
DNA Damage Response Inhibitors in Cholangiocarcinoma: Current Progress and Perspectives

 and
and
Abstract
1. Introduction
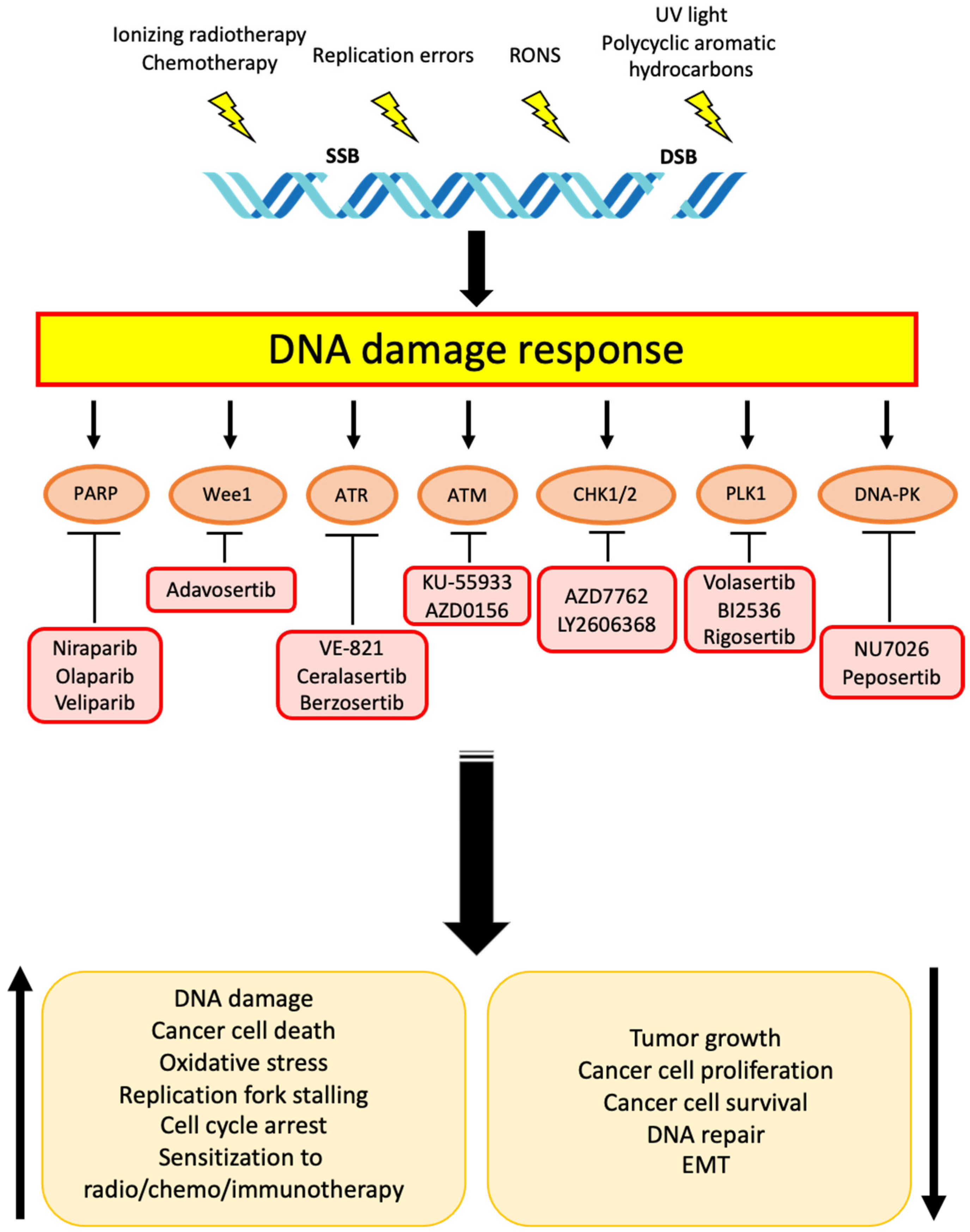
2. Targets and Related Therapies for DDR in Cholangiocarcinoma
2.1. PARP
2.2. Wee1
2.3. ATR and ATM
2.4. CHK1 and CHK2
2.5. Other Targets
2.5.1. DNA-PK
2.5.2. PLK1

{kind=link}
| Target | Treatments | Primary Endpoints | Phase | Study Identifier (ClinicalTrials.gov, accessed on 8 March 2022) |
|---|---|---|---|---|
| PARP | Rucaparib + Irinotecan/5-FU/Leucovorin calcium | MTD; DCR | I/II | NCT03337087 [178] |
| PARP | Niraparib | ORR | II | NCT03207347 [123] |
| PARP | Olaparib | ORR | II | NCT03212274 [112] |
| PARP; ATR | Olaparib + Ceralasertib (AZD6738) | ORR | II | NCT03878095 [114] |
| PARP; PD-L1 | Olaparib + Durvalamab | ORR; DCR | II | NCT03991832 [116] |
| PARP | Olaparib | ORR | II | NCT04042831 [109] |
| PARP; PD-1 | Olaparib + Pembrolizumab | ORR | II | NCT04306367 [117] |
| PARP; ATRATR; PD-L1 | Olaparib + Ceralasertib (AZD6738) Ceralasertib (AZD6738) + Durvalamab | DCR | II | NCT04298021 [115] |
| PARP; PD-1 | Rucaparib + Nivolumab | PFS at 4 months | II | NCT03639935 [121] |
| Wee1 | Adavosertib (AZD1775) | ORR | II | NCT02465060 [138] |
| DNA-PK; PD-L1 | Peposertib + Avelumab | MTD; ORR | I/II | NCT04068194 [166] |
2.5.3. Evidence and Future Prospects of DDR Inhibition-Based Combination Therapy in CCA
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Florio, A.A.; Ferlay, J.; Znaor, A.; Ruggieri, D.; Alvarez, C.S.; Laversanne, M.; Bray, F.; McGlynn, K.A.; Petrick, J.L. Global Incidence and Trends in Intra- and Extrahepatic Cholangiocarcinoma from 1993 to 2012. Cancer 2020, 126, 2666–2678. [Google Scholar] [CrossRef] [PubMed]
- Benavides, M.; Antón, A.; Gallego, J.; Gómez, M.A.; Jiménez-Gordo, A.; La Casta, A.; Laquente, B.; Macarulla, T.; Rodríguez-Mowbray, J.R.; Maurel, J. Biliary Tract Cancers: SEOM Clinical Guidelines. Clin. Transl. Oncol. 2015, 17, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Nakeeb, A.; Pitt, H.A.; Sohn, T.A.; Coleman, J.; Abrams, R.A.; Piantadosi, S.; Hruban, R.H.; Lillemoe, K.D.; Yeo, C.J.; Cameron, J.L. Cholangiocarcinoma: A Spectrum of Intrahepatic, Perihilar, and Distal Tumors. Ann. Surg. 1996, 224, 463–475. [Google Scholar] [CrossRef]
- Su, C.-H.; Tsay, S.-H.; Wu, C.-C.; Shyr, Y.-M.; King, K.-L.; Lee, C.-H.; Lui, W.-Y.; Liu, T.-J.; P’eng, F.-K. Factors Influencing Postoperative Morbidity, Mortality, and Survival After Resection for Hilar Cholangiocarcinoma. Ann. Surg. 1996, 223, 384–394. [Google Scholar] [CrossRef]
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef]
- Choi, C.W.; Choi, K.; Seo, J.H.; Kim, B.S.; Kim, J.S.; Kim, C.D.; Um, S.H.; Kim, J.S.; Kim, Y.H. Effects of 5-Fluorouracil and Leucovorin in the Treatment of Pancreatic–Biliary Tract Adenocarcinomas. Am. J. Clin. Oncol. 2000, 23, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Oh, S.-Y.; Kim, S.-H.; Kwon, H.-C.; Kim, J.-S.; Jin-Kim, H.; Kim, Y.-H. Single-Agent Gemcitabine in the Treatment of Advanced Biliary Tract Cancers: A Phase II Study. Jpn. J. Clin. Oncol. 2005, 35, 68–73. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Giuliani, F.; Gebbia, V.; Maiello, E.; Borsellino, N.; Bajardi, E.; Colucci, G. Gemcitabine and Cisplatin for Inoperable and/or Metastatic Biliary Tree Carcinomas: A Multicenter Phase II Study of the Gruppo Oncologico Dell’Italia Meridionale (GOIM). Ann. Oncol. 2006, 17, vii73–vii77. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Jani, C.R.; Savarese, D.M.; O’Donnell, J.L.; Stuart, K.E.; Rocha Lima, C.M. Gemcitabine and Irinotecan in Locally Advanced or Metastatic Biliary Cancer: Preliminary Report. Oncology 2003, 17, 23–26. [Google Scholar] [PubMed]
- Knox, J.J.; Hedley, D.; Oza, A.; Feld, R.; Siu, L.L.; Chen, E.; Nematollahi, M.; Pond, G.R.; Zhang, J.; Moore, M.J. Combining Gemcitabine and Capecitabine in Patients with Advanced Biliary Cancer: A Phase II Trial. J. Clin. Oncol. 2005, 23, 2332–2338. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Mahoney, M.R.; Allmer, C.; Thomas, J.; Pitot, H.C.; Kim, G.; Donehower, R.C.; Fitch, T.; Picus, J.; Erlichman, C. Phase II Study of Erlotinib in Patients with Advanced Biliary Cancer. J. Clin. Oncol. 2006, 24, 3069–3074. [Google Scholar] [CrossRef]
- Bonet Beltrán, M.; Allal, A.S.; Gich, I.; Solé, J.M.; Carrió, I. Is Adjuvant Radiotherapy Needed after Curative Resection of Extrahepatic Biliary Tract Cancers? A Systematic Review with a Meta-Analysis of Observational Studies. Cancer Treat. Rev. 2012, 38, 111–119. [Google Scholar] [CrossRef]
- Bridgewater, J.A.; Goodman, K.A.; Kalyan, A.; Mulcahy, M.F. Biliary Tract Cancer: Epidemiology, Radiotherapy, and Molecular Profiling. Am Soc. Clin. Oncol. Educ. Book 2016, 35, e194–e203. [Google Scholar] [CrossRef]
- Li, H.; Chen, L.; Zhu, G.-Y.; Yao, X.; Dong, R.; Guo, J.-H. Interventional Treatment for Cholangiocarcinoma. Front. Oncol.. 2021, 11, 671327. [Google Scholar] [CrossRef]
- Renzulli, M.; Ramai, D.; Singh, J.; Sinha, S.; Brandi, N.; Ierardi, A.M.; Albertini, E.; Sacco, R.; Facciorusso, A.; Golfieri, R. Locoregional Treatments in Cholangiocarcinoma and Combined Hepatocellular Cholangiocarcinoma. Cancers 2021, 13, 3336. [Google Scholar] [CrossRef]
- Li, Y.; Song, Y.; Liu, S. The New Insight of Treatment in Cholangiocarcinoma. J. Cancer 2022, 13, 450–464. [Google Scholar] [CrossRef]
- Manne, A.; Woods, E.; Tsung, A.; Mittra, A. Biliary Tract Cancers: Treatment Updates and Future Directions in the Era of Precision Medicine and Immuno-Oncology. Front. Oncol. 2021, 11, 768009. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Hu, J.; Liu, S.; Meric-Bernstam, F.; Abdel-Wahab, R.; Xu, J.; Li, Q.; Yan, M.; Feng, Y.; Lin, J.; et al. Intrahepatic Cholangiocarcinoma: Genomic Heterogeneity Between Eastern and Western Patients. JCO Precis. Oncol. 2020, 4, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Gong, J.; Zhong, G.; Hu, J.; Cai, D.; Zhao, L.; Zhao, Z. Identification of Mutator-Derived Alternative Splicing Signatures of Genomic Instability for Improving the Clinical Outcome of Cholangiocarcinoma. Front. Oncol. 2021, 11, 666847. [Google Scholar] [CrossRef] [PubMed]
- Brandi, G.; Farioli, A.; Astolfi, A.; Biasco, G.; Tavolari, S. Genetic Heterogeneity in Cholangiocarcinoma: A Major Challenge for Targeted Therapies. Oncotarget 2015, 6, 14744–14753. [Google Scholar] [CrossRef] [PubMed]
- Calkins, G.N. Zur Frage Der Entstehung Maligner Tumoren. By Th. Boveri. Jena, Gustav Fischer. 1914. 64 Pages. Science 1914, 40, 857–859. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.-A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal Instability Drives Metastasis through a Cytosolic DNA Response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Chambers, T.; Lopez, J.I.; Nicol, D.; O’Brien, T.; Larkin, J.; Horswell, S.; et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell 2018, 173, 581–594.e12. [Google Scholar] [CrossRef]
- Goh, J.Y.; Feng, M.; Wang, W.; Oguz, G.; Yatim, S.M.J.M.; Lee, P.L.; Bao, Y.; Lim, T.H.; Wang, P.; Tam, W.L.; et al. Chromosome 1q21.3 Amplification Is a Trackable Biomarker and Actionable Target for Breast Cancer Recurrence. Nat. Med. 2017, 23, 1319–1330. [Google Scholar] [CrossRef]
- Cahill, D.P.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Genetic Instability and Darwinian Selection in Tumours. Trends Cell Biol. 1999, 9, M57–M60. [Google Scholar] [CrossRef]
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic Alterations in Colorectal Cancer. Gastrointest Cancer Res. 2012, 5, 19–27. [Google Scholar]
- Pikor, L.; Thu, K.; Vucic, E.; Lam, W. The Detection and Implication of Genome Instability in Cancer. Cancer Metastasis Rev. 2013, 32, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Giam, M.; Rancati, G. Aneuploidy and Chromosomal Instability in Cancer: A Jackpot to Chaos. Cell Div. 2015, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Cibulskis, K.; Helman, E.; McKenna, A.; Shen, H.; Zack, T.; Laird, P.W.; Onofrio, R.C.; Winckler, W.; Weir, B.A.; et al. Absolute Quantification of Somatic DNA Alterations in Human Cancer. Nat. Biotechnol 2012, 30, 413–421. [Google Scholar] [CrossRef]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome Doubling Shapes the Evolution and Prognosis of Advanced Cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689.e3. [Google Scholar] [CrossRef]
- Thompson, S.L.; Bakhoum, S.F.; Compton, D.A. Mechanisms of Chromosomal Instability. Curr. Biol. 2010, 20, R285–R295. [Google Scholar] [CrossRef]
- Laughney, A.M.; Elizalde, S.; Genovese, G.; Bakhoum, S.F. Dynamics of Tumor Heterogeneity Derived from Clonal Karyotypic Evolution. Cell Rep. 2015, 12, 809–820. [Google Scholar] [CrossRef]
- Burkard, M.E.; Weaver, B.A. Tuning Chromosomal Instability to Optimize Tumor Fitness. Cancer Discov. 2017, 7, 134–136. [Google Scholar] [CrossRef]
- Lee, H.-S.; Lee, N.C.O.; Kouprina, N.; Kim, J.-H.; Kagansky, A.; Bates, S.; Trepel, J.B.; Pommier, Y.; Sackett, D.; Larionov, V. Effects of Anticancer Drugs on Chromosome Instability and New Clinical Implications for Tumor-Suppressing Therapies. Cancer Res. 2016, 76, 902–911. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Kabeche, L.; Wood, M.D.; Laucius, C.D.; Qu, D.; Laughney, A.M.; Reynolds, G.E.; Louie, R.J.; Phillips, J.; Chan, D.A.; et al. Numerical Chromosomal Instability Mediates Susceptibility to Radiation Treatment. Nat. Commun. 2015, 6, 5990. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA Damage Response in Cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Colin, D.J.; Limagne, E.; Ragot, K.; Lizard, G.; Ghiringhelli, F.; Solary, É.; Chauffert, B.; Latruffe, N.; Delmas, D. The Role of Reactive Oxygen Species and Subsequent DNA-Damage Response in the Emergence of Resistance towards Resveratrol in Colon Cancer Models. Cell Death Dis. 2014, 5, e1533. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Lee, S.; Seo, D.; Kim, D.; Kim, K.; Kim, E.; Kang, J.; Seong, K.M.; Youn, H.; Youn, B. Cellular Stress Responses in Radiotherapy. Cells 2019, 8, E1105. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.-X.; Zhou, P.-K. DNA Damage Response Signaling Pathways and Targets for Radiotherapy Sensitization in Cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Dai, J.; Jiang, M.; He, K.; Wang, H.; Chen, P.; Guo, H.; Zhao, W.; Lu, H.; He, Y.; Zhou, C. DNA Damage Response and Repair Gene Alterations Increase Tumor Mutational Burden and Promote Poor Prognosis of Advanced Lung Cancer. Front. Oncol. 2021, 11, 708294. [Google Scholar] [CrossRef]
- Jin, J.; Tao, Z.; Cao, J.; Li, T.; Hu, X. DNA Damage Response Inhibitors: An Avenue for TNBC Treatment. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188521. [Google Scholar] [CrossRef]
- Minchom, A.; Aversa, C.; Lopez, J. Dancing with the DNA Damage Response: Next-Generation Anti-Cancer Therapeutic Strategies. Ther. Adv. Med. Oncol. 2018, 10, 1758835918786658. [Google Scholar] [CrossRef]
- Lozano, R.; Castro, E.; Aragón, I.M.; Cendón, Y.; Cattrini, C.; López-Casas, P.P.; Olmos, D. Genetic Aberrations in DNA Repair Pathways: A Cornerstone of Precision Oncology in Prostate Cancer. Br. J. Cancer 2021, 124, 552–563. [Google Scholar] [CrossRef]
- Perkhofer, L.; Gout, J.; Roger, E.; de Almeida, F.K.; Wiesmüller, L.; Seufferlein, T.; Kleger, A. DNA Damage Repair as a Target in Pancreatic Cancer: State-of-the-Art and Future Perspectives. Gut 2021, 70, 606–617. [Google Scholar] [CrossRef]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef] [PubMed]
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in Synthetic Lethality for Cancer Therapy: Cellular Mechanism and Clinical Translation. J. Hematol. Oncol. 2020, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.; Guo, M. Epigenetic Based Synthetic Lethal Strategies in Human Cancers. Biomark. Res. 2020, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Bekaii-Saab, T.S.; Bridgewater, J.; Normanno, N. Practical Considerations in Screening for Genetic Alterations in Cholangiocarcinoma. Ann. Oncol. 2021, 32, 1111–1126. [Google Scholar] [CrossRef]
- Miller, G.; Socci, N.D.; Dhall, D.; D’Angelica, M.; DeMatteo, R.P.; Allen, P.J.; Singh, B.; Fong, Y.; Blumgart, L.H.; Klimstra, D.S.; et al. Genome Wide Analysis and Clinical Correlation of Chromosomal and Transcriptional Mutations in Cancers of the Biliary Tract. J. Exp. Clin. Cancer Res. 2009, 28, 62. [Google Scholar] [CrossRef]
- Andersen, J.B.; Thorgeirsson, S.S. Genetic Profiling of Intrahepatic Cholangiocarcinoma. Curr. Opin. Gastroenterol. 2012, 28, 266–272. [Google Scholar] [CrossRef]
- International Network of Cancer Genome Projects. Nature 2010, 464, 993–998. [CrossRef]
- Zhang, J.; Baran, J.; Cros, A.; Guberman, J.M.; Haider, S.; Hsu, J.; Liang, Y.; Rivkin, E.; Wang, J.; Whitty, B.; et al. International Cancer Genome Consortium Data Portal—a One-Stop Shop for Cancer Genomics Data. Database 2011, 2011, bar026. [Google Scholar] [CrossRef]
- Cancer Projects | ICGC Data Portal. Available online: https://dcc.icgc.org/projects (accessed on 16 April 2022).
- The Cancer Genome Atlas Program—National Cancer Institute. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 16 April 2022).
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate Produced by Neomorphic IDH Mutations Suppresses Homologous Recombination and Induces PARP Inhibitor Sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef]
- Lin, J.; Shi, J.; Guo, H.; Yang, X.; Jiang, Y.; Long, J.; Bai, Y.; Wang, D.; Yang, X.; Wan, X.; et al. Alterations in DNA Damage Repair Genes in Primary Liver Cancer. Clin. Cancer Res. 2019, 25, 4701–4711. [Google Scholar] [CrossRef]
- Ahn, D.H.; Bekaii-Saab, T. Biliary Tract Cancer and Genomic Alterations in Homologous Recombinant Deficiency: Exploiting Synthetic Lethality with PARP Inhibitors. Chin. Clin. Oncol. 2020, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Biliary Tract Cancer: State of the Art and Potential Role of DNA Damage Repair. Cancer Treat. Rev. 2018, 70, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Kapacee, Z.; Breeze, M.; Bell, C.; Belcher, D.; Staiger, H.; Taylor, C.; McNamara, M.G.; Hubner, R.A.; Valle, J.W. Molecular Profiling in Daily Clinical Practice: Practicalities in Advanced Cholangiocarcinoma and Other Biliary Tract Cancers. J. Clin. Med. 2020, 9, 2854. [Google Scholar] [CrossRef] [PubMed]
- Bezrookove, V.; Patino, J.M.; Nosrati, M.; Desprez, P.-Y.; McAllister, S.; Soroceanu, L.; Baron, A.; Osorio, R.; Kashani-Sabet, M.; Dar, A.A. Niraparib Suppresses Cholangiocarcinoma Tumor Growth by Inducing Oxidative and Replication Stress. Cancers 2021, 13, 4405. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.; Kim, D.; Yoo, C.; Kim, K.-P.; Jeong, J.H.; Chang, H.-M.; Lee, S.S.; Park, D.H.; Song, T.J.; Hwang, S.; et al. Therapeutic Relevance of Targeted Sequencing in Management of Patients with Advanced Biliary Tract Cancer: DNA Damage Repair Gene Mutations as a Predictive Biomarker. Eur J. Cancer 2019, 120, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Cao, Y.; Yang, X.; Li, G.; Shi, Y.; Wang, D.; Long, J.; Song, Y.; Mao, J.; Xie, F.; et al. Mutational Spectrum and Precision Oncology for Biliary Tract Carcinoma. Theranostics 2021, 11, 4585–4598. [Google Scholar] [CrossRef]
- Boerner, T.; Drill, E.; Pak, L.M.; Nguyen, B.; Sigel, C.S.; Doussot, A.; Shin, P.; Goldman, D.A.; Gonen, M.; Allen, P.J.; et al. Genetic Determinants of Outcome in Intrahepatic Cholangiocarcinoma. Hepatology 2021, 74, 1429–1444. [Google Scholar] [CrossRef]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert Consensus Document: Cholangiocarcinoma: Current Knowledge and Future Perspectives Consensus Statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef]
- Kohya, N.; Miyazaki, K.; Matsukura, S.; Yakushiji, H.; Kitajima, Y.; Kitahara, K.; Fukuhara, M.; Nakabeppu, Y.; Sekiguchi, M. Deficient Expression of O(6)-Methylguanine-DNA Methyltransferase Combined with Mismatch-Repair Proteins HMLH1 and HMSH2 Is Related to Poor Prognosis in Human Biliary Tract Carcinoma. Ann. Surg. Oncol. 2002, 9, 371–379. [Google Scholar] [CrossRef]
- Ahn, D.H.; Javle, M.; Ahn, C.W.; Jain, A.; Mikhail, S.; Noonan, A.M.; Ciombor, K.; Wu, C.; Shroff, R.T.; Chen, J.L.; et al. Next-Generation Sequencing Survey of Biliary Tract Cancer Reveals the Association between Tumor Somatic Variants and Chemotherapy Resistance. Cancer 2016, 122, 3657–3666. [Google Scholar] [CrossRef]
- Hwang, I.G.; Jang, J.S.; Do, J.H.; Kang, J.H.; Lee, G.W.; Oh, S.Y.; Kwon, H.C.; Jun, H.J.; Lim, H.Y.; Lee, S.; et al. Different Relation between ERCC1 Overexpression and Treatment Outcomes of Two Platinum Agents in Advanced Biliary Tract Adenocarcinoma Patients. Cancer Chemother Pharm. 2011, 68, 935–944. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, W.-Y.; Andreotti, G.; Gao, Y.-T.; Rashid, A.; Chen, J.; Sakoda, L.C.; Shen, M.-C.; Wang, B.-S.; Chanock, S.; et al. Variants of DNA Repair Genes and the Risk of Biliary Tract Cancers and Stones: A Population-Based Study in China. Cancer Epidemiol. Biomark. Prev. 2008, 17, 2123–2127. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic Lethality and Cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A. A Synthetic Lethal Therapeutic Approach: Poly(ADP) Ribose Polymerase Inhibitors for the Treatment of Cancers Deficient in DNA Double-Strand Break Repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.D.; Rizzo, A.; Bonucci, C.; Tober, N.; Palloni, A.; Mollica, V.; Maggio, I.; Deserti, M.; Tavolari, S.; Brandi, G. PARP Inhibitors in Biliary Tract Cancer: A New Kid on the Block? Medicines 2020, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. The Concept of Synthetic Lethality in the Context of Anticancer Therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef]
- Mehta, R.; Wood, A.C.; Yu, J.; Kim, R. Investigational PARP Inhibitors for the Treatment of Biliary Tract Cancer: Spotlight on Preclinical and Clinical Studies. Expert Opin. Investig. Drugs 2021, 30, 451–461. [Google Scholar] [CrossRef]
- Hottiger, M.O.; Hassa, P.O.; Lüscher, B.; Schüler, H.; Koch-Nolte, F. Toward a Unified Nomenclature for Mammalian ADP-Ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef]
- Kamaletdinova, T.; Fanaei-Kahrani, Z.; Wang, Z.-Q. The Enigmatic Function of PARP1: From PARylation Activity to PAR Readers. Cells 2019, 8, 1625. [Google Scholar] [CrossRef]
- Amé, J.-C.; Spenlehauer, C.; de Murcia, G. The PARP Superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.; Robert, I.; Reina-San-Martin, B.; Schreiber, V.; Dantzer, F. Poly(ADP-Ribose) Polymerases in Double-Strand Break Repair: Focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 2014, 329, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Gonzalez, R.; Jacobson, M.K. Characterization of Polymers of Adenosine Diphosphate Ribose Generated in Vitro and in Vivo. Biochemistry 1987, 26, 3218–3224. [Google Scholar] [CrossRef] [PubMed]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-Ribosyl)Ation by PARP1: Reaction Mechanism and Regulatory Proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The Multifaceted Roles of PARP1 in DNA Repair and Chromatin Remodelling. Nat. Rev. Mol. Cell Biol 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Hanzlikova, H.; Caldecott, K.W. Perspectives on PARPs in S Phase. Trends Genet. 2019, 35, 412–422. [Google Scholar] [CrossRef]
- Azarm, K.; Smith, S. Nuclear PARPs and Genome Integrity. Genes Dev. 2020, 34, 285–301. [Google Scholar] [CrossRef]
- Hanzlikova, H.; Kalasova, I.; Demin, A.A.; Pennicott, L.E.; Cihlarova, Z.; Caldecott, K.W. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell 2018, 71, 319–331.e3. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.; Matulonis, U.; Gourley, C.; du Bois, A.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Long-Term Efficacy, Tolerability and Overall Survival in Patients with Platinum-Sensitive, Recurrent High-Grade Serous Ovarian Cancer Treated with Maintenance Olaparib Capsules Following Response to Chemotherapy. Br. J. Cancer 2018, 119, 1075–1085. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib Monotherapy in Patients with Advanced Cancer and a Germline BRCA1/2 Mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib Tablets as Maintenance Therapy in Patients with Platinum-Sensitive, Relapsed Ovarian Cancer and a BRCA1/2 Mutation (SOLO2/ENGOT-Ov21): A Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Robson, M.E.; Tung, N.; Conte, P.; Im, S.-A.; Senkus, E.; Xu, B.; Masuda, N.; Delaloge, S.; Li, W.; Armstrong, A.; et al. OlympiAD Final Overall Survival and Tolerability Results: Olaparib versus Chemotherapy Treatment of Physician’s Choice in Patients with a Germline BRCA Mutation and HER2-Negative Metastatic Breast Cancer. Ann. Oncol. 2019, 30, 558–566. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Peyraud, F.; Italiano, A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers 2020, 12, E1502. [Google Scholar] [CrossRef]
- Fehling, S.C.; Miller, A.L.; Garcia, P.L.; Vance, R.B.; Yoon, K.J. The Combination of BET and PARP Inhibitors Is Synergistic in Models of Cholangiocarcinoma. Cancer Lett. 2020, 468, 48–58. [Google Scholar] [CrossRef]
- Mao, Y.; Huang, X.; Shuang, Z.; Lin, G.; Wang, J.; Duan, F.; Chen, J.; Li, S. PARP Inhibitor Olaparib Sensitizes Cholangiocarcinoma Cells to Radiation. Cancer Med. 2018, 7, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wild, A.T.; Turcan, S.; Wu, W.H.; Sigel, C.; Klimstra, D.S.; Ma, X.; Gong, Y.; Holland, E.C.; Huse, J.T.; et al. Targeting Therapeutic Vulnerabilities with PARP Inhibition and Radiation in IDH-Mutant Gliomas and Cholangiocarcinomas. Sci. Adv. 2020, 6, eaaz3221. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Raitses-Gurevich, M.; Kelley, R.K.; Bocobo, A.G.; Borgida, A.; Shroff, R.T.; Holter, S.; Gallinger, S.; Ahn, D.H.; Aderka, D.; et al. Overall Survival and Clinical Characteristics of BRCA-Associated Cholangiocarcinoma: A Multicenter Retrospective Study. Oncologist 2017, 22, 804–810. [Google Scholar] [CrossRef]
- Xie, Y.; Jiang, Y.; Yang, X.-B.; Wang, A.-Q.; Zheng, Y.-C.; Wan, X.-S.; Sang, X.-T.; Wang, K.; Zhang, D.-D.; Xu, J.-J.; et al. Response of BRCA1-Mutated Gallbladder Cancer to Olaparib: A Case Report. World J. Gastroenterol 2016, 22, 10254–10259. [Google Scholar] [CrossRef] [PubMed]
- Hanna, D.; Chopra, N.; Hochhauser, D.; Khan, K. The Role of PARP Inhibitors in Gastrointestinal Cancers. Crit. Rev. Oncol. Hematol 2022, 171, 103621. [Google Scholar] [CrossRef] [PubMed]
- Academic and Community Cancer Research United. A Phase II Study of Olaparib in Patients with Advanced Biliary Tract Cancer with Aberrant DNA Repair Gene Mutations. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04042831 (accessed on 8 March 2022).
- Makawita, S.; Borad, M.J.; Carapeto, F.; Kwong, L.; Bekaii-Saab, T.S.; Murugesan, K.; Ross, J.S.; Danziger, N.; Israel, M.A.; McGregor, K.; et al. IDH1 and IDH2 Driven Intrahepatic Cholangiocarcinoma (IHCC): A Comprehensive Genomic and Immune Profiling Study. J. Clin. Oncol. 2021, 39, 4009. [Google Scholar] [CrossRef]
- IDH-Mutant Tumors Vulnerable to PARP Inhibition. Cancer Discov. 2017, 7, OF4. [CrossRef]
- National Cancer Institute (NCI). A Phase 2 Study of the PARP Inhibitor Olaparib (AZD2281) in IDH1 and IDH2 Mutant Advanced Solid Tumors. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03212274 (accessed on 8 March 2022).
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- National Cancer Institute (NCI). A Phase II Study of Olaparib and AZD6738 in Isocitrate Dehydrogenase (IDH) Mutant Solid Tumors. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03878095 (accessed on 8 March 2022).
- Oh, D.-Y. DDR-Umbrella Study of DDR (DNA-Damage Response) Targeting Agents in Advanced Biliary Tract Cancer. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04298021 (accessed on 8 March 2022).
- University Health Network, Toronto. A Phase II Study of Olaparib and Durvalumab (MEDI 4736) in Patients With IDH-Mutated Solid Tumors. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03991832 (accessed on 8 March 2022).
- Georgetown University. A Phase II, Single-Arm Study of Combination Pembrolizumab and Olaparib in the Treatment of Patients With Advanced Cholangiocarcinoma. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04306367 (accessed on 8 March 2022).
- Xie, H.; Wang, W.; Qi, W.; Jin, W.; Xia, B. Targeting DNA Repair Response Promotes Immunotherapy in Ovarian Cancer: Rationale and Clinical Application. Front. Immunol. 2021, 12, 661115. [Google Scholar] [CrossRef]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA Damage Response Pathway: Biomarker and Therapeutic Strategy for Cancer Immunotherapy. Acta Pharm. Sin. B 2021, 11, 2983–2994. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA Damaging Therapeutics with Immunotherapy: More Haste, Less Speed. Br. J. Cancer 2018, 118, 312–324. [Google Scholar] [CrossRef] [PubMed]
- University of Michigan Rogel Cancer Center. Phase II Multi-Center Study of PARP Inhibitor Rucaparib in Combination with Anti-PD-1 Antibody Nivolumab in Patients with Advanced or Metastatic Biliary Tract Cancer Following Platinum Therapy. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03639935 (accessed on 8 March 2022).
- National Cancer Institute (NCI). Phase I Study of Veliparib (ABT-888) in Combination with Cisplatin Plus Gemcitabine in Advanced Biliary, Pancreatic, Urothelial, and Non-Small Cell Lung Cancer. 2013. Available online: https://clinicaltrials.gov/ct2/show/NCT01282333 (accessed on 8 March 2022).
- University of Florida. A Phase II Trial of the PARP Inhibitor, Niraparib, in BAP1 and Other DNA Damage Response (DDR) Pathway Deficient Neoplasms (UF-STO-ETI-001). 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03207347 (accessed on 8 March 2022).
- Schmidt, M.; Rohe, A.; Platzer, C.; Najjar, A.; Erdmann, F.; Sippl, W. Regulation of G2/M Transition by Inhibition of WEE1 and PKMYT1 Kinases. Molecules 2017, 22, E2045. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Ando, H.; Watanabe, N.; Kitamura, K.; Ito, K.; Okayama, H.; Miyamoto, T.; Agui, T.; Sasaki, M. Identification and Characterization of Human Wee1B, a New Member of the Wee1 Family of Cdk-Inhibitory Kinases. Genes Cells 2000, 5, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Asquith, C.R.M.; Laitinen, T.; East, M.P. PKMYT1: A Forgotten Member of the WEE1 Family. Nat. Rev. Drug Discov. 2020, 19, 157. [Google Scholar] [CrossRef]
- Liu, Y.; Qi, J.; Dou, Z.; Hu, J.; Lu, L.; Dai, H.; Wang, H.; Yang, W. Systematic Expression Analysis of WEE Family Kinases Reveals the Importance of PKMYT1 in Breast Carcinogenesis. Cell Prolif. 2020, 53, e12741. [Google Scholar] [CrossRef]
- Jeong, D.; Kim, H.; Kim, D.; Ban, S.; Oh, S.; Ji, S.; Kang, D.; Lee, H.; Ahn, T.S.; Kim, H.J.; et al. Protein Kinase, Membrane-associated Tyrosine/Threonine 1 Is Associated with the Progression of Colorectal Cancer. Oncol. Rep. 2018, 39, 2829–2836. [Google Scholar] [CrossRef]
- Ghelli Luserna di Rorà, A.; Cerchione, C.; Martinelli, G.; Simonetti, G. A WEE1 Family Business: Regulation of Mitosis, Cancer Progression, and Therapeutic Target. J. Hematol Oncol. 2020, 13, 126. [Google Scholar] [CrossRef]
- Seo, H.-R.; Nam, A.-R.; Bang, J.-H.; Oh, K.-S.; Kim, J.-M.; Yoon, J.; Kim, T.-Y.; Oh, D.-Y. Inhibition of WEE1 Potentiates Sensitivity to PARP Inhibitor in Biliary Tract Cancer. Cancer Res. Treat. 2022, 54, 541–553. [Google Scholar] [CrossRef]
- Nam, A.-R.; Jin, M.-H.; Bang, J.-H.; Oh, K.-S.; Seo, H.-R.; Oh, D.-Y.; Bang, Y.-J. Inhibition of ATR Increases the Sensitivity to WEE1 Inhibitor in Biliary Tract Cancer. Cancer Res. Treat. 2020, 52, 945–956. [Google Scholar] [CrossRef]
- Jin, M.-H.; Nam, A.-R.; Bang, J.-H.; Oh, K.-S.; Seo, H.-R.; Kim, J.-M.; Yoon, J.; Kim, T.-Y.; Oh, D.-Y. WEE1 Inhibition Reverses Trastuzumab Resistance in HER2-Positive Cancers. Gastric Cancer 2021, 24, 1003–1020. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Naqash, A.R.; O’Sullivan Coyne, G.; Kummar, S.; Do, K.; Bruns, A.; Juwara, L.; Zlott, J.; Rubinstein, L.; Piekarz, R.; et al. Safety, Antitumor Activity, and Biomarker Analysis in a Phase I Trial of the Once-Daily Wee1 Inhibitor Adavosertib (AZD1775) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3834–3844. [Google Scholar] [CrossRef] [PubMed]
- Någård, M.; Ah-See, M.-L.; So, K.; Vermunt, M.; Thistlethwaite, F.; Labots, M.; Roxburgh, P.; Ravaud, A.; Campone, M.; Valkenburg-van Iersel, L.; et al. Effect of Food on the Pharmacokinetics of the WEE1 Inhibitor Adavosertib (AZD1775) in Patients with Advanced Solid Tumors. Cancer Chemother. Pharm. 2020, 86, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Cristea, M.C.; Bruce, J.P.; Garg, S.; Cabanero, M.; Mantia-Smaldone, G.; Olawaiye, A.B.; Ellard, S.L.; Weberpals, J.I.; Wahner Hendrickson, A.E.; et al. Adavosertib plus Gemcitabine for Platinum-Resistant or Platinum-Refractory Recurrent Ovarian Cancer: A Double-Blind, Randomised, Placebo-Controlled, Phase 2 Trial. Lancet 2021, 397, 281–292. [Google Scholar] [CrossRef]
- Lu, Y.-L.; Huang, Y.-T.; Wu, M.-H.; Chou, T.-C.; J Wong, R.; Lin, S.-F. Efficacy of Adavosertib Therapy against Anaplastic Thyroid Cancer. Endocr. Relat. Cancer 2021, 28, 311–324. [Google Scholar] [CrossRef]
- Liu, J.F.; Xiong, N.; Campos, S.M.; Wright, A.A.; Krasner, C.; Schumer, S.; Horowitz, N.; Veneris, J.; Tayob, N.; Morrissey, S.; et al. Phase II Study of the WEE1 Inhibitor Adavosertib in Recurrent Uterine Serous Carcinoma. J. Clin. Oncol. 2021, 39, 1531–1539. [Google Scholar] [CrossRef]
- National Cancer Institute (NCI). Molecular Analysis for Therapy Choice (MATCH). 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT02465060 (accessed on 8 March 2022).
- Choi, W.; Lee, E.S. Therapeutic Targeting of DNA Damage Response in Cancer. Int J. Mol. Sci. 2022, 23, 1701. [Google Scholar] [CrossRef]
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs Kinases-the Lessons from the Mouse Models: Inhibition ≠ Deletion. Cell BioSci. 2020, 10, 8. [Google Scholar] [CrossRef]
- Bradbury, A.; Hall, S.; Curtin, N.; Drew, Y. Targeting ATR as Cancer Therapy: A New Era for Synthetic Lethality and Synergistic Combinations? Pharmacol. Ther. 2020, 207, 107450. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- You, Z.; Chahwan, C.; Bailis, J.; Hunter, T.; Russell, P. ATM Activation and Its Recruitment to Damaged DNA Require Binding to the C Terminus of Nbs1. Mol. Cell Biol. 2005, 25, 5363–5379. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. ATM and Related Protein Kinases: Safeguarding Genome Integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F. Ataxia-Telangiectasia: From a Rare Disorder to a Paradigm for Cell Signalling and Cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 759–769. [Google Scholar] [CrossRef]
- Chapman, J.R.; Jackson, S.P. Phospho-Dependent Interactions between NBS1 and MDC1 Mediate Chromatin Retention of the MRN Complex at Sites of DNA Damage. EMBO Rep. 2008, 9, 795–801. [Google Scholar] [CrossRef]
- Bouquet, F.; Muller, C.; Salles, B. The Loss of GammaH2AX Signal Is a Marker of DNA Double Strand Breaks Repair Only at Low Levels of DNA Damage. Cell Cycle 2006, 5, 1116–1122. [Google Scholar] [CrossRef]
- Banáth, J.P.; Macphail, S.H.; Olive, P.L. Radiation Sensitivity, H2AX Phosphorylation, and Kinetics of Repair of DNA Strand Breaks in Irradiated Cervical Cancer Cell Lines. Cancer Res. 2004, 64, 7144–7149. [Google Scholar] [CrossRef]
- Xie, A.; Puget, N.; Shim, I.; Odate, S.; Jarzyna, I.; Bassing, C.H.; Alt, F.W.; Scully, R. Control of Sister Chromatid Recombination by Histone H2AX. Mol. Cell 2004, 16, 1017–1025. [Google Scholar] [CrossRef]
- Moolmuang, B.; Ruchirawat, M. The Antiproliferative Effects of Ataxia-Telangiectasia Mutated and ATM- and Rad3-Related Inhibitions and Their Enhancements with the Cytotoxicity of DNA Damaging Agents in Cholangiocarcinoma Cells. J. Pharm. Pharmacol. 2021, 73, 40–51. [Google Scholar] [CrossRef]
- Nam, A.-R.; Yoon, J.; Jin, M.-H.; Bang, J.-H.; Oh, K.-S.; Seo, H.-R.; Kim, J.-M.; Kim, T.-Y.; Oh, D.-Y. ATR Inhibition Amplifies Antitumor Effects of Olaparib in Biliary Tract Cancer. Cancer Lett. 2021, 516, 38–47. [Google Scholar] [CrossRef]
- Nam, A.-R.; Jin, M.H.; Park, J.E.; Bang, J.-H.; Oh, D.-Y.; Bang, Y.-J. Therapeutic Targeting of the DNA Damage Response Using an ATR Inhibitor in Biliary Tract Cancer. Cancer Res. Treat. 2019, 51, 1167–1179. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.-R.; Wu, C.-E.; Yeh, C.-N. ATM Inhibitor Suppresses Gemcitabine-Resistant BTC Growth in a Polymerase θ Deficiency-Dependent Manner. Biomolecules 2020, 10, E1529. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; de Miguel Luken, M.J.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination With Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; Wesolowski, R.; Devoe, C.; Lord, S.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R.; Yap, T.A. Phase 1 Study of the ATR Inhibitor Berzosertib in Combination with Cisplatin in Patients with Advanced Solid Tumours. Br. J. Cancer 2021, 125, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Barnieh, F.M.; Loadman, P.M.; Falconer, R.A. Progress towards a Clinically-Successful ATR Inhibitor for Cancer Therapy. Curr Res. Pharmacol. Drug Discov. 2021, 2, 100017. [Google Scholar] [CrossRef] [PubMed]
- Testing the Addition of an Anti-Cancer Drug, BAY 1895344, to the Usual Chemotherapy Treatment (Cisplatin, or Cisplatin and Gemcitabine) for Advanced Solid Tumors with Emphasis on Urothelial Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04491942 (accessed on 8 March 2022).
- Reinhardt, H.C.; Yaffe, M.B. Kinases That Control the Cell Cycle in Response to DNA Damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol. 2009, 21, 245–255. [Google Scholar] [CrossRef]
- Hematulin, A.; Sagan, D.; Sawanyawisuth, K.; Seubwai, W.; Wongkham, S. Association between Cellular Radiosensitivity and G1/G2 Checkpoint Proficiencies in Human Cholangiocarcinoma Cell Lines. Int J. Oncol. 2014, 45, 1159–1166. [Google Scholar] [CrossRef]
- Moore, K.N.; Hong, D.S.; Patel, M.R.; Pant, S.; Ulahannan, S.V.; Jones, S.; Meric-Bernstam, F.; Wang, J.S.; Aljumaily, R.; Hamilton, E.P.; et al. A Phase 1b Trial of Prexasertib in Combination with Standard-of-Care Agents in Advanced or Metastatic Cancer. Target. Oncol. 2021, 16, 569–589. [Google Scholar] [CrossRef]
- Mohiuddin, I.S.; Kang, M.H. DNA-PK as an Emerging Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 635. [Google Scholar] [CrossRef]
- Yu, Z.; Sui, J.; Ding, Y.; Cao, Z.; Zhou, P.; Wu, D. Expression of DNA-PK in hepato- and cholangio-neoplasms and its significance. Zhonghua Gan Zang Bing Za Zhi 2004, 12, 652–655. [Google Scholar]
- Medová, M.; Medo, M.; Hovhannisyan, L.; Muñoz-Maldonado, C.; Aebersold, D.M.; Zimmer, Y. DNA-PK in Human Malignant Disorders: Mechanisms and Implications for Pharmacological Interventions. Pharmacol. Ther. 2020, 215, 107617. [Google Scholar] [CrossRef]
- Lustri, A.M.; Di Matteo, S.; Fraveto, A.; Costantini, D.; Cantafora, A.; Napoletano, C.; Bragazzi, M.C.; Giuliante, F.; De Rose, A.M.; Berloco, P.B.; et al. TGF-β Signaling Is an Effective Target to Impair Survival and Induce Apoptosis of Human Cholangiocarcinoma Cells: A Study on Human Primary Cell Cultures. PLoS ONE 2017, 12, e0183932. [Google Scholar] [CrossRef]
- van Bussel, M.T.J.; Awada, A.; de Jonge, M.J.A.; Mau-Sørensen, M.; Nielsen, D.; Schöffski, P.; Verheul, H.M.W.; Sarholz, B.; Berghoff, K.; El Bawab, S.; et al. A First-in-Man Phase 1 Study of the DNA-Dependent Protein Kinase Inhibitor Peposertib (Formerly M3814) in Patients with Advanced Solid Tumours. Br. J. Cancer 2021, 124, 728–735. [Google Scholar] [CrossRef]
- National Cancer Institute (NCI). A Phase I/II Study of M3814 and Avelumab in Combination With Hypofractionated Radiation in Patients With Advanced/Metastatic Solid Tumors and Hepatobiliary Malignancies. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04068194 (accessed on 8 March 2022).
- Lens, S.M.A.; Voest, E.E.; Medema, R.H. Shared and Separate Functions of Polo-like Kinases and Aurora Kinases in Cancer. Nat. Rev. Cancer 2010, 10, 825–841. [Google Scholar] [CrossRef] [PubMed]
- Mundt, K.E.; Golsteyn, R.M.; Lane, H.A.; Nigg, E.A. On the Regulation and Function of Human Polo-like Kinase 1 (PLK1): Effects of Overexpression on Cell Cycle Progression. Biochem. Biophys. Res. Commun. 1997, 239, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A Potential Target for Cancer Therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, Y.; Yang, X.; Chen, T.; Han, T. Analysis of Differentially Expressed MRNAs and the Prognosis of Cholangiocarcinoma Based on TCGA Database. Transl. Cancer Res. 2020, 9, 4739–4749. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Zhong, X.-Z.; Wang, X.-D.; Li, J.-J.; Zhao, R.-Q.; He, Y.; Jiang, Y.-Q.; Huang, X.-W.; Chen, G.; He, Y.; et al. Survival Analysis of Genome-Wide Profiles Coupled with Connectivity Map Database Mining to Identify Potential Therapeutic Targets for Cholangiocarcinoma. Oncol. Rep. 2018, 40, 3189–3198. [Google Scholar] [CrossRef]
- Fingas, C.D.; Mertens, J.C.; Razumilava, N.; Sydor, S.; Bronk, S.F.; Christensen, J.D.; Rizvi, S.H.; Canbay, A.; Treckmann, J.W.; Paul, A.; et al. Polo-like Kinase 2 Is a Mediator of Hedgehog Survival Signaling in Cholangiocarcinoma. Hepatology 2013, 58, 1362–1374. [Google Scholar] [CrossRef]
- Sydor, S.; Jafoui, S.; Wingerter, L.; Swoboda, S.; Mertens, J.C.; Gerken, G.; Canbay, A.; Paul, A.; Fingas, C.D. Bcl-2 Degradation Is an Additional pro-Apoptotic Effect of Polo-like Kinase Inhibition in Cholangiocarcinoma Cells. World J. Gastroenterol 2017, 23, 4007–4015. [Google Scholar] [CrossRef]
- Thrum, S.; Lorenz, J.; Mössner, J.; Wiedmann, M. Polo-like Kinase 1 Inhibition as a New Therapeutic Modality in Therapy of Cholangiocarcinoma. Anticancer Res. 2011, 31, 3289–3299. [Google Scholar]
- Malacrida, A.; Rigolio, R.; Celio, L.; Damian, S.; Cavaletti, G.; Mazzaferro, V.; Miloso, M. In Vitro Evaluation of Rigosertib Antitumoral and Radiosensitizing Effects against Human Cholangiocarcinoma Cells. Int. J. Mol. Sci. 2021, 22, 8230. [Google Scholar] [CrossRef] [PubMed]
- Nokihara, H.; Yamada, Y.; Fujiwara, Y.; Yamamoto, N.; Wakui, H.; Nakamichi, S.; Kitazono, S.; Inoue, K.; Harada, A.; Taube, T.; et al. Phase I Trial of Volasertib, a Polo-like Kinase Inhibitor, in Japanese Patients with Advanced Solid Tumors. Invest New Drugs 2016, 34, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Su, W.-C.; Yen, C.-J.; Hsu, C.-H.; Su, W.-P.; Yeh, K.-H.; Lu, Y.-S.; Cheng, A.-L.; Huang, D.C.-L.; Fritsch, H.; et al. A Phase I Study of Two Dosing Schedules of Volasertib (BI 6727), an Intravenous Polo-like Kinase Inhibitor, in Patients with Advanced Solid Malignancies. Br. J. Cancer 2014, 110, 2434–2440. [Google Scholar] [CrossRef] [PubMed]
- Academic and Community Cancer Research United. Phase I Study of Irinotecan Liposome (Nal-IRI), Fluorouracil, Leucovorin and Rucaparib in the Treatment of Select Gastrointestinal Metastatic Malignancies Followed by a Phase Ib of First and Second Line Treatment of Both Unselected and Selected (for BRCA 1/2 and PALB2 Mutations) Patients with Metastatic Adenocarcinoma of the Pancreas Then Followed by a Phase II Study of First Line Treatment of Selected Patients with Metastatic Adenocarcinoma of the Pancreas with Genomic Markers (Signature) of Homologous Recombination Deficiency (HRD). 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03337087 (accessed on 8 March 2022).
- Cleary, J.M.; Aguirre, A.J.; Shapiro, G.I.; D’Andrea, A.D. Biomarker-Guided Development of DNA Repair Inhibitors. Mol. Cell 2020, 78, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef]
- Spizzo, G.; Puccini, A.; Xiu, J.; Goldberg, R.M.; Grothey, A.; Shields, A.F.; Arora, S.P.; Khushman, M.; Salem, M.E.; Battaglin, F.; et al. Molecular Profile of BRCA-Mutated Biliary Tract Cancers. ESMO Open 2020, 5, e000682. [Google Scholar] [CrossRef]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness Revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Bajrami, I.; Frankum, J.R.; Konde, A.; Miller, R.E.; Rehman, F.L.; Brough, R.; Campbell, J.; Sims, D.; Rafiq, R.; Hooper, S.; et al. Genome-Wide Profiling of Genetic Synthetic Lethality Identifies CDK12 as a Novel Determinant of PARP1/2 Inhibitor Sensitivity. Cancer Res. 2014, 74, 287–297. [Google Scholar] [CrossRef]
- Sabbatino, F.; Liguori, L.; Malapelle, U.; Schiavi, F.; Tortora, V.; Conti, V.; Filippelli, A.; Tortora, G.; Ferrone, C.R.; Pepe, S. Case Report: BAP1 Mutation and RAD21 Amplification as Predictive Biomarkers to PARP Inhibitor in Metastatic Intrahepatic Cholangiocarcinoma. Front. Oncol. 2020, 10, 567289. [Google Scholar] [CrossRef]
- Kuznetsov, J.N.; Aguero, T.H.; Owens, D.A.; Kurtenbach, S.; Field, M.G.; Durante, M.A.; Rodriguez, D.A.; King, M.L.; Harbour, J.W. BAP1 Regulates Epigenetic Switch from Pluripotency to Differentiation in Developmental Lineages Giving Rise to BAP1-Mutant Cancers. Sci. Adv. 2019, 5, eaax1738. [Google Scholar] [CrossRef]
- Kuznetsoff, J.N.; Owens, D.A.; Lopez, A.; Rodriguez, D.A.; Chee, N.T.; Kurtenbach, S.; Bilbao, D.; Roberts, E.R.; Volmar, C.-H.; Wahlestedt, C.; et al. Dual Screen for Efficacy and Toxicity Identifies HDAC Inhibitor with Distinctive Activity Spectrum for BAP1-Mutant Uveal Melanoma. Mol. Cancer Res. 2021, 19, 215–222. [Google Scholar] [CrossRef]
- Sacco, J.J.; Kenyani, J.; Butt, Z.; Carter, R.; Chew, H.Y.; Cheeseman, L.P.; Darling, S.; Denny, M.; Urbé, S.; Clague, M.J.; et al. Loss of the Deubiquitylase BAP1 Alters Class I Histone Deacetylase Expression and Sensitivity of Mesothelioma Cells to HDAC Inhibitors. Oncotarget 2015, 6, 13757–13771. [Google Scholar] [CrossRef]
- Pant, K.; Peixoto, E.; Richard, S.; Gradilone, S.A. Role of Histone Deacetylases in Carcinogenesis: Potential Role in Cholangiocarcinoma. Cells 2020, 9, E780. [Google Scholar] [CrossRef]
- Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins. A Phase 2 Clinical Trial of Entinostat in Combination with Nivolumab for Patients with Previously Treated Unresectable or Metastatic Cholangiocarcinoma and Pancreatic Adenocarcinoma. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03250273 (accessed on 8 March 2022).
- Xu, Y.-Y.; Ren, Z.-L.; Liu, X.-L.; Zhang, G.-M.; Huang, S.-S.; Shi, W.-H.; Ye, L.-X.; Luo, X.; Liu, S.-W.; Li, Y.-L.; et al. BAP1 Loss Augments Sensitivity to BET Inhibitors in Cancer Cells. Acta Pharmacol. Sin. 2021. [Google Scholar] [CrossRef]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-Induced DNA Damage, Mutations and Cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef] [PubMed]
- Loeuillard, E.; Conboy, C.B.; Gores, G.J.; Rizvi, S. Immunobiology of Cholangiocarcinoma. JHEP Rep. 2019, 1, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zhang, Q.; Wang, R.; Li, Y.; Sun, Y.; Yang, L. Targeting DNA Damage Repair for Immune Checkpoint Inhibition: Mechanisms and Potential Clinical Applications. Front. Oncol. 2021, 11, 648687. [Google Scholar] [CrossRef] [PubMed]
- Tommelein, J.; De Vlieghere, E.; Verset, L.; Melsens, E.; Leenders, J.; Descamps, B.; Debucquoy, A.; Vanhove, C.; Pauwels, P.; Gespach, C.P.; et al. Radiotherapy-Activated Cancer-Associated Fibroblasts Promote Tumor Progression through Paracrine IGF1R Activation. Cancer Res. 2018, 78, 659–670. [Google Scholar] [CrossRef]
- Gomez-Sarosi, L.; Sun, Y.; Coleman, I.; Bianchi-Frias, D.; Nelson, P.S. DNA Damage Induces a Secretory Program in the Quiescent TME That Fosters Adverse Cancer Phenotypes. Mol. Cancer Res. 2017, 15, 842–851. [Google Scholar] [CrossRef]
- Banerjee, J.; Mishra, R.; Li, X.; Jackson, R.S.; Sharma, A.; Bhowmick, N.A. A Reciprocal Role of Prostate Cancer on Stromal DNA Damage. Oncogene 2014, 33, 4924–4931. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.; Edeline, J.; Coulouarn, C. Targeting the Tumor Microenvironment in Cholangiocarcinoma: Implications for Therapy. Expert Opin. Ther. Targets 2021, 25, 153–162. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gönül Geyik, Ö.; Anichini, G.; Ulukaya, E.; Marra, F.; Raggi, C. DNA Damage Response Inhibitors in Cholangiocarcinoma: Current Progress and Perspectives. Cells 2022, 11, 1463. https://doi.org/10.3390/cells11091463
Gönül Geyik Ö, Anichini G, Ulukaya E, Marra F, Raggi C. DNA Damage Response Inhibitors in Cholangiocarcinoma: Current Progress and Perspectives. Cells. 2022; 11(9):1463. https://doi.org/10.3390/cells11091463
Chicago/Turabian StyleGönül Geyik, Öykü, Giulia Anichini, Engin Ulukaya, Fabio Marra, and Chiara Raggi. 2022. "DNA Damage Response Inhibitors in Cholangiocarcinoma: Current Progress and Perspectives" Cells 11, no. 9: 1463. https://doi.org/10.3390/cells11091463
APA StyleGönül Geyik, Ö., Anichini, G., Ulukaya, E., Marra, F., & Raggi, C. (2022). DNA Damage Response Inhibitors in Cholangiocarcinoma: Current Progress and Perspectives. Cells, 11(9), 1463. https://doi.org/10.3390/cells11091463