z.; Zhang, M.; Duan, A.; Qin, W.; et al. Dissection of Glomerular Transcriptional Profile in Patients with Diabetic Nephropathy: SRGAP2a Protects Podocyte Structure and Function. Diabetes 2018, 67, 717–730. [Google Scholar] [CrossRef]
- Gojo, A.; Utsunomiya, K.; Taniguchi, K.; Yokota, T.; Ishizawa, S.; Kanazawa, Y.; Kurata, H.; Tajima, N. The Rho-kinase inhibitor, fasudil, attenuates diabetic nephropathy in streptozotocin-induced diabetic rats. Eur. J. Pharmacol. 2007, 568, 242–247. [Google Scholar] [CrossRef]
- Komers, R.; Oyama, T.T.; Beard, D.R.; Tikellis, C.; Xu, B.; Lotspeich, D.F.; Anderson, S. Rho kinase inhibition protects kidneys from diabetic nephropathy without reducing blood pressure. Kidney Int. 2011, 79, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, T.; Baldwin, C.; Aoudjit, L.; Nezvitsky, L.; Robins, R.; Jiang, R.; Takano, T. Protein tyrosine phosphatase 1B inhibition protects against podocyte injury and proteinuria. Am. J. Pathol. 2014, 184, 2211–2224. [Google Scholar] [CrossRef]
- Ma, H.; Togawa, A.; Soda, K.; Zhang, J.; Lee, S.; Ma, M.; Yu, Z.; Ardito, T.; Czyzyk, J.; Diggs, L.; et al. Inhibition of podocyte FAK protects against proteinuria and foot process effacement. J. Am. Soc. Nephrol. 2010, 21, 1145–1156. [Google Scholar] [CrossRef]
- Sever, S.; Schiffer, M. Actin dynamics at focal adhesions: A common endpoint and putative therapeutic target for proteinuric kidney diseases. Kidney Int. 2018, 93, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Oyarzún, C.; Garrido, W.; Alarcón, S.; Yáñez, A.; Sobrevia, L.; Quezada, C.; San Martín, R. Adenosine contribution to normal renal physiology and chronic kidney disease. Mol. Asp. Med. 2017, 55, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Schnermann, J.; Levine, D.Z. Paracrine factors in tubuloglomerular feedback: Adenosine, ATP, and nitric oxide. Annu. Rev. Physiol. 2003, 65, 501–529. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.S.; Rouse, M.; Liu, L.; Vergis, A.L.; Rosin, D.L.; Linden, J.; Sedor, J.R.; Okusa, M.D. Activation of adenosine 2A receptors preserves structure and function of podocytes. J. Am. Soc. Nephrol. 2008, 19, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.F.; Liang, Q.L.; Hu, P.; Wang, Y.M.; Li, P.; Luo, G.A. Correlations of six related purine metabolites and diabetic nephropathy in Chinese type 2 diabetic patients. Clin. Biochem. 2009, 42, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Pak, E.S.; Cha, J.J.; Cha, D.R.; Kanasaki, K.; Ha, H. Adenosine receptors as emerging therapeutic targets for diabetic kidney disease. Kidney Res. Clin. Pract. 2022, 41 (Suppl. 2), S74–S88. [Google Scholar] [CrossRef] [PubMed]
- Kretschmar, C.; Oyarzún, C.; Villablanca, C.; Jaramillo, C.; Alarcón, S.; Perez, G.; Díaz-Encarnación, M.M.; Pastor-Anglada, M.; Garrido, W.; Quezada, C.; et al. Reduced Adenosine Uptake and Its Contribution to Signaling that Mediates Profibrotic Activation in Renal Tubular Epithelial Cells: Implication in Diabetic Nephropathy. PLoS ONE 2016, 11, e0147430. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, S.; Garrido, W.; Vega, G.; Cappelli, C.; Suárez, R.; Oyarzún, C.; Quezada, C.; San Martín, R. Deficient Insulin-mediated Upregulation of the Equilibrative Nucleoside Transporter 2 Contributes to Chronically Increased Adenosine in Diabetic Glomerulopathy. Sci. Rep. 2017, 7, 9439. [Google Scholar] [CrossRef] [PubMed]
- Suarez, R.; Villarreal, C.; Nahuelpán, Y.; Jara, C.; Oyarzún, C.; Alarcón, S.; Díaz-Encarnación, M.M.; Guillén-Gómez, E.; Quezada, C.; San Martín, R. Defective insulin-stimulated equilibrative nucleoside transporter-2 activity and altered subcellular transporter distribution drive the loss of adenosine homeostasis in diabetic kidney disease progression. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 166890. [Google Scholar] [CrossRef]
- Roa, H.; Gajardo, C.; Troncoso, E.; Fuentealba, V.; Escudero, C.; Yáñez, A.; Sobrevia, L.; Pastor-Anglada, M.; Quezada, C.; San Martin, R. Adenosine mediates transforming growth factor-beta 1 release in kidney glomeruli of diabetic rats. FEBS Lett. 2009, 583, 3192–3198. [Google Scholar] [CrossRef]
- Torres, Á.; Muñoz, K.; Nahuelpán, Y.; Saez, A.P.R.; Mendoza, P.; Jara, C.; Cappelli, C.; Suarez, R.; Oyarzún, C.; Quezada, C.; et al. Intraglomerular Monocyte/Macrophage Infiltration and Macrophage-Myofibroblast Transition during Diabetic Nephropathy Is Regulated by the A2B Adenosine Receptor. Cells 2020, 9, 1051. [Google Scholar] [CrossRef]
- Cárdenas, A.; Toledo, C.; Oyarzún, C.; Sepúlveda, A.; Quezada, C.; Guillén-Gómez, E.; Díaz-Encarnación, M.M.; Pastor-Anglada, M.; San Martín, R. Adenosine A2B receptor-mediated VEGF induction promotes diabetic glomerulopathy. Lab. Invest. 2013, 93, 135–144. [Google Scholar] [CrossRef]
- Torres-Arévalo, Á.; Nahuelpán, Y.; Muñoz, K.; Jara, C.; Cappelli, C.; Taracha-Wiśniewska, A.; Quezada-Monrás, C.; Martín, R.S. A2BAR Antagonism Decreases the Glomerular Expression and Secretion of Chemoattractants for Monocytes and the Pro-Fibrotic M2 Macrophages Polarization during Diabetic Nephropathy. Int. J. Mol. Sci. 2023, 24, 10829. [Google Scholar] [CrossRef]
- Reiser, J.; Sever, S. Podocyte biology and pathogenesis of kidney disease. Annu. Rev. Med. 2013, 64, 357–366. [Google Scholar] [CrossRef]
- Saleem, M.A.; O’Hare, M.J.; Reiser, J.; Coward, R.J.; Inward, C.D.; Farren, T.; Xing, C.Y.; Ni, L.; Mathieson, P.W.; Mundel, P. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J. Am. Soc. Nephrol. 2002, 13, 630–638. [Google Scholar] [CrossRef]
- Yaoita, E.; Yoshida, Y.; Nameta, M.; Takimoto, H.; Fujinaka, H. Induction of interdigitating cell processes in podocyte culture. Kidney Int. 2018, 93, 519–524. [Google Scholar] [CrossRef]
- Pasapera, A.M.; Schneider, I.C.; Rericha, E.; Schlaepfer, D.D.; Waterman, C.M. Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J. Cell Biol. 2010, 188, 877–890. [Google Scholar] [CrossRef]
- Fischer, R.S.; Myers, K.A.; Gardel, M.L.; Waterman, C.M. Stiffness-controlled three-dimensional extracellular matrices for high-resolution imaging of cell behavior. Nat. Protoc. 2012, 7, 2056–2066. [Google Scholar] [CrossRef]
- Levental, I.; Levental, K.R.; Klein, E.A.; Assoian, R.; Miller, R.T.; Wells, R.G.; Janmey, P.A. A simple indentation device for measuring micrometer-scale tissue stiffness. J. Phys. Condens. Matter 2010, 22, 194120. [Google Scholar] [CrossRef] [PubMed]
- Wyss, H.M.; Henderson, J.M.; Byfield, F.J.; Bruggeman, L.A.; Ding, Y.; Huang, C.; Suh, J.H.; Franke, T.; Mele, E.; Pollak, M.R.; et al. Biophysical properties of normal and diseased renal glomeruli. Am. J. Physiol. Cell Physiol. 2011, 300, C397–C405. [Google Scholar] [CrossRef] [PubMed]
- Embry, A.E.; Mohammadi, H.; Niu, X.; Liu, L.; Moe, B.; Miller-Little, W.A.; Lu, C.Y.; Bruggeman, L.A.; McCulloch, C.A.; Janmey, P.A.; et al. Biochemical and Cellular Determinants of Renal Glomerular Elasticity. PLoS ONE 2016, 11, e0167924. [Google Scholar] [CrossRef]
- Elosegui-Artola, A.; Oria, R.; Chen, Y.; Kosmalska, A.; Pérez-González, C.; Castro, N.; Zhu, C.; Trepat, X.; Roca-Cusachs, P. Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity. Nat. Cell Biol. 2016, 18, 540–548. [Google Scholar] [CrossRef]
- Castrop, H. Mediators of tubuloglomerular feedback regulation of glomerular filtration: ATP and adenosine. Acta Physiol. 2007, 189, 3–14. [Google Scholar] [CrossRef]
- Thomson, S.; Bao, D.; Deng, A.; Vallon, V. Adenosine formed by 5′-nucleotidase mediates tubuloglomerular feedback. J. Clin. Investig. 2000, 106, 289–298. [Google Scholar] [CrossRef]
- Peti-Peterdi, J. Calcium wave of tubuloglomerular feedback. Am. J. Physiol. Renal Physiol. 2006, 291, F473–F480. [Google Scholar] [CrossRef]
- Thomson, S.C.; Vallon, V.; Blantz, R.C. Kidney function in early diabetes: The tubular hypothesis of glomerular filtration. Am. J. Physiol. Renal Physiol. 2004, 286, F8–F15. [Google Scholar] [CrossRef]
- Guo, K.; Pan, P.; Wu, M.; Ma, Y.; Lu, J.; Chen, H. Hyposialylated angiopoietin-like-4 induces apoptosis of podocytes via β1 Integrin/FAK signaling in diabetic nephropathy. Mol. Cell. Endocrinol. 2020, 505, 110730. [Google Scholar] [CrossRef]
- Mundel, P.; Reiser, J. Proteinuria: An enzymatic disease of the podocyte? Kidney Int. 2010, 77, 571–580. [Google Scholar] [CrossRef]
- Greka, A.; Mundel, P. Cell biology and pathology of podocytes. Annu. Rev. Physiol. 2012, 74, 299–323. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, J.; Asano-Matsuda, K.; Kitzler, T.M.; Takano, T. Rho GTPase regulatory proteins in podocytes. Kidney Int. 2021, 99, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Feoktistov, I.; Biaggioni, I. Adenosine A2b receptors evoke interleukin-8 secretion in human mast cells. An enprofylline-sensitive mechanism with implications for asthma. J. Clin. Investig. 1995, 96, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Somlyo, A.P.; Somlyo, A.V. Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J. Physiol. 2000, 522, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Somlyo, A.P.; Somlyo, A.V. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: Modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 2003, 83, 1325–1358. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef] [PubMed]
- McGeough, M.D.; Wree, A.; Inzaugarat, M.E.; Haimovich, A.; Johnson, C.D.; Peña, C.A.; Goldbach-Mansky, R.; Broderick, L.; Feldstein, A.E.; Hoffman, H.M. TNF regulates transcription of NLRP3 inflammasome components and inflammatory molecules in cryopyrinopathies. J. Clin. Investig. 2017, 127, 4488–4497. [Google Scholar] [CrossRef] [PubMed]
- Amaral, F.A.; Bastos, L.F.; Oliveira, T.H.; Dias, A.C.; Oliveira, V.L.; Tavares, L.D.; Costa, V.V.; Galvão, I.; Soriani, F.M.; Szymkowski, D.E.; et al. Transmembrane TNF-α is sufficient for articular inflammation and hypernociception in a mouse model of gout. Eur. J. Immunol. 2016, 46, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Liang, R.; Huang, B.; Hou, J.; Yin, J.; Zhao, T.; Zhou, L.; Wu, R.; Qian, Y.; Wang, F. Tumor necrosis factor-α blockade ameliorates diabetic nephropathy in rats. Clin. Kidney J. 2019, 14, 301–308. [Google Scholar] [CrossRef] [PubMed]
- St Hilaire, C.; Koupenova, M.; Carroll, S.H.; Smith, B.D.; Ravid, K. TNF-α upregulates the A2B adenosine receptor gene: The role of NAD(P)H oxidase 4. Biochem. Biophys. Res. Commun. 2008, 375, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Coca, S.G.; Nadkarni, G.N.; Huang, Y.; Moledina, D.G.; Rao, V.; Zhang, J.; Ferket, B.; Crowley, S.T.; Fried, L.F.; Parikh, C.R. Plasma Biomarkers and Kidney Function Decline in Early and Established Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2017, 28, 2786–2793. [Google Scholar] [CrossRef]
- Niewczas, M.A.; Gohda, T.; Skupien, J.; Smiles, A.M.; Walker, W.H.; Rosetti, F.; Cullere, X.; Eckfeldt, J.H.; Doria, A.; Mayadas, T.N.; et al. Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J. Am. Soc. Nephrol. 2012, 23, 507–515. [Google Scholar] [CrossRef]
- Awad, A.S.; You, H.; Gao, T.; Cooper, T.K.; Nedospasov, S.A.; Vacher, J.; Wilkinson, P.F.; Farrell, F.X.; Brian Reeves, W. Macrophage-derived tumor necrosis factor-α mediates diabetic renal injury. Kidney Int. 2015, 88, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Garrido, W.; Jara, C.; Torres, A.; Suarez, R.; Cappelli, C.; Oyarzún, C.; Quezada, C.; San Martín, R. Blockade of the Adenosine A3 Receptor Attenuates Caspase 1 Activation in Renal Tubule Epithelial Cells and Decreases Interleukins IL-1β and IL-18 in Diabetic Rats. Int. J. Mol. Sci. 2019, 20, 4531. [Google Scholar] [CrossRef] [PubMed]
- He, F.F.; Bao, D.; Su, H.; Wang, Y.M.; Lei, C.T.; Zhang, C.Y.; Ye, C.; Tang, H.; Wan, C.; You, C.Q.; et al. IL-6 increases podocyte motility via MLC-mediated focal adhesion impairment and cytoskeleton disassembly. J. Cell. Physiol. 2018, 233, 7173–7181. [Google Scholar] [CrossRef] [PubMed]
- Cuvelier, D.; Théry, M.; Chu, Y.S.; Dufour, S.; Thiéry, J.P.; Bornens, M.; Nassoy, P.; Mahadevan, L. The universal dynamics of cell spreading. Curr. Biol. 2007, 17, 694–699. [Google Scholar] [CrossRef]
- Murrell, M.; Pontani, L.L.; Guevorkian, K.; Cuvelier, D.; Nassoy, P.; Sykes, C. Spreading dynamics of biomimetic actin cortices. Biophys. J. 2011, 100, 1400–1409. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendoza-Soto, P.; Jara, C.; Torres-Arévalo, Á.; Oyarzún, C.; Mardones, G.A.; Quezada-Monrás, C.; San Martín, R. Pharmacological Blockade of the Adenosine A2B Receptor Is Protective of Proteinuria in Diabetic Rats, through Affecting Focal Adhesion Kinase Activation and the Adhesion Dynamics of Podocytes. Cells 2024, 13, 846. https://doi.org/10.3390/cells13100846
Mendoza-Soto P, Jara C, Torres-Arévalo Á, Oyarzún C, Mardones GA, Quezada-Monrás C, San Martín R. Pharmacological Blockade of the Adenosine A2B Receptor Is Protective of Proteinuria in Diabetic Rats, through Affecting Focal Adhesion Kinase Activation and the Adhesion Dynamics of Podocytes. Cells. 2024; 13(10):846. https://doi.org/10.3390/cells13100846
Chicago/Turabian StyleMendoza-Soto, Pablo, Claudia Jara, Ángelo Torres-Arévalo, Carlos Oyarzún, Gonzalo A. Mardones, Claudia Quezada-Monrás, and Rody San Martín. 2024. "Pharmacological Blockade of the Adenosine A2B Receptor Is Protective of Proteinuria in Diabetic Rats, through Affecting Focal Adhesion Kinase Activation and the Adhesion Dynamics of Podocytes" Cells 13, no. 10: 846. https://doi.org/10.3390/cells13100846
APA StyleMendoza-Soto, P., Jara, C., Torres-Arévalo, Á., Oyarzún, C., Mardones, G. A., Quezada-Monrás, C., & San Martín, R. (2024). Pharmacological Blockade of the Adenosine A2B Receptor Is Protective of Proteinuria in Diabetic Rats, through Affecting Focal Adhesion Kinase Activation and the Adhesion Dynamics of Podocytes. Cells, 13(10), 846. https://doi.org/10.3390/cells13100846