
MarvelD3 Is Upregulated in Ulcerative Colitis and Has Attenuating Effects during Colitis Indirectly Stabilizing the Intestinal Barrier

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients Features

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Control (n = 8) | UC (n = 8) | CD (n = 8) |
|---|---|---|---|
| Age (median, range) | 48, 20–72 | 37, 23–79 | 37, 21–61 |
| Gender (male/female) | 2/6 | 5/3 | 2/6 |
| Mayo endoscopic subscore (median, range) | - | 2, 1–3 | - |
| SES-CD (median, range) | - | - | 5.3, 1–12 |
2.2. Cell Lines
2.3. Cytokines and Inhibitors Experiments
2.4. Protein Isolation and Western Blotting
2.5. Immunofluorescent Staining
2.6. Generation of Mice with Intestinal Overexpression of MD3
2.7. Animal Housing, Handling, and Induction of DSS Colitis
2.8. Dilution Potentials and Impedance Spectroscopy
2.9. Flux Measurements
2.10. Statistical Analysis
3. Results
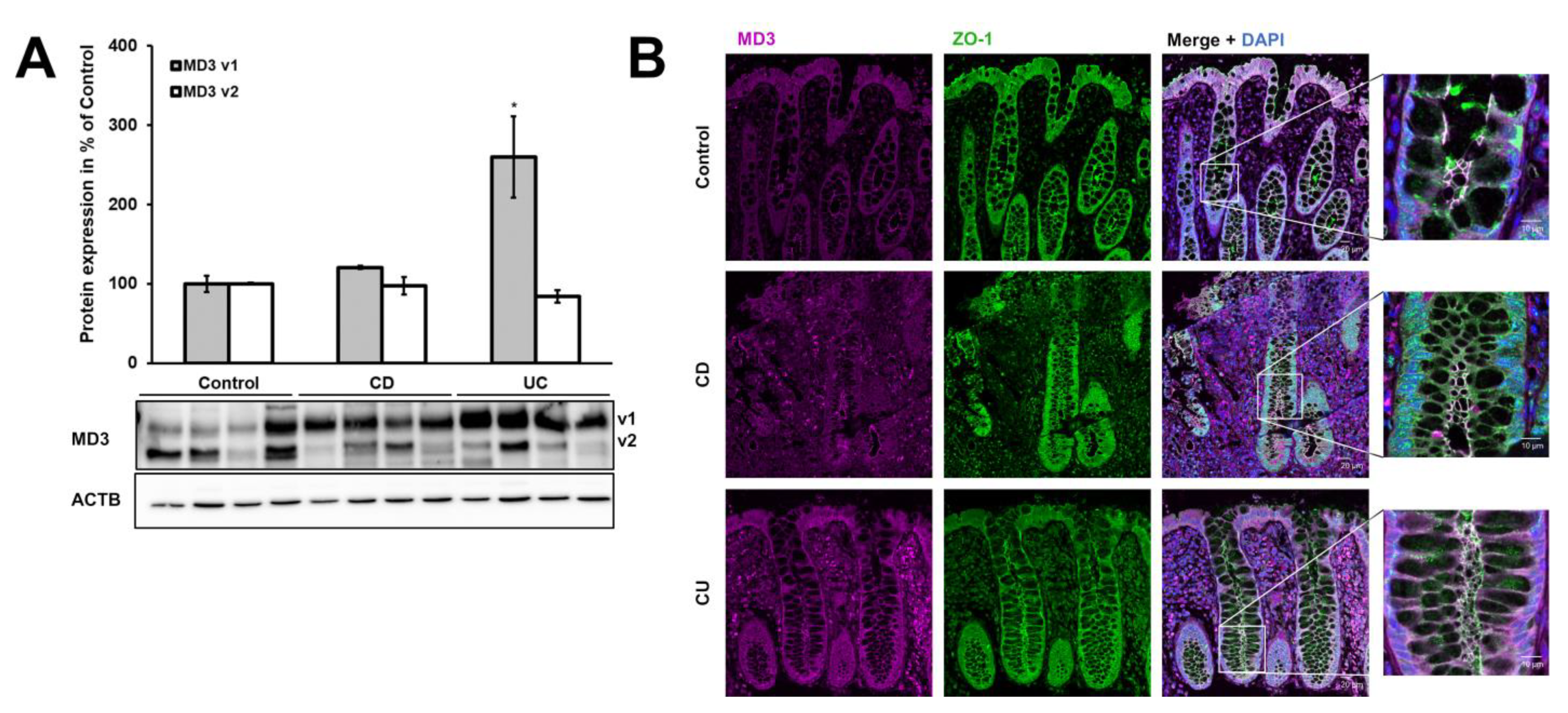
3.1. Expression of MD3 in Intestinal Epithelium of IBD Patients
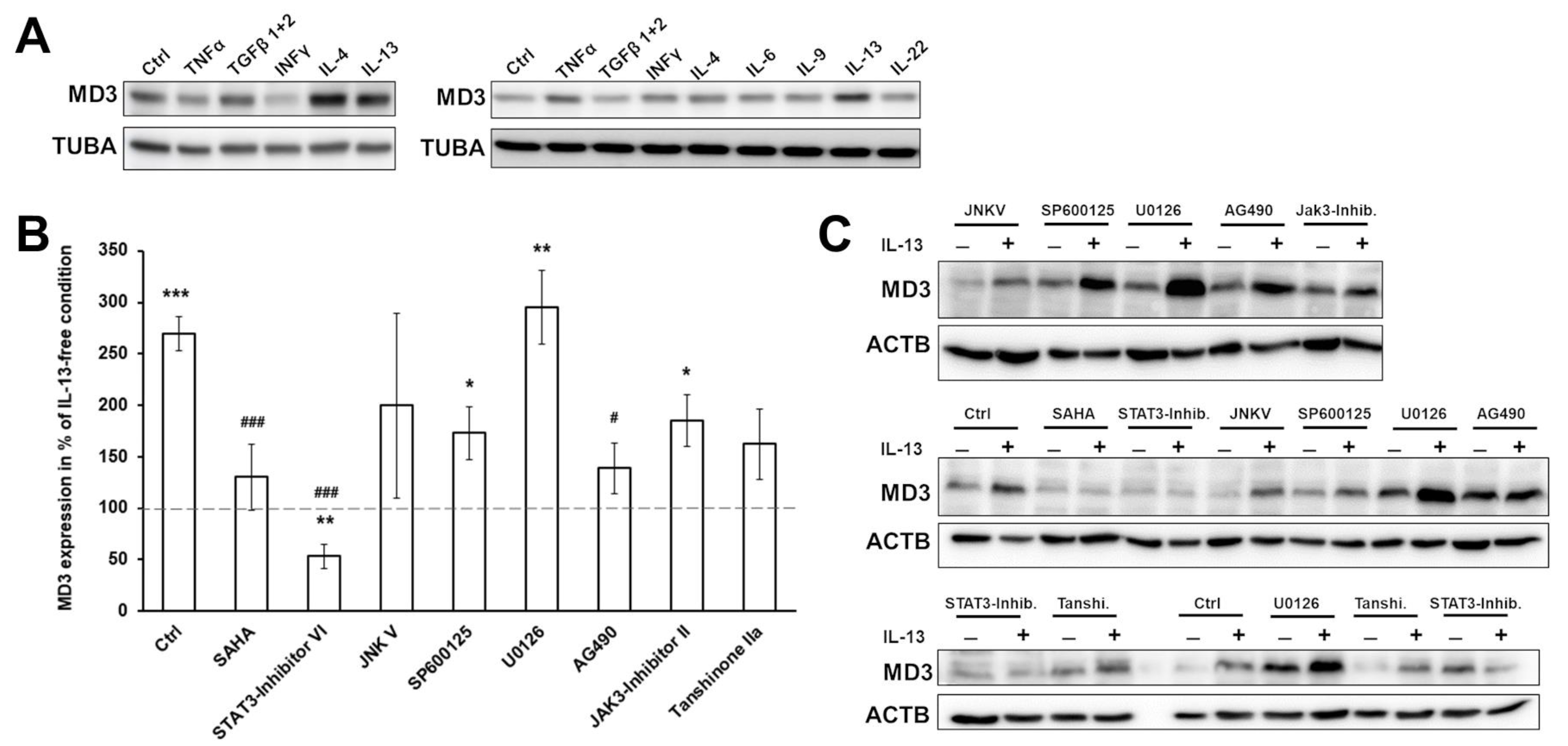
3.2. Signaling Pathways Involved in MD3 Upregulation
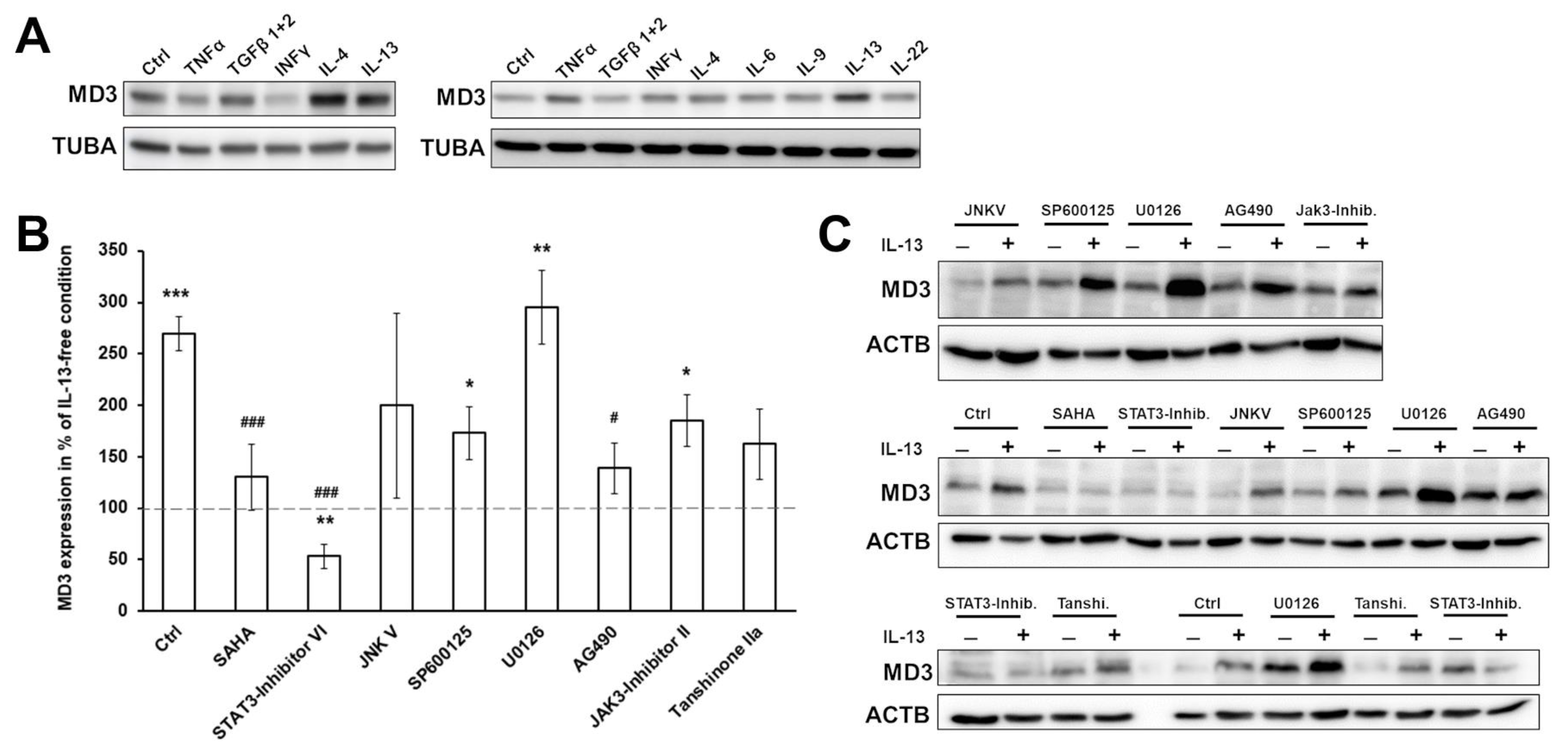
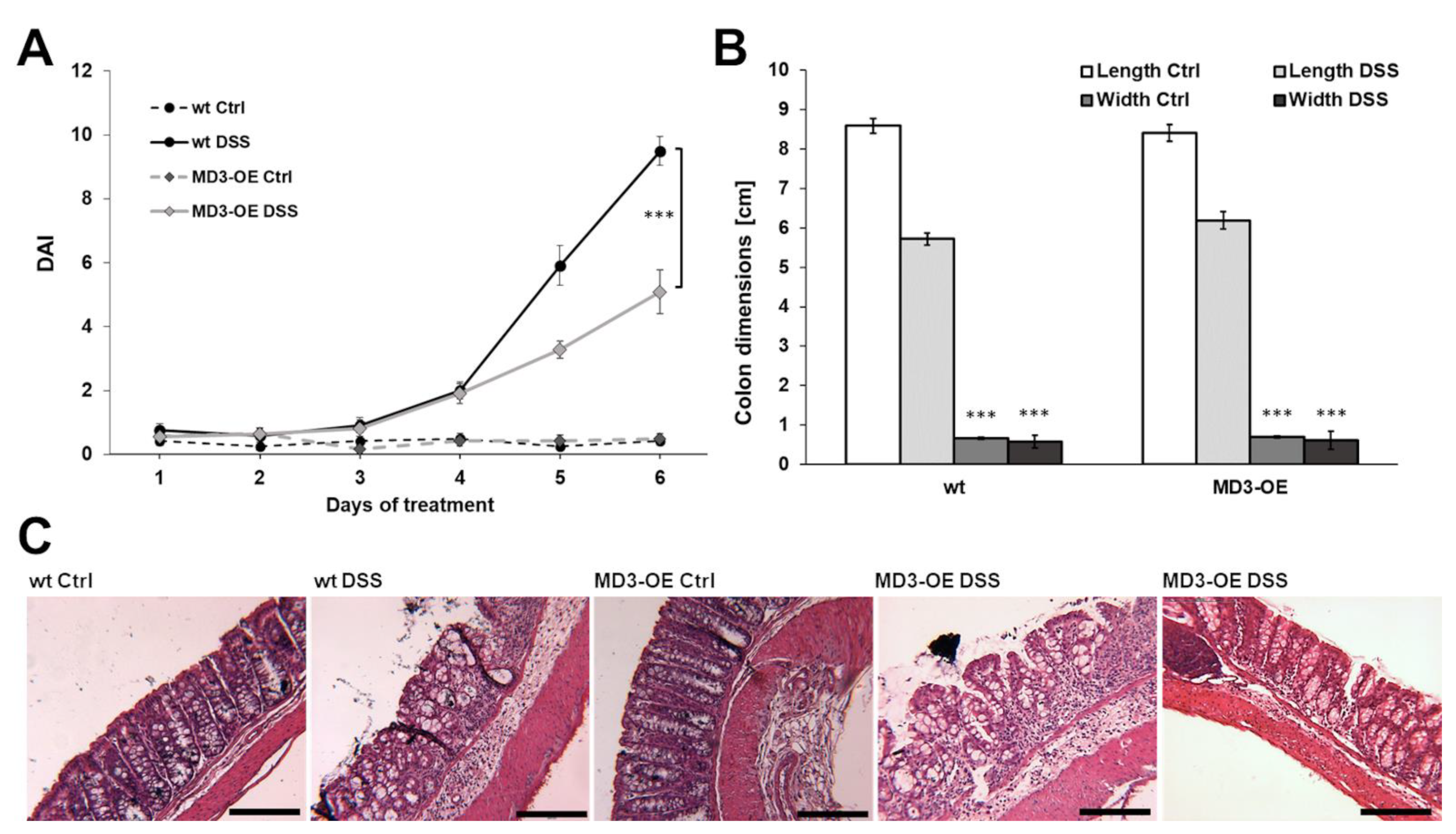
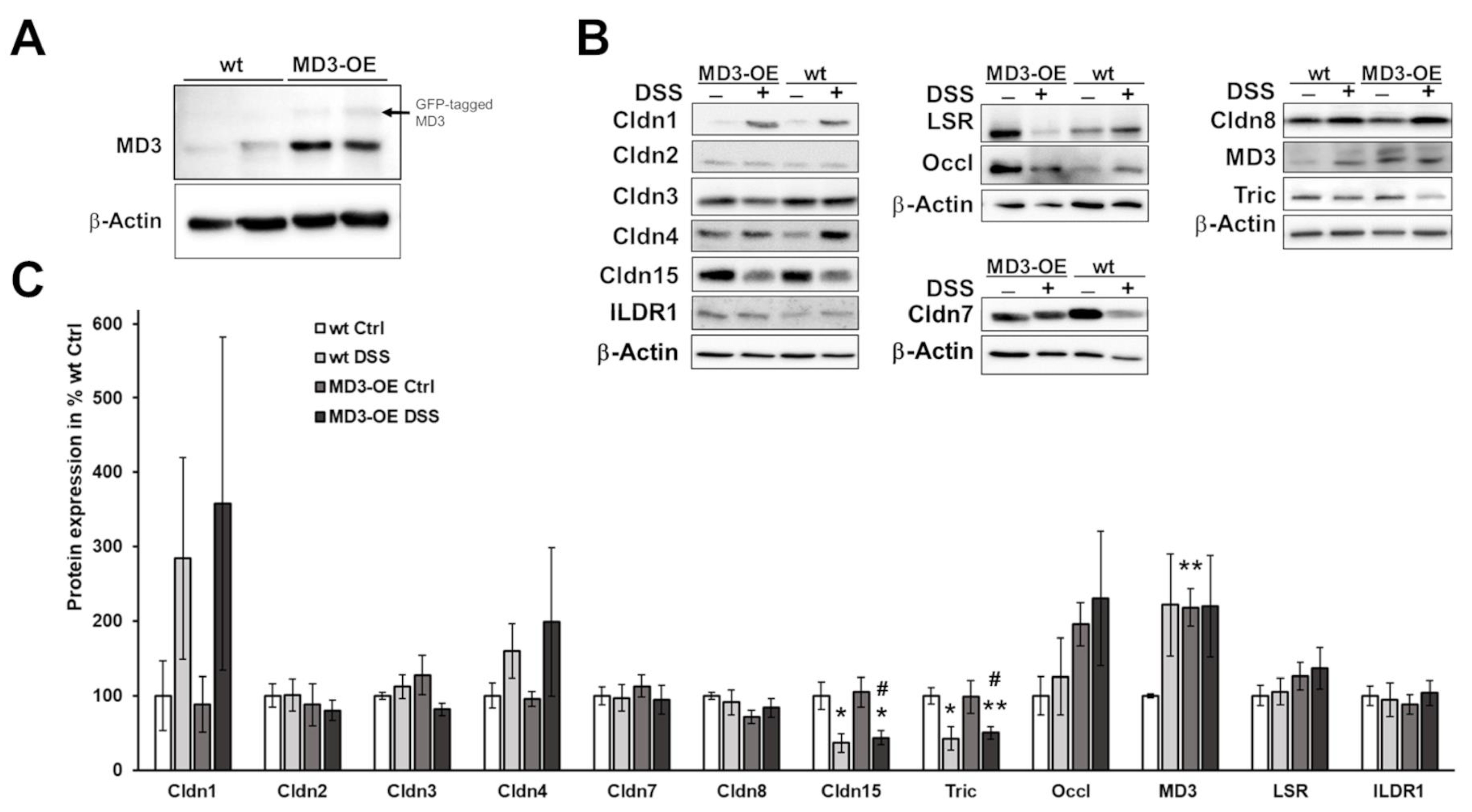
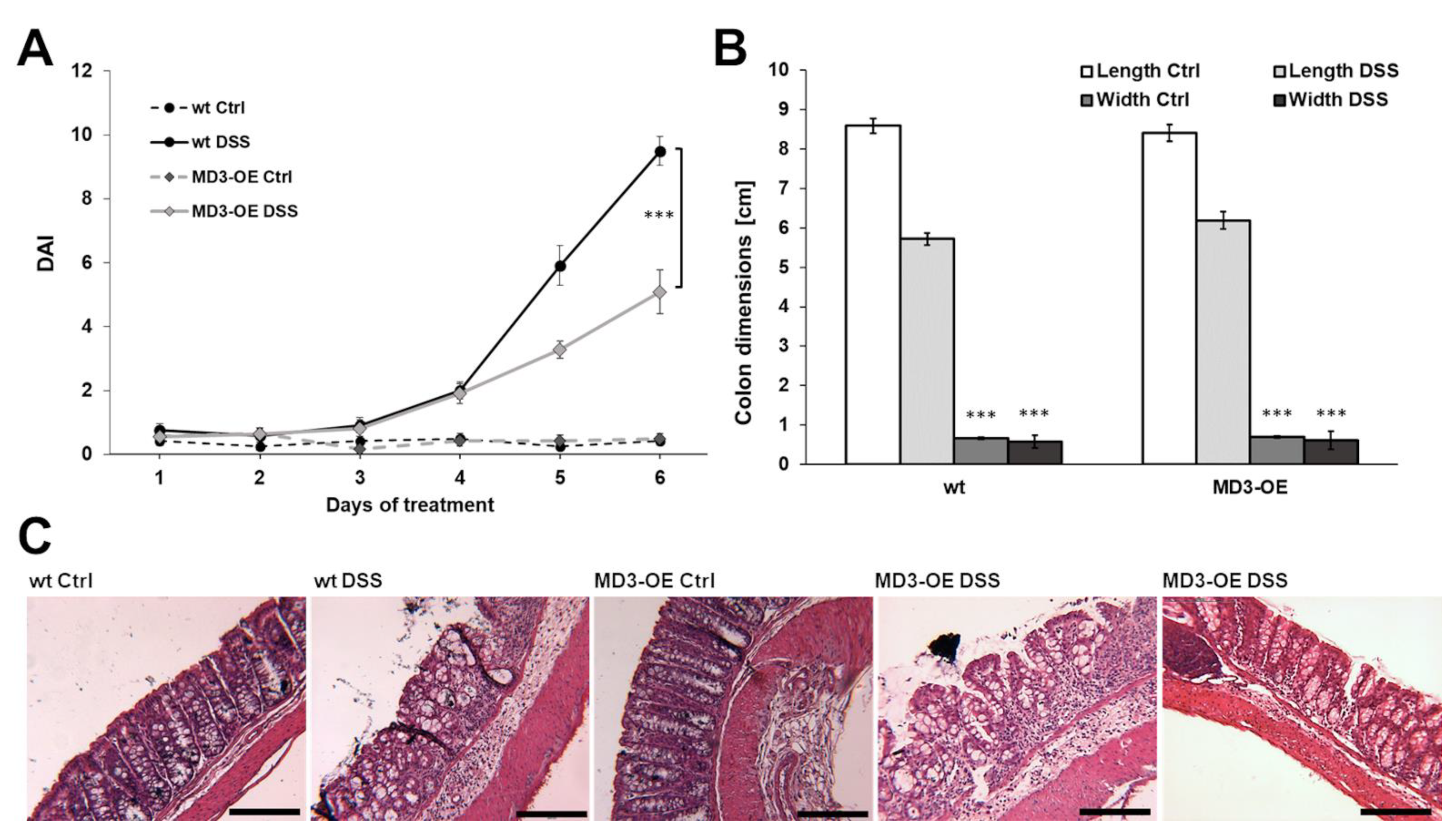
3.3. Effects of MD3 in DSS-Induced Colitis


4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Kaplan, G.G.; Ng, S.C. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology 2017, 152, 313–321.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burisch, J.; Munkholm, P. The epidemiology of inflammatory bowel disease. Scand J. Gastroenterol. 2015, 50, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Martini, E.; Krug, S.M.; Siegmund, B.; Neurath, M.F.; Becker, C. Mend Your Fences: The Epithelial Barrier and its Relationship With Mucosal Immunity in Inflammatory Bowel Disease. Cell Mol. Gastroenterol Hepatol. 2017, 23, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Günzel, D.; Fromm, M. Claudins and Other Tight Junction Proteins. Compr. Physiol. 2012, 2, 1819–1852. [Google Scholar] [CrossRef]
- Staehelin, L.A.; Mukherjee, T.M.; Williams, A.W. Freeze-etch appearance of the tight junctions in the epithelium of small and large intestine of mice. Protoplasma 1969, 67, 165–184. [Google Scholar] [CrossRef]
- Staehelin, L.A. Further observations on the fine structure of freeze-cleaved tight junctions. J. Cell Sci. 1973, 13, 763–786. [Google Scholar] [CrossRef]
- Mineta, K.; Yamamoto, Y.; Yamazaki, Y.; Tanaka, H.; Tada, Y.; Saito, K.; Tamura, A.; Igarashi, M.; Endo, T.; Takeuchi, K.; et al. Predicted expansion of the claudin multigene family. FEBS Lett. 2011, 585, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Martìn-Padura, I.; Lostaglio, S.; Schneemann, M.; Williams, L.; Romano, M.; Fruscella, P.; Panzeri, M.C.; Stoppacciaro, A.; Ruco, L.; Villa, A.; et al. Junctional Adhesion Molecule, a Novel Member of the Immunoglobulin Superfamily That Distributes at Intercellular Junctions and Modulates Monocyte Transmigration. J. Cell Biol. 1998, 142, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Higashi, T.; Tokuda, S.; Kitajiri, S.; Masuda, S.; Nakamura, H.; Oda, Y.; Furuse, M. Analysis of the ‘angulin’ proteins LSR, ILDR1 and ILDR2—Tricellulin recruitment, epithelial barrier function and implication in deafness pathogenesis. J. Cell Sci. 2013, 15, 966–977. [Google Scholar] [CrossRef] [Green Version]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S. Occludin: A novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar] [CrossRef]
- Ikenouchi, J.; Furuse, M.; Furuse, K.; Sasaki, H.; Tsukita, S.; Tsukita, S. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J. Cell Biol. 2005, 171, 939–945. [Google Scholar] [CrossRef]
- Steed, E.; Rodrigues, N.T.; Balda, M.S.; Matter, K. Identification of MarvelD3 as a tight junction-associated transmembrane protein of the occludin family. BMC Cell Biol. 2009, 10, 95. [Google Scholar] [CrossRef] [Green Version]
- Fromm, M.; Raleigh, D.R.; Marchiando, A.M.; Zhang, Y.; Shen, L.; Sasaki, H.; Wang, Y.; Long, M.; Turner, J.R. Faculty Opinions recommendation of Tight junction-associated MARVEL proteins marveld3, tricellulin, and occludin have distinct but overlapping functions. Mol. Biol. Cell 2010, 21, 1200–1213. [Google Scholar] [CrossRef]
- Oshima, T.; Miwa, H.; Joh, T. Changes in the expression of claudins in active ulcerative colitis. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. S2), S146–S150. [Google Scholar] [CrossRef]
- Zeissig, S.; Bürgel, N.; Günzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Bürgel, N.; Fromm, M.; et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef]
- Krug, S.M.; Bojarski, C.; Fromm, A.; Lee, I.M.; Dames, P.; Richter, J.F.; Turner, J.R.; Fromm, M.; Schulzke, J.D. Tricellulin is regulated via interleukin-13-receptor alpha2, affects macromolecule uptake, and is decreased in ulcerative colitis. Mucosal Immunol. 2018, 11, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.-C.E.; Bojarski, C.; Branchi, F.; Fromm, M.; Krug, S.M. Leptin Downregulates Angulin-1 in Active Crohn’s Disease via STAT3. Int. J. Mol. Sci. 2020, 21, 7824. [Google Scholar] [CrossRef]
- Steed, E.; Elbediwy, A.; Vacca, B.; Dupasquier, S.; Hemkemeyer, S.A.; Suddason, T.; Costa, A.C.; Beaudry, J.-B.; Zihni, C.; Gallagher, E.; et al. MarvelD3 couples tight junctions to the MEKK1–JNK pathway to regulate cell behavior and survival. J. Cell Biol. 2014, 204, 821–838. [Google Scholar] [CrossRef] [Green Version]
- Vacca, B.; Sanchez-Heras, E.; Steed, E.; Balda, M.; Ohnuma, S.-I.; Sasai, N.; Mayor, R.; Matter, K. MarvelD3 regulates the c-Jun N-terminal kinase pathway during eye development in Xenopus. Biol. Open 2016, 5, 1631–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacca, B.; Sanchez-Heras, E.; Steed, E.; Busson, S.L.; Balda, M.S.; Ohnuma, S.-I.; Sasai, N.; Mayor, R.; Matter, K. Control of neural crest induction by MarvelD3-mediated attenuation of JNK signalling. Sci. Rep. 2018, 8, 1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daperno, M.; D’Haens, G.; Van Assche, G.; Baert, F.; Bulois, P.; Maunoury, V.; Sostegni, R.; Rocca, R.; Pera, A.; Gevers, A.; et al. Development and validation of a new, simplified endoscopic activity score for Crohn’s disease: The SES-CD. Gastrointest Endosc. 2004, 60, 505–512. [Google Scholar] [CrossRef]
- Schroeder, K.W.; Tremaine, W.J.; Ilstrup, D.M. Coated Oral 5-Aminosalicylic Acid Therapy for Mildly to Moderately Active Ulcerative Colitis. N. Engl. J. Med. 1987, 317, 1625–1629. [Google Scholar] [CrossRef]
- Kreusel, K.M.; Fromm, M.; Schulzke, J.D.; Hegel, U. Cl- secretion in epithelial monolayers of mucus-forming human colon cells (HT-29/B6). Am. J. Physiol. 1991, 261, C574–C582. [Google Scholar] [CrossRef]
- Jun-Ichi, M.; Satoshi, T.; Kimi, A.; Fumi, T.; Akira, T.; Kiyoshi, T.; Ken-Ichi, Y. Expression vector system based on the chicken β-actin promoter directs efficient production of interleukin-5. Gene 1989, 79, 269–277. [Google Scholar] [CrossRef]
- el Marjou, F.; Janssen, K.P.; Chang, B.H.; Li, M.; Hindie, V.; Chan, L.; Louvard, D.; Chambon, P.; Metzger, D.; Robine, S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 2004, 39, 186–193. [Google Scholar] [CrossRef]
- Amasheh, S.; Meiri, N.; Gitter, A.H.; Schöneberg, T.; Mankertz, J.; Schulzke, J.D.; Fromm, M. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J. Cell Sci. 2002, 115, 4969–4976. [Google Scholar] [CrossRef] [Green Version]
- Günzel, D.; Amasheh, S.; Pfaffenbach, S.; Richter, J.F.; Kausalya, P.J.; Hunziker, W.; Fromm, M. Claudin-16 affects transcellular Cl- secretion in MDCK cells. J. Physiol. 2009, 587, 3777–3793. [Google Scholar] [CrossRef]
- Yu, A.S.; Cheng, M.H.; Angelow, S.; Günzel, D.; Kanzawa, S.A.; Schneeberger, E.E.; Fromm, M.; Coalson, R.D. Molecular Basis for Cation Selectivity in Claudin-2–based Paracellular Pores: Identification of an Electrostatic Interaction Site. J. Gen. Physiol. 2008, 133, 111–127. [Google Scholar] [CrossRef] [Green Version]
- A West, G.; Matsuura, T.; Levine, A.D.; Klein, J.S.; Fiocchi, C. Interleukin 4 in inflammatory bowel disease and mucosal immune reactivity. Gastroenterology 1996, 110, 1683–1695. [Google Scholar] [CrossRef]
- Alex, P.; Zachos, N.C.; Nguyen, T.; Gonzales, L.; Chen, T.E.; Conklin, L.S.; Centola, M.; Li, X. Distinct Cytokine Patterns Identified from Multiplex Profiles of Murine DSS and TNBS-Induced Colitis. Inflamm. Bowel Dis. 2009, 15, 341–352. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, L.; Ding, M.; Luo, Z.; Yuan, S.; Bansal, M.B.; Gilkeson, G.; Lang, R.; Jiang, W. Estrogen decreases tight junction protein ZO-1 expression in human primary gut tissues. Clin. Immunol. 2017, 183, 174–180. [Google Scholar] [CrossRef] [Green Version]
- DeVoss, J.; Diehl, L. Murine Models of Inflammatory Bowel Disease (IBD): Challenges of Modeling Human Disease. Toxicol. Pathol. 2013, 42, 99–110. [Google Scholar] [CrossRef]
- Nemeth, Z.H.; Bogdanovski, D.A.; Barratt-Stopper, P.; Paglinco, S.R.; Antonioli, L.; Rolandelli, R.H. Crohn’s Disease and Ulcerative Colitis Show Unique Cytokine Profiles. Cureus 2017, 9, e1177. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Hashimoto, K.; Uchikawa, R.; Tegoshi, T.; Yamada, M.; Arizono, N. Direct effects of IL-4/IL-13 and the nematode Nippostrongylus brasiliensis on intestinal epithelial cells in vitro. Parasite Immunol. 2010, 32, 420–429. [Google Scholar] [CrossRef]
- McCormick, S.M.; Heller, N.M. Commentary: IL-4 and IL-13 receptors and signaling. Cytokine 2015, 75, 38–50. [Google Scholar] [CrossRef] [Green Version]
- LaPorte, S.L.; Juo, Z.S.; Vaclavikova, J.; Colf, L.A.; Qi, X.; Heller, N.M.; Keegan, A.D.; Garcia, K.C. Molecular and structural basis of cytokine receptor pleiotropy in the interleukin-4/13 system. Cell 2008, 132, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, M.H.; Schindler, U.; Smiley, S.T.; Grusby, M.J. Stat6 Is Required for Mediating Responses to IL-4 and for the Development of Th2 Cells. Immunity 1996, 4, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Shajib, M.S.; Wang, H.; Kim, J.J.; Sunjic, I.; Ghia, J.E.; Denou, E.; Collins, M.; Denburg, J.A.; Khan, W.I. Interleukin 13 and serotonin: Linking the immune and endocrine systems in murine models of intestinal inflammation. PLoS ONE 2013, 8, e72774. [Google Scholar] [CrossRef] [Green Version]
- Schulzke, J.; Gitter, A.; Mankertz, J.; Spiegel, S.; Seidler, U.; Amasheh, S.; Saitou, M.; Tsukita, S.; Fromm, M. Epithelial transport and barrier function in occludin-deficient mice. Biochim. Biophys. Acta BBA Biomembr. 2005, 1669, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Ikenouchi, J.; Sasaki, H.; Tsukita, S.; Furuse, M.; Tsukita, S. Loss of Occludin Affects Tricellular Localization of Tricellulin. Mol. Biol. Cell 2008, 19, 4687–4693. [Google Scholar] [CrossRef] [Green Version]
- Butera, A.; Di Paola, M.; Vitali, F.; De Nitto, D.; Covotta, F.; Borrini, F.; Pica, R.; De Filippo, C.; Cavalieri, D.; Giuliani, A.; et al. IL-13 mRNA Tissue Content Identifies Two Subsets of Adult Ulcerative Colitis Patients With Different Clinical and Mucosa-Associated Microbiota Profiles. J. Crohns Colitis 2019, 14, 369–380. [Google Scholar] [CrossRef]
- Reinisch, W.; Panes, J.; Khurana, S.; Toth, G.; Hua, F.; Comer, G.M.; Hinz, M.; Page, K.; O’Toole, M.; Moorehead, T.M.; et al. Anrukinzumab, an anti-interleukin 13 monoclonal antibody, in active UC: Efficacy and safety from a phase IIa randomised multicentre study. Gut 2015, 64, 894–900. [Google Scholar] [CrossRef]
- Danese, S.; Rudziński, J.; Brandt, W.; Dupas, J.-L.; Peyrin-Biroulet, L.; Bouhnik, Y.; Kleczkowski, D.; Uebel, P.; Lukas, M.; Knutsson, M.; et al. Tralokinumab for moderate-to-severe UC: A randomised, double-blind, placebo-controlled, phase IIa study. Gut 2014, 64, 243–249. [Google Scholar] [CrossRef] [Green Version]
- Keely, S.; Feighery, L.; Campion, D.P.; O’Brien, L.; Brayden, D.J.; Baird, A.W. Chloride-led Disruption of the Intestinal Mucous Layer Impedes Salmonella Invasion: Evidence for an ‘Enteric Tear’ Mechanism. Cell Physiol. Biochem. 2011, 28, 743–752. [Google Scholar] [CrossRef]
- Kawakami, K.; Taguchi, J.; Murata, T.; Puri, R.K. The interleukin-13 receptor α2 chain: An essential component for binding and internalization but not for interleukin-13–induced signal transduction through the STAT6 pathway. Blood 2001, 97, 2673–2679. [Google Scholar] [CrossRef] [Green Version]
- Abbas, A.K.; Murphy, K.M.; Sher, A. Functional diversity of helper T lymphocytes. Nature 1996, 383, 787–793. [Google Scholar] [CrossRef]
- Gieseck, R.L.; Wilson, M.S.; Wynn, T.A. Type 2 immunity in tissue repair and fibrosis. Nat. Rev. Immunol. 2018, 18, 62–76. [Google Scholar] [CrossRef]
- Andrews, A.-L.; Holloway, J.; Puddicombe, S.M.; Holgate, S.T.; Davies, D. Kinetic Analysis of the Interleukin-13 Receptor Complex. J. Biol. Chem. 2002, 277, 46073–46078. [Google Scholar] [CrossRef] [Green Version]
- Karmele, E.P.; Pasricha, T.S.; Ramalingam, T.R.; Thompson, R.W.; Gieseck, R.L.; Knilans, K.J.; Hegen, M.; Farmer, M.; Jin, F.; Kleinman, A.; et al. Anti-IL-13Ralpha2 therapy promotes recovery in a murine model of inflammatory bowel disease. Mucosal. Immunol. 2019, 12, 1174–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, R.; Chaturvedi, R.; Olivares-Villagómez, D.; Habib, T.; Asim, M.; Shivesh, P.; Polk, D.B.; Wilson, K.; Washington, M.K.; Van Kaer, L.; et al. Targeted colonic claudin-2 expression renders resistance to epithelial injury, induces immune suppression, and protects from colitis. Mucosal Immunol. 2014, 7, 1340–1353. [Google Scholar] [CrossRef] [PubMed]

| Concentration | Source | ||
|---|---|---|---|
| Antibodies | |||
| mouse anti-ACTB | WB | 1:10,000 | Sigma-Aldrich, Schnelldorf, Germany, CAT. A5441 |
| rabbit anti-CLDN1 | WB | 1:1000 | Invitrogen, ThermoFisher, Waltham, MA, USA Cat. 519000 |
| rabbit anti-CLDN2 (used for mouse tissue analysis) | WB | 1:1000 | Invitrogen, ThermoFisher, Waltham, MA, USA Cat. 516100 |
| mouse anti-CLDN2 | WB | 1:1000 | Invitrogen, ThermoFisher, Waltham, MA, USA, Cat. 325600 |
| rabbit anti-CLDN3 | WB | 1:1000 | Invitrogen, ThermoFisher, Waltham, MA, USA, Cat. 341700 |
| mouse anti-CLDN4 | WB | 1:1000 | Invitrogen, ThermoFisher, Waltham, MA, USA, Cat. 329400 |
| mouse anti-CLDN7 | WB | 1:1000 | Invitrogen, ThermoFisher, Waltham, MA, USA, Cat. 349100 |
| rabbit anti-CLDN8 | WB | 1:1000 | Invitrogen, ThermoFisher, Waltham, MA, USA Cat. 400700Z |
| rabbit anti-CLDN15 | WB | 1:1000 | Invitrogen, ThermoFisher, Waltham, MA, USA, Cat. 364200 |
| rabbit anti-ILDR1 | WB | 1:1000 | Bioss, ThermoFisher, Waltham, MA, USA, Cat: bs-11013R |
| rabbit anti-LSR | WB | 1:1000 | Atlas antibodies, Cat. HPA007270 |
| rabbit anti-MD3 | IF WB | 1:200 1:1000 | Proteintech, Rosemont, IL, USA, Cat. 25567-1-AP |
| rabbit anti-MD3 (used for patient tissue and some mouse tissue analysis) | WB | 1:1000 | Kind gift of Jerrold Turner, Laboratory of Mucosal Barrier Pathobiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA, [13,14] |
| rabbit anti-OCCL | WB | 1:1000 | Invitrogen, ThermoFisher, Waltham, MA, USA, Cat. 711500 |
| rabbit anti-TRIC | WB | 1:1000 | Invitrogen, ThermoFisher, Waltham, MA, USA, Cat. 700191 |
| mouse anti-TUBA | WB | 1:4000 | Sigma-Aldrich, Schnellendorf, Germany, CAT. T9026 |
| mouse anti-ZO-1 647 | IF | 1:500 | Invitrogen, ThermoFisher, Waltham, MA, USA, Cat. MA3-39100-A647 |
| DAPI | IF | 1:1000 | Roche Diagnositics, Mannheim, Germany, Cat. 10 236 276 001 |
| goat anti-rabbit IgG (H+L) secondary antibody Alexa Flour 488 | IF | 1:500 | Invitrogen, ThermoFisher, Waltham, MA, USA, Cat. A11034 |
| goat anti-mouse IgG (H+L) antibody Alexa Flour 594 | IF | 1:500 | Invitrogen, ThermoFisher, Waltham, MA, USA, Cat. A11032 |
| secondary peroxidase-conjugated antibodies anti-rabbit | WB | 1:10000 | Jackson ImmunoResearch, Cambridge House, UK, Cat. 111-036-003 |
| secondary peroxidase-conjugated antibodies anti-mouse | WB | 1:10000 | Jackson ImmunoResearch, Cambridge House, UK, Cat. 115-036-003 |
| Cytokines | |||
| TNFα | 1000 | U/mL | PeproTech, Hamburg, Germany |
| IFNγ | 50 | U/mL | PeproTech, Hamburg, Germany |
| IL-1α | 10 | ng/mL | PeproTech, Hamburg, Germany |
| TGFβ 1 | 10 | ng/mL | Miltenyi Biotec, Bergisch Gladbach, Germany |
| TGFβ 2 | 10 | ng/mL | Miltenyi Biotec, Bergisch Gladbach, Germany |
| IL-4 | 100 | ng/mL | PeproTech, Hamburg, Germany |
| IL-5 | 50 | ng/mL | Miltenyi Biotec, Bergisch Gladbach, Germany |
| IL-6 | 50 | ng/mL | Miltenyi Biotec, Bergisch Gladbach, Germany |
| IL-9 | 100 | ng/mL | Miltenyi Biotec, Bergisch Gladbach, Germany |
| IL-13 | 100 | ng/mL | PeproTech, Hamburg, Germany |
| IL-22 | 100 | ng/mL | Miltenyi Biotec, Bergisch Gladbach, Germany |
| Inhibitors | |||
| U0126 | 10 | µM | Cell signaling Technology, Frankfurt am Main, Germany |
| SAHA | 5 | µM | Sigma-Aldrich, Schnelldorf, Germany |
| AG490 | 50 | µM | Sigma-Aldrich, Schnelldorf, Germany |
| Tanshinone IIa | 10 | µM | Sigma-Aldrich, Schnelldorf, Germany |
| STAT3-Inhibitor VI | 10 | µM | Calbiochem, Darmstadt, Germany |
| JAK3-Inhibitor II | 50 | µM | Calbiochem, Darmstadt, Germany |
| JNK V | 10 | µM | Calbiochem, Darmstadt, Germany |
| SP600125 | 10 | µM | Cell Signaling Technology, Frankfurt am Main, Germany |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiß, F.; Czichos, C.; Knobe, L.; Voges, L.; Bojarski, C.; Michel, G.; Fromm, M.; Krug, S.M. MarvelD3 Is Upregulated in Ulcerative Colitis and Has Attenuating Effects during Colitis Indirectly Stabilizing the Intestinal Barrier. Cells 2022, 11, 1541. https://doi.org/10.3390/cells11091541
Weiß F, Czichos C, Knobe L, Voges L, Bojarski C, Michel G, Fromm M, Krug SM. MarvelD3 Is Upregulated in Ulcerative Colitis and Has Attenuating Effects during Colitis Indirectly Stabilizing the Intestinal Barrier. Cells. 2022; 11(9):1541. https://doi.org/10.3390/cells11091541
Chicago/Turabian StyleWeiß, Franziska, Carolina Czichos, Lukas Knobe, Lena Voges, Christian Bojarski, Geert Michel, Michael Fromm, and Susanne M. Krug. 2022. "MarvelD3 Is Upregulated in Ulcerative Colitis and Has Attenuating Effects during Colitis Indirectly Stabilizing the Intestinal Barrier" Cells 11, no. 9: 1541. https://doi.org/10.3390/cells11091541
APA StyleWeiß, F., Czichos, C., Knobe, L., Voges, L., Bojarski, C., Michel, G., Fromm, M., & Krug, S. M. (2022). MarvelD3 Is Upregulated in Ulcerative Colitis and Has Attenuating Effects during Colitis Indirectly Stabilizing the Intestinal Barrier. Cells, 11(9), 1541. https://doi.org/10.3390/cells11091541