
Dynamics of Metabolic Pathways and Stress Response Patterns during Human Neural Stem Cell Proliferation and Differentiation
 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. lt-NES Cell Culture Maintenance and Differentiation
2.3. Gene Expression Analysis
2.4. Compound Treatments
2.5. Immunofluorescence Imaging
2.6. SDS-PAGE and Western Blotting
2.7. ATPlite Assay
2.8. Autophagosomal Activity Assay
2.9. Proteasome Activity Assay
2.10. Statistical Analysis
3. Results
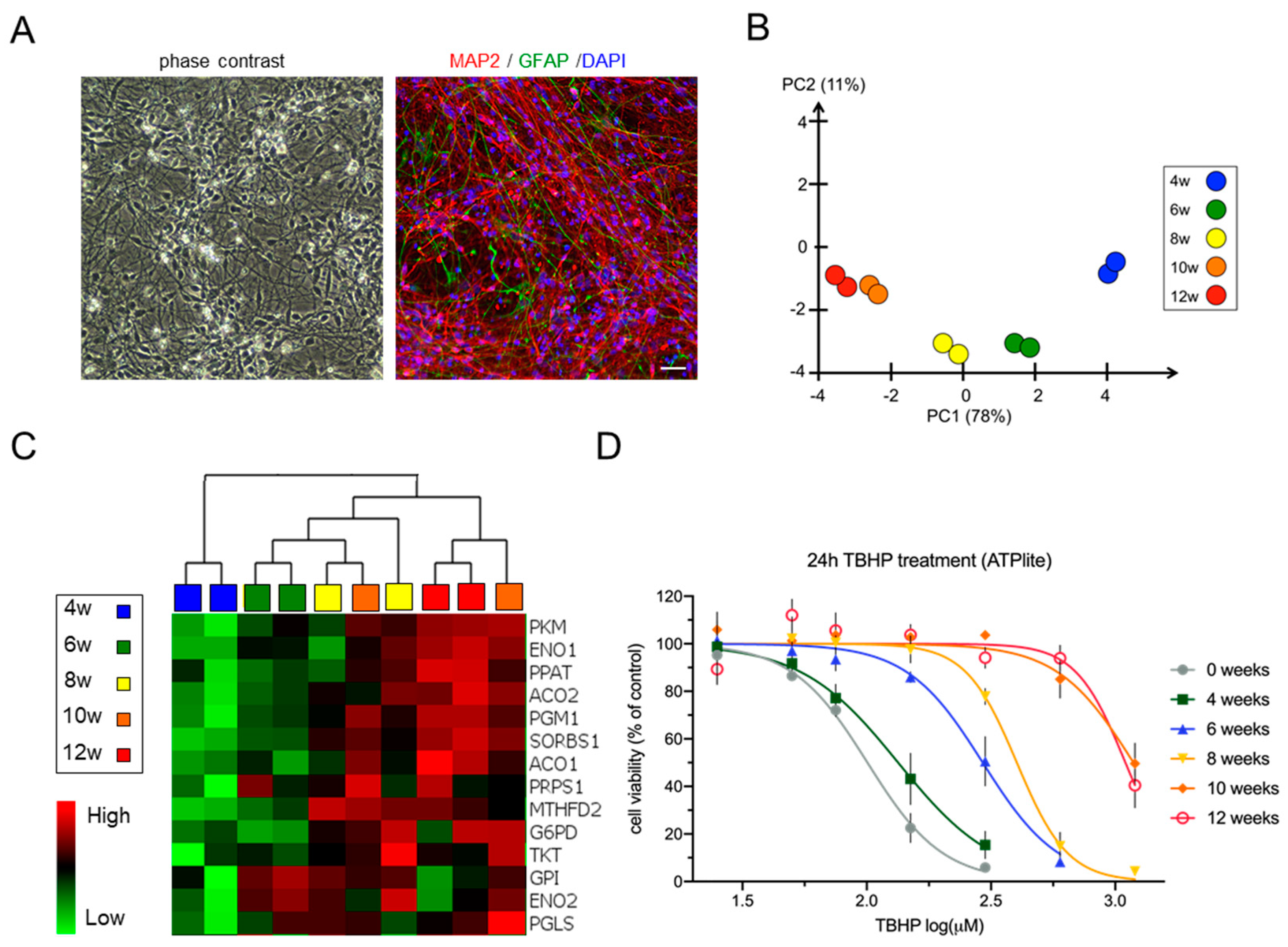
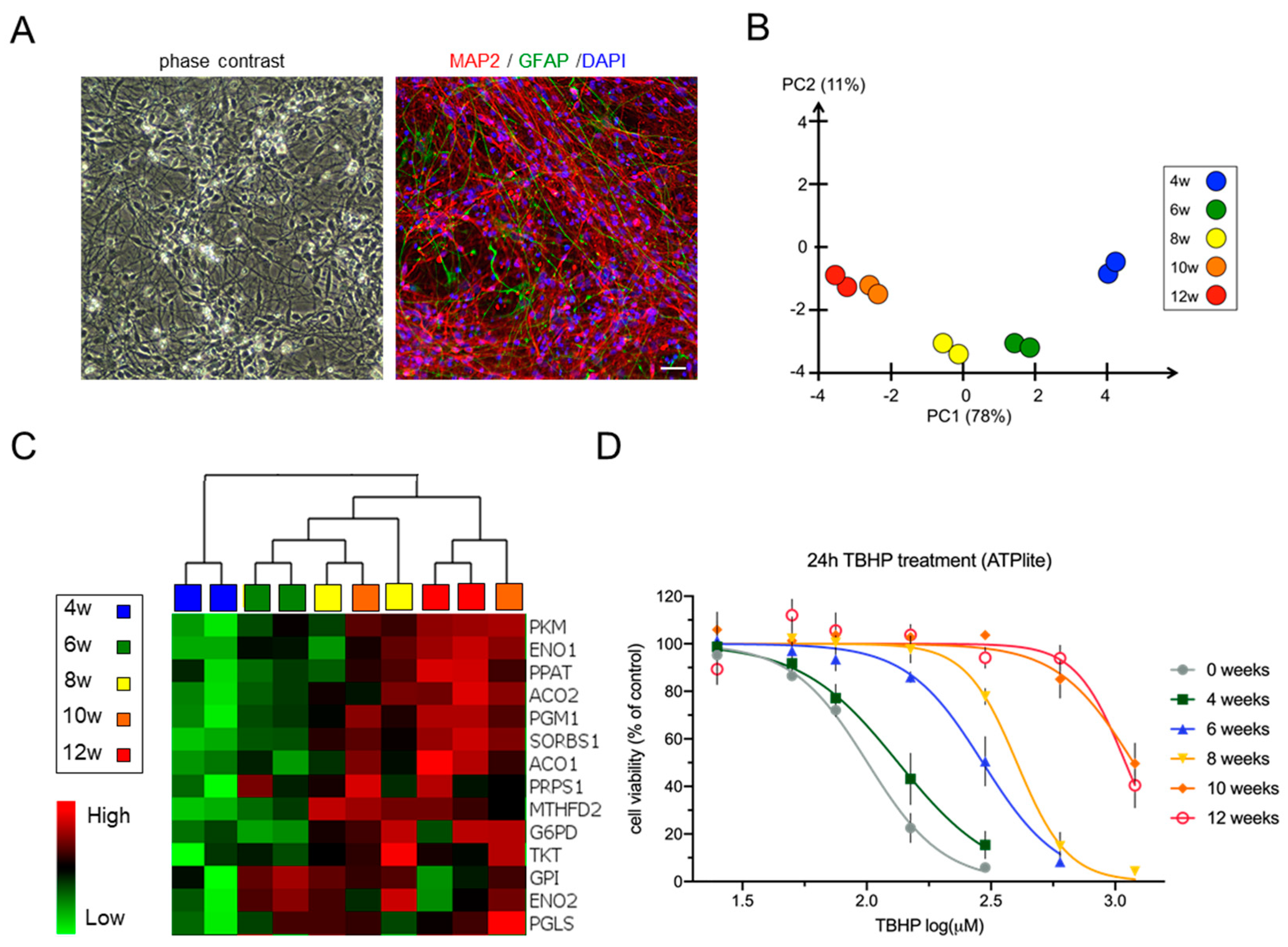
3.1. Differentiation of Neural Stem Cell Cultures Is Accompanied by Changes in the Expression of Metabolic Pathway Genes and an Increase in Stress Resistance
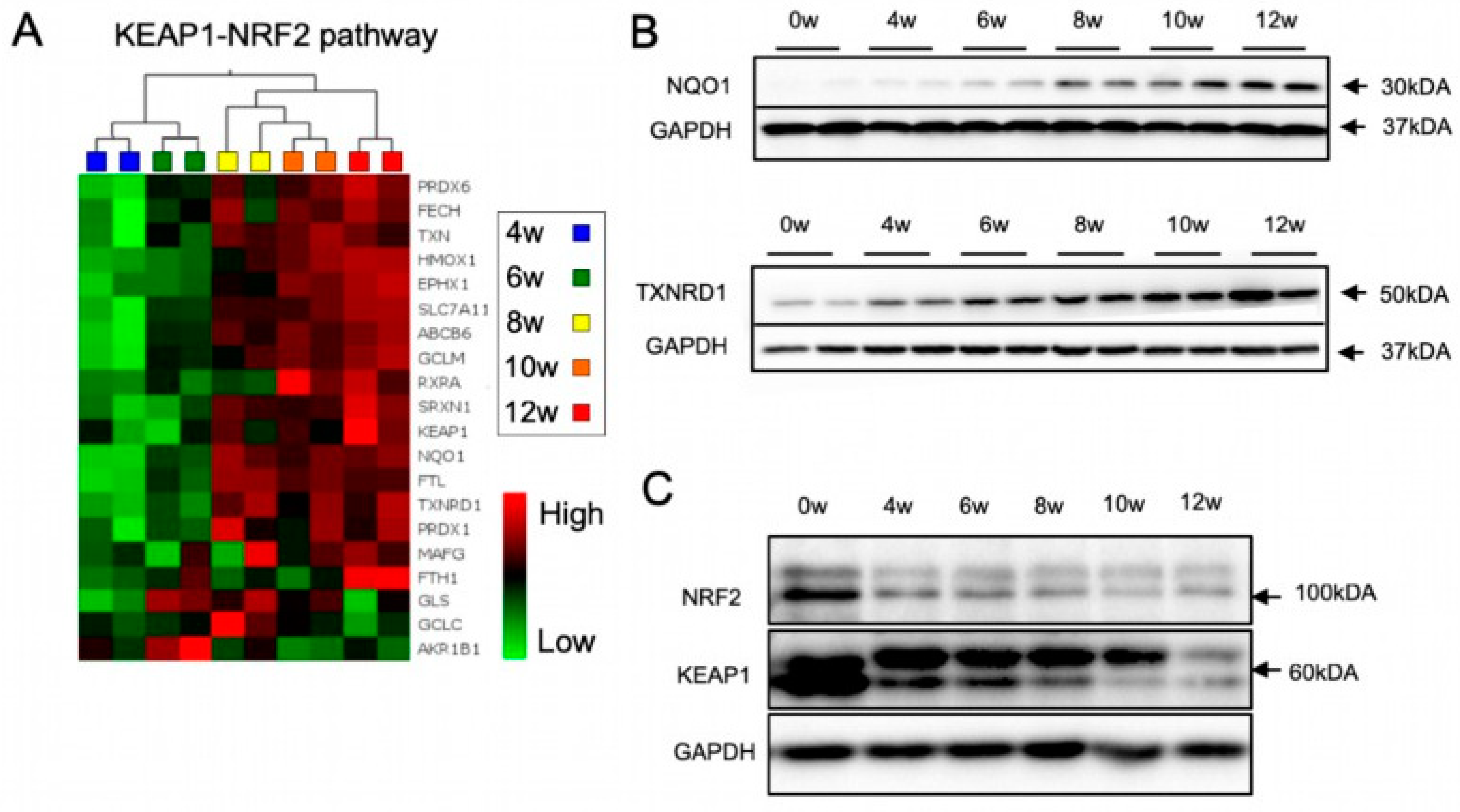
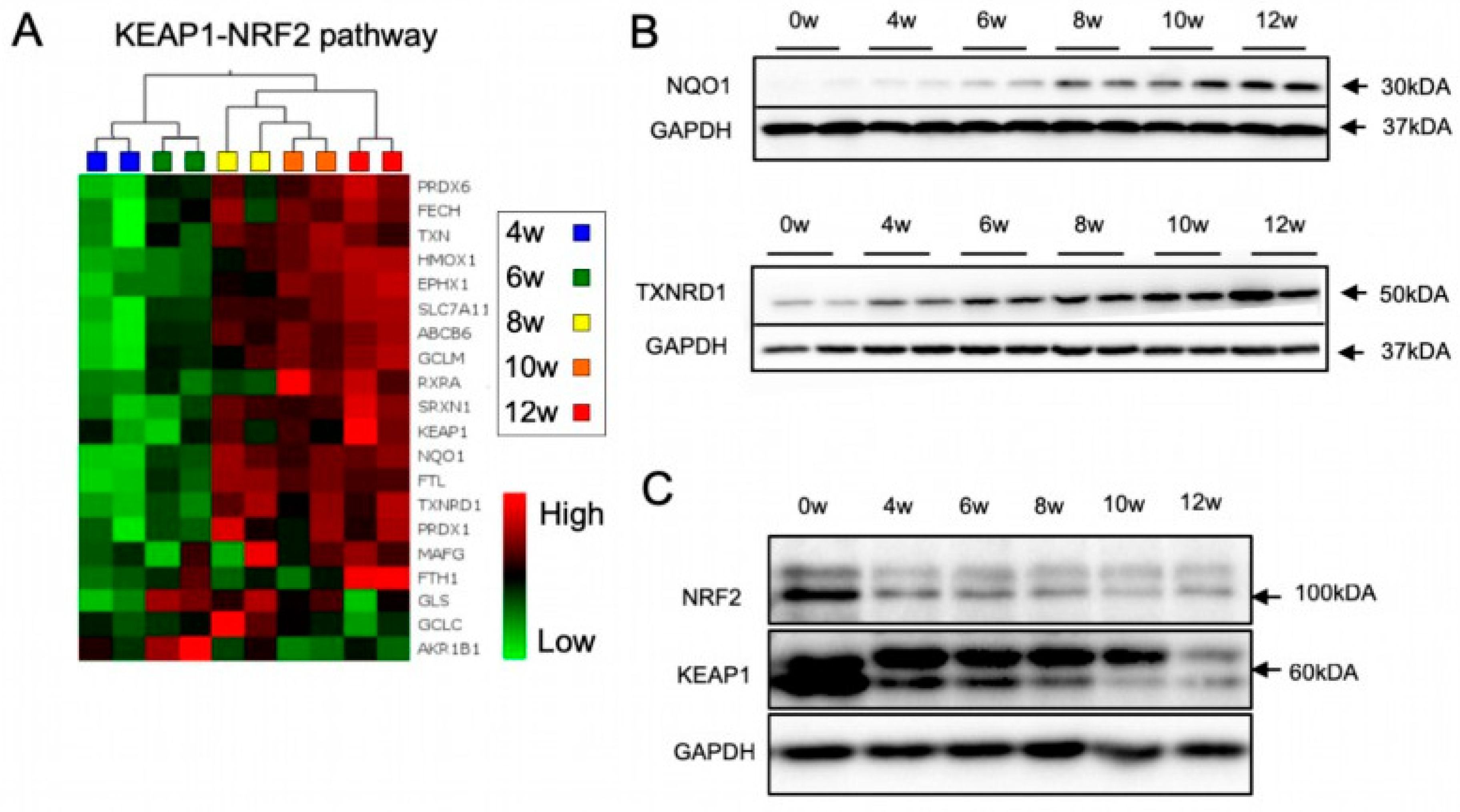
3.2. The KEAP1-NRF2 Pathway Is Dynamically Regulated during lt-NES Cell Differentiation
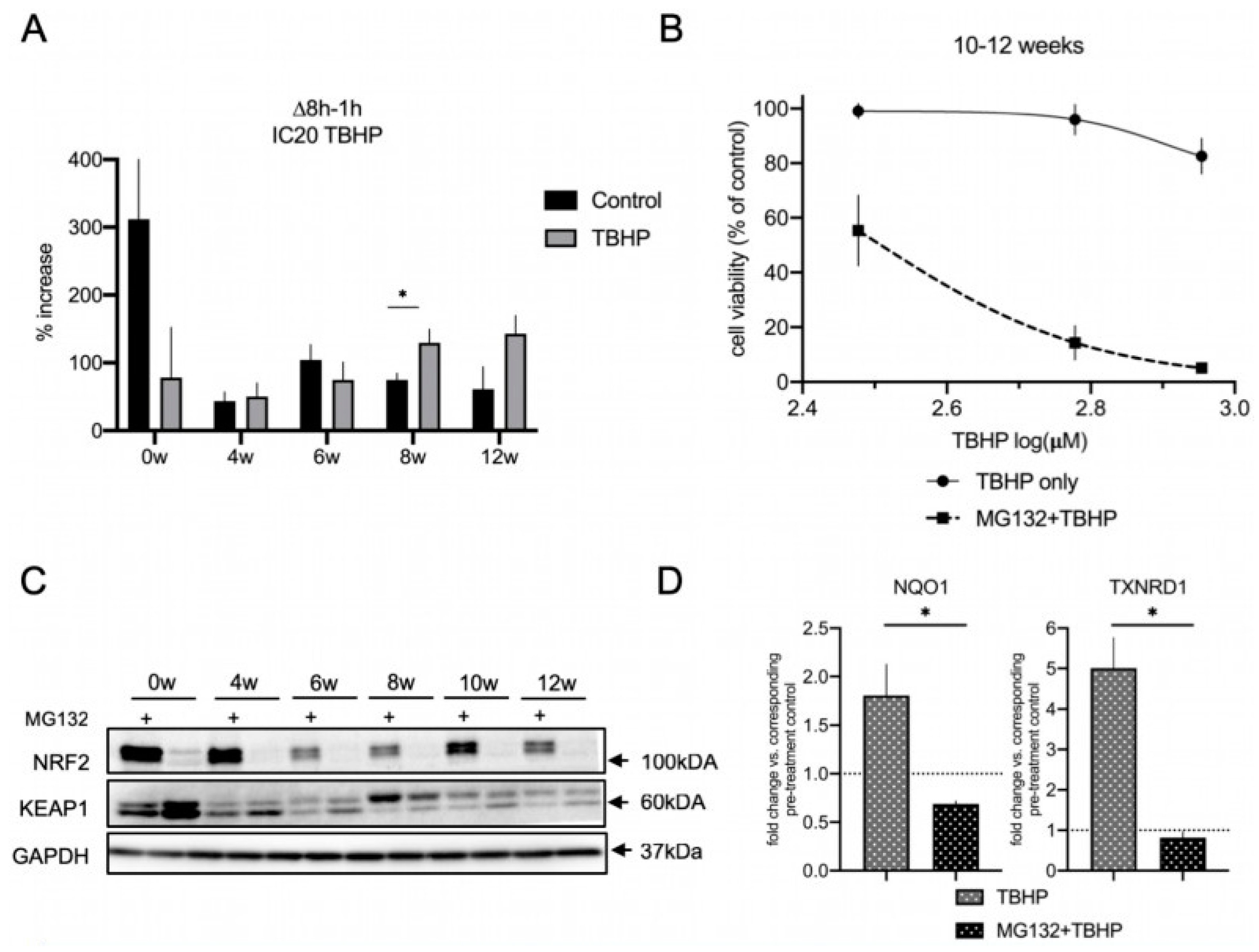
3.3. The NRF2-KEAP1 Signaling Pathway Contributes to Increasing Resistance to Oxidative Stress with Neural Differentiation
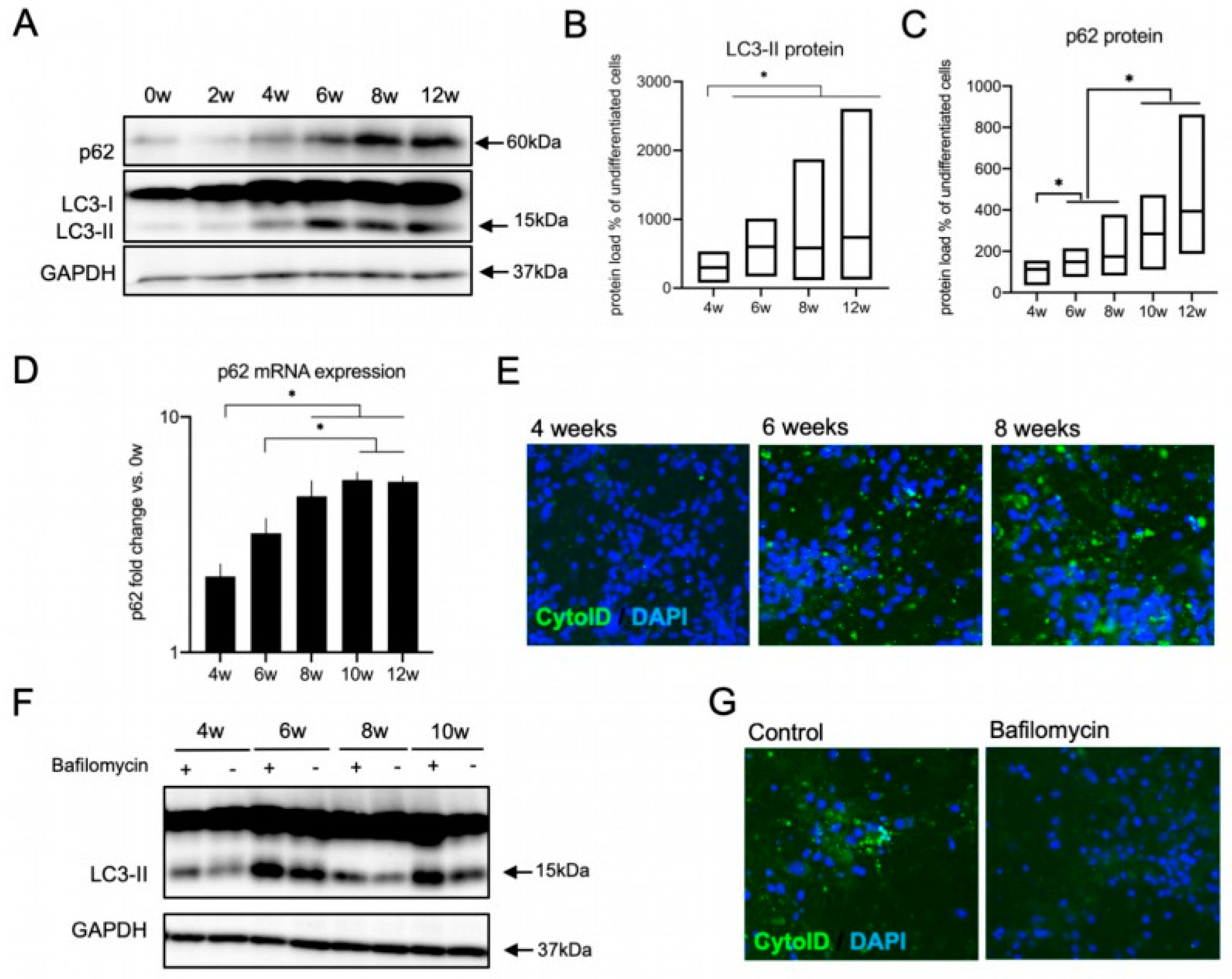
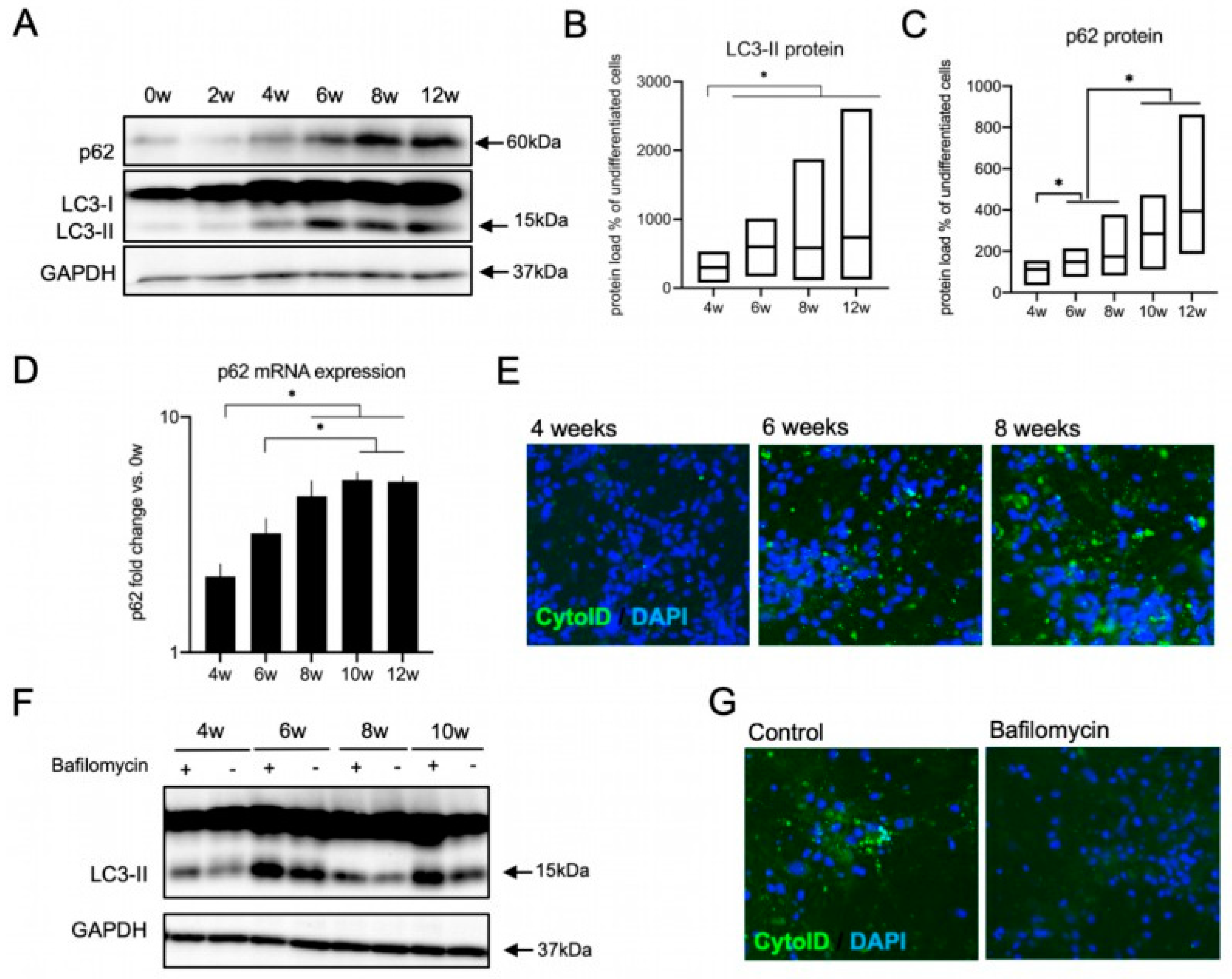
3.4. The Autophagy Pathway Is Upregulated during Neural Differentiation
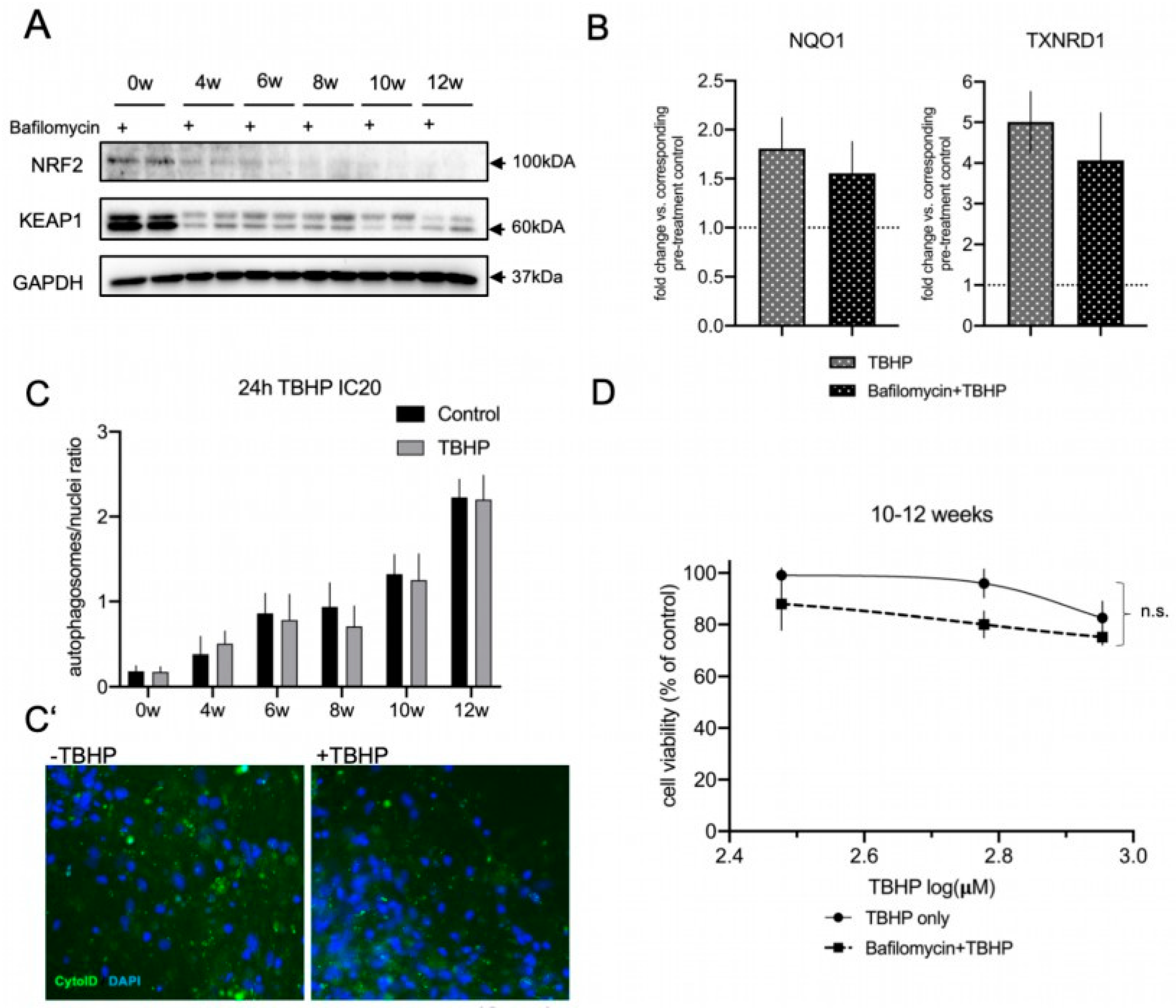
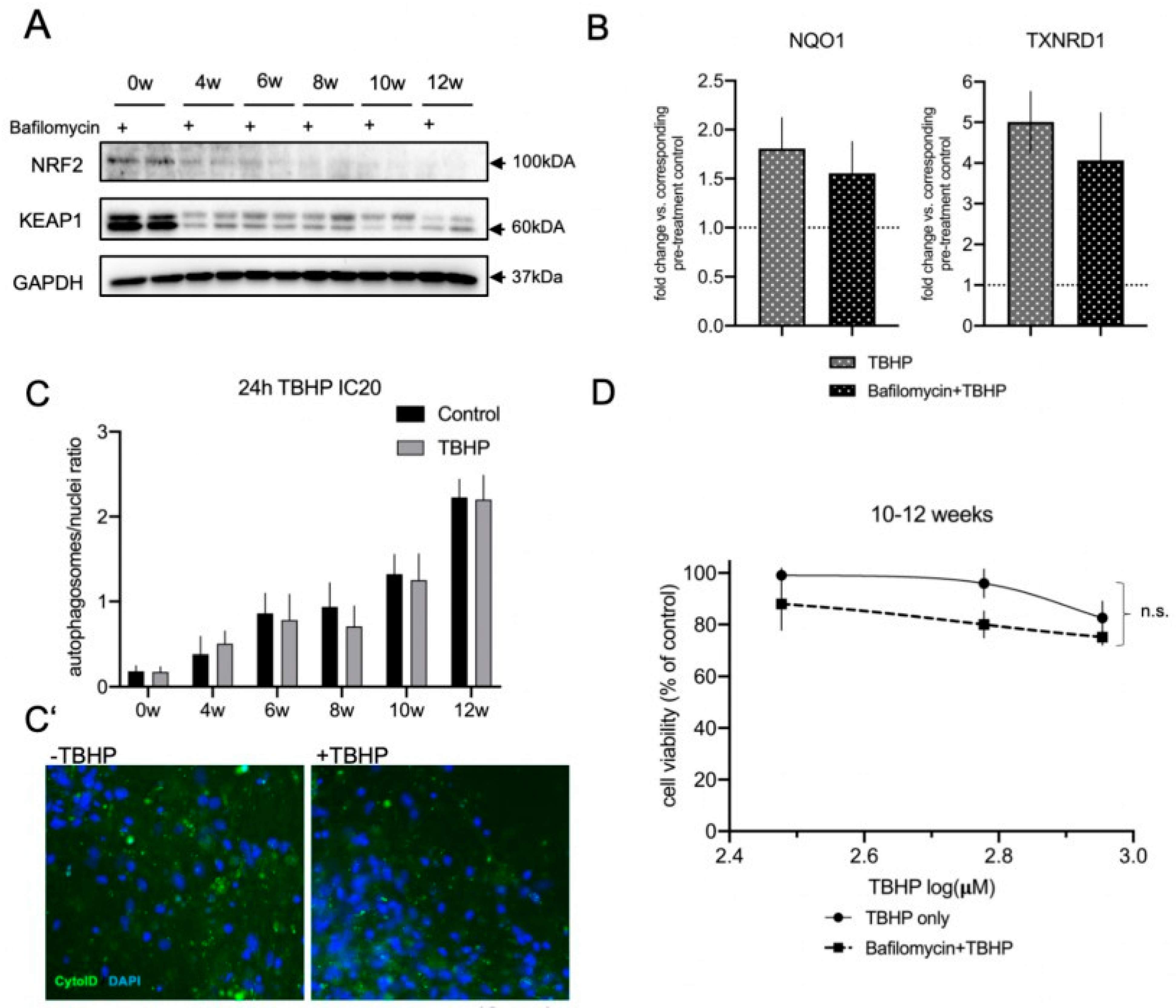
3.5. Autophagic Flux Does Not Contribute to the Acute Oxidative Stress Response in Differentiated lt-NES Cells
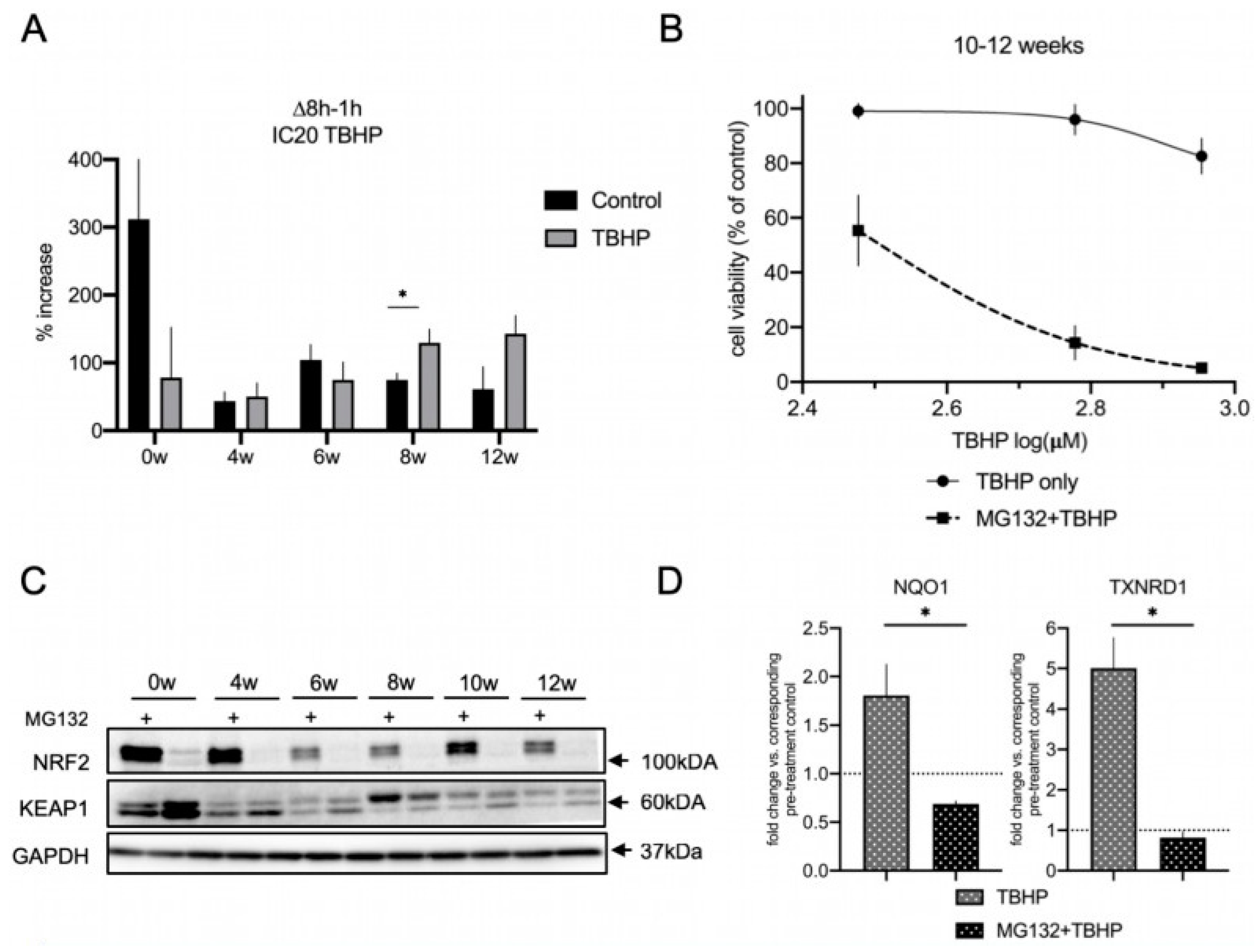
3.6. The Proteasome Is a Major Contributor to the Acute Oxidative Stress Response in Differentiating lt-NES Cells and Regulates the NRF2-KEAP1 Pathway Response
4. Discussion
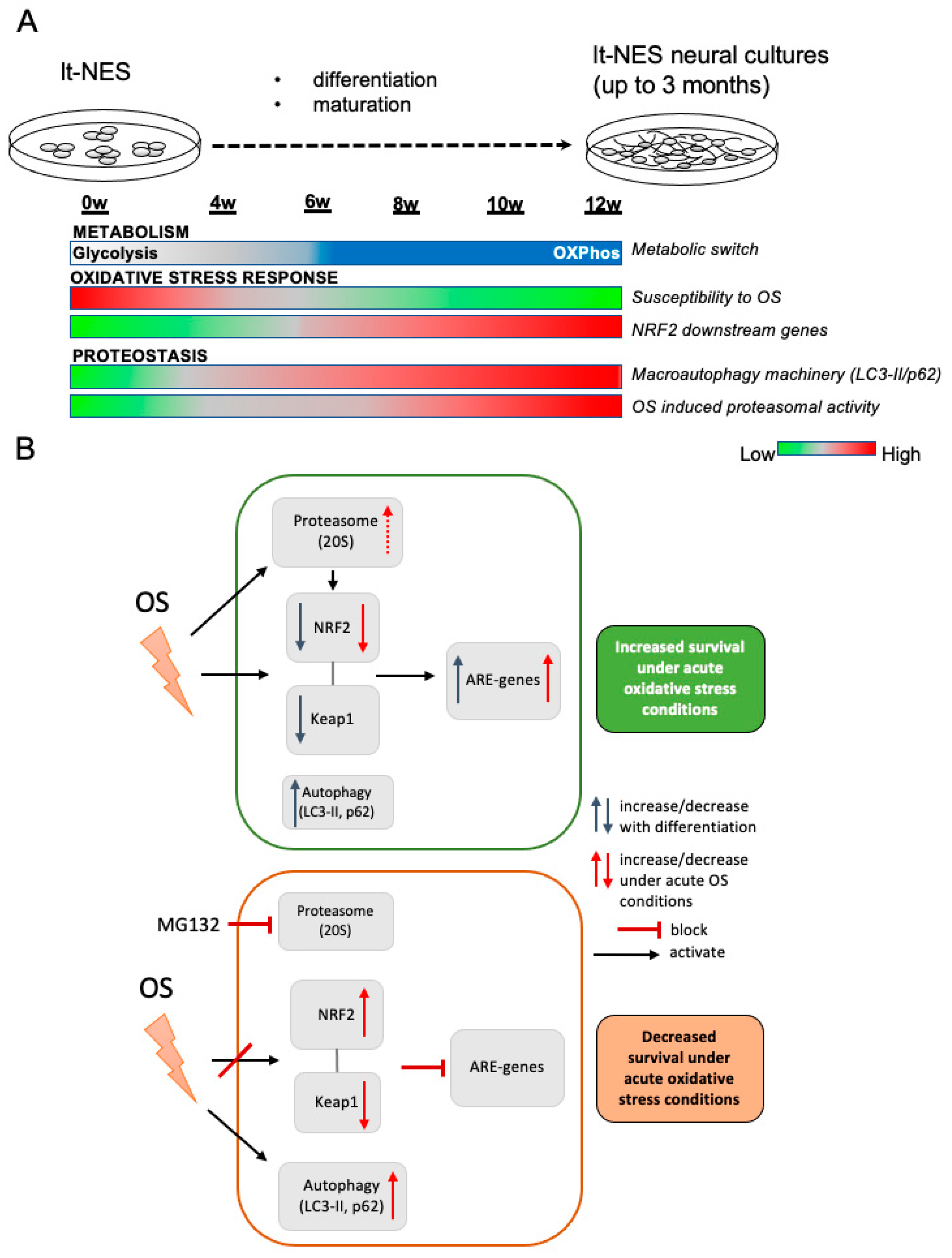
4.1. A Model of Dynamic Changes in Stress Response Patterns during Neural Differentiation
4.2. Changes in Metabolism during Neural Differentiation Are Associated with Increased Resistance to Stress
4.3. Dynamic Activation of NRF2 Pathway along lt-NES Cell Differentiation
4.4. Modeling of Autophagic Degradation during Neurodevelopment and Its Role in the Acute Response to Stress in Differentiating lt-NES Cells
4.5. Proteasomal Degradation as a Key Regulator of the NRF2 Pathway Response under Oxidative Stress Conditions
4.6. lt-NES Cultures as a Proxy for Human Neural Development In Vitro
5. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ARE | antioxidant response element |
| DNT | developmental neurotoxicity |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| NRF2 | nuclear factor (erythroid-derived 2)-like-2 factor |
| OS | oxidative stress |
| OXPHOS | oxidative phosphorylation |
| ROS | reactive oxygen species |
| SNP | single-nucleotide polymorphism |
| TBHP | tert-butyl hydrogenperoxide |
| UPS | ubiquitin-proteasome system |
| hESC | human embryonic stem cells |
| iPS | induced pluripotent stem cell |
| lt-NES cells | long-term self-renewing neuroepithelial stem cells |
| hPSC | human pluripotent stem cell |
References
- Grandjean, P.; Landrigan, P.J. Developmental neurotoxicity of industrial chemicals: A silent pandemic. Lancet 2006, 368, 2167–2178. [Google Scholar] [CrossRef]
- Rice, D.; Barone, S., Jr. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ. Health Perspect. 2000, 108, 511–533. [Google Scholar] [PubMed]
- Woodruff, T.J.; Zota, A.; Schwartz, J.M. Environmental chemicals in pregnant women in the United States: NHANES 2003–2004. Environ. Health Perspect. 2011, 119, 878–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Z.; Misra, H.P. Reactive oxygen species in in vitro pesticide-induced neuronal cell (SH-SY5Y) cytotoxicity: Role of NFκB and caspase-3. Free Radic. Biol. Med. 2007, 42, 288–298. [Google Scholar] [CrossRef]
- Zhu, Y.; Mapuskar, K.A.; Marek, R.; Xu, W.; Lehmler, H.-J.; Robertson, L.W.; Hornbuckle, K.; Spitz, D.; Aykin-Burns, N. A new player in environmentally induced oxidative stress: Polychlorinated Biphenyl Congener, 3,3′-Dichlorobiphenyl (PCB11). Toxicol. Sci. 2013, 136, 39–50. [Google Scholar] [CrossRef]
- Uchendu, C. The organophosphate, chlorpyrifos, oxidative stress and the role of some antioxidants: A review. Afr. J. Agric. Res. 2012, 7, 2720–2728. [Google Scholar] [CrossRef]
- Dos Santos, A.A.; Ferrer, B.; Gonçalves, F.M.; Tsatsakis, A.M.; Renieri, E.A.; Skalny, A.V.; Farina, M.; Rocha, J.B.T.; Aschner, M. Oxidative Stress in Methylmercury-Induced Cell Toxicity. Toxics 2018, 6, 47. [Google Scholar] [CrossRef] [Green Version]
- Rauh, V.A.; Garfinkel, R.; Perera, F.P.; Andrews, H.F.; Hoepner, L.; Barr, D.B.; Whitehead, R.; Tang, D.; Whyatt, R.W. Impact of prenatal chlorpyrifos exposure on neurodevelopment in the first 3 years of life among inner-city children. Pediatrics 2006, 118, e1845–e1859. [Google Scholar] [CrossRef] [Green Version]
- Rauh, V.; Arunajadai, S.; Horton, M.; Perera, F.; Hoepner, L.; Barr, D.B.; Whyatt, R. Seven-year neurodevelopmental scores and prenatal exposure to chlorpyrifos, a common agricultural pesticide. In Everyday Environmental Toxins: Childrens Exposure Risks; Apple Academic Press: New York, NY, USA, 2015. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, M.F.; Chevrier, J.; Harley, K.G.; Kogut, K.; Vedar, M.; Calderon, N.; Trujillo, C.; Johnson, C.; Bradman, A.; Barr, D.B.; et al. Prenatal exposure to organophosphate pesticides and IQ in 7-Year-old children. Environ. Health Perspect. 2011, 119, 1189–1195. [Google Scholar] [CrossRef] [Green Version]
- Grandjean, P.; Weihe, P.; White, R.F.; Debes, F.; Araki, S.; Yokoyama, K.; Murata, K.; Sørensen, N.; Dahl, R.; Jørgensen, P.J. cognitive deficit in 7-year-old children with prenatal exposure to Methylmercury. Neurotoxicol. Teratol. 1997, 19, 417–428. [Google Scholar] [CrossRef]
- Davidson, P.W.; Strain, J.; Myers, G.J.; Thurston, S.W.; Bonham, M.P.; Shamlaye, C.F.; Stokes-Riner, A.; Wallace, J.M.; Robson, P.J.; Duffy, E.M.; et al. Neurodevelopmental effects of maternal nutritional status and exposure to methylmercury from eating fish during pregnancy. Neurotoxicology 2008, 29, 767–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, P.W.; Lonky, E.; Reihman, J.; Pagano, J.; Gump, B.B.; Darvill, T. The relationship between prenatal PCB exposure and intelligence (IQ) in 9-year-old children. Environ. Health Perspect. 2008, 116, 1416–1422. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, L.; Hogberg, H.T.; Leist, M.; Hartung, T. Developmental neurotoxicity–challenges in the 21st Century and in vitro opportunities. ALTEX 2014, 31, 129–156. [Google Scholar] [CrossRef] [Green Version]
- Chiapella, G.; Flores-Martín, J.; Ridano, M.; Reyna, L.; de Potas, G.M.; Panzetta-Dutari, G.; Genti-Raimondi, S. The organophosphate chlorpyrifos disturbs redox balance and triggers antioxidant defense mechanisms in JEG-3 cells. Placenta 2013, 34, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Osburn, W.O.; Kensler, T.W. Nrf2 signaling: An adaptive response pathway for protection against environmental toxic insults. Mutat. Res. Mutat. Res. 2008, 659, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyama, T.; Sumi, D.; Shinkai, Y.; Yasutake, A.; Taguchi, K.; Tong, K.I.; Yamamoto, M.; Kumagai, Y. Cytoprotective role of Nrf2/Keap1 system in methylmercury toxicity. Biochem. Biophys. Res. Commun. 2007, 363, 645–650. [Google Scholar] [CrossRef]
- Lacher, S.E.; Lee, J.S.; Wang, X.; Campbell, M.R.; Bell, D.; Slattery, M. Beyond antioxidant genes in the ancient Nrf2 regulatory network. Free Radic. Biol. Med. 2015, 88, 452–465. [Google Scholar] [CrossRef] [Green Version]
- Hong, F.; Sekhar, K.R.; Freeman, M.L.; Liebler, D.C. Specific Patterns of Electrophile Adduction Trigger Keap1 Ubiquitination and Nrf2 Activation. J. Biol. Chem. 2005, 280, 31768–31775. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J. Keap1-dependent Proteasomal Degradation of Transcription Factor Nrf2 Contributes to the Negative Regulation of Antioxidant Response Element-driven Gene Expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [Green Version]
- Bell, K.F.; Al-Mubarak, B.; Martel, M.-A.; McKay, S.; Wheelan, N.; Hasel, P.; Márkus, N.M.; Baxter, P.; Deighton, R.F.; Serio, A.; et al. Neuronal development is promoted by weakened intrinsic antioxidant defences due to epigenetic repression of Nrf2. Nat. Commun. 2015, 6, 7066. [Google Scholar] [CrossRef] [Green Version]
- Son, G.; Han, J. Roles of mitochondria in neuronal development. BMB Rep. 2018, 51, 549–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agathocleous, M.; Love, N.K.; Randlett, O.; Harris, J.J.; Liu, J.; Murray, A.J.; Harris, W.A. Metabolic differentiation in the embryonic retina. Nature 2012, 14, 859–864. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. eLife 2016, 5, e13374. [Google Scholar] [CrossRef] [PubMed]
- Le Belle, J.E.; Orozco, N.M.; Paucar, A.A.; Saxe, J.P.; Mottahedeh, J.; Pyle, A.D.; Wu, H.; Kornblum, H.I. Proliferative Neural Stem Cells Have High Endogenous ROS Levels that Regulate Self-Renewal and Neurogenesis in a PI3K/Akt-Dependant Manner. Cell Stem Cell 2011, 8, 59–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, N.M.; Shin, R.; Tajinda, K.; Heusner, C.L.; Kogan, J.H.; Miyake, S.; Chen, Q.; Tamura, K.; Matsumoto, M. Adult Neurogenesis Transiently Generates Oxidative Stress. PLoS ONE 2012, 7, e35264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Götz, M.; Huttner, W.B. The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 777–788. [Google Scholar] [CrossRef]
- Sakakibara, A.; Ando, R.; Sapir, T.; Tanaka, T. Microtubule dynamics in neuronal morphogenesis. Open Biol. 2013, 3, 130061. [Google Scholar] [CrossRef] [Green Version]
- Dragich, J.; Kuwajima, T.; Hirose-Ikeda, M.; Yoon, M.; Eenjes, E.; Bosco, J.R.; Fox, L.M.; Lystad, A.H.; Oo, T.F.; Yarygina, O.; et al. Autophagy linked FYVE (Alfy/WDFY3) is required for establishing neuronal connectivity in the mammalian brain. eLife 2016, 5, e14810. [Google Scholar] [CrossRef]
- Stavoe, A.; Hill, S.; Hall, D.H.; Colón-Ramos, D.A. KIF1A/UNC-104 Transports ATG-9 to Regulate Neurodevelopment and Autophagy at Synapses. Dev. Cell 2016, 38, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Schäffner, I.; Minakaki, G.; Khan, M.A.; Balta, E.-A.; Schlötzer-Schrehardt, U.; Schwarz, T.J.; Beckervordersandforth, R.; Winner, B.; Webb, A.E.; DePinho, R.; et al. FoxO Function Is Essential for Maintenance of Autophagic Flux and Neuronal Morphogenesis in Adult Neurogenesis. Neuron 2018, 99, 1188–1203.e6. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Khodosevich, K.; Monyer, H. Dendrite Development Regulated by the Schizophrenia-Associated Gene FEZ1 Involves the Ubiquitin Proteasome System. Cell Rep. 2014, 7, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Hsu, M.-T.; Guo, C.-L.; Liou, A.Y.; Chang, T.-Y.; Ng, M.-C.; Florea, B.I.; Overkleeft, H.S.; Wu, Y.-L.; Liao, J.-C.; Cheng, P.-L. Stage-Dependent Axon Transport of Proteasomes Contributes to Axon Development. Dev. Cell 2015, 35, 418–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, M.K.; Sheng, M. USP8 Deubiquitinates SHANK3 to Control Synapse Density and SHANK3 Activity-Dependent Protein Levels. J. Neurosci. 2018, 38, 5289–5301. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [Green Version]
- Pajares, M.; Rojo, A.I.; Arias, E.; Díaz-Carretero, A.; Cuervo, A.M.; Cuadrado, A. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 2018, 14, 1310–1322. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A Noncanonical Mechanism of Nrf2 Activation by Autophagy Deficiency: Direct Interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.; Wang, Y.; Kim, H.-S.; Lalli, M.A.; Kosik, K.S. Nrf2, a Regulator of the Proteasome, Controls Self-Renewal and Pluripotency in Human Embryonic Stem Cells. Stem Cells 2014, 32, 2616–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickering, A.M.; Linder, R.A.; Zhang, H.; Forman, H.J.; Davies, K.J.A. Nrf2-dependent Induction of Proteasome and Pa28αβ Regulator Are Required for Adaptation to Oxidative Stress. J. Biol. Chem. 2012, 287, 10021–10031. [Google Scholar] [CrossRef] [Green Version]
- Kwak, M.-K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants Enhance Mammalian Proteasome Expression through the Keap1-Nrf2 Signaling Pathway. Mol. Cell. Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef] [Green Version]
- Koch, P.; Opitz, T.; Steinbeck, J.A.; Ladewig, J.; Brüstle, O. A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc. Natl. Acad. Sci. USA 2009, 106, 3225–3230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, A.; Koch, P.; Kesavan, J.; Takashima, Y.; Ladewig, J.; Alexander, M.; Wiskow, O.; Tailor, J.; Trotter, M.; Pollard, S.; et al. Capture of Neuroepithelial-Like Stem Cells from Pluripotent Stem Cells Provides a Versatile System for In Vitro Production of Human Neurons. PLoS ONE 2012, 7, e29597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, P.; Breuer, P.; Peitz, M.; Jungverdorben, J.; Kesavan, J.; Poppe, D.; Doerr, J.; Ladewig, J.; Mertens, J.; Tüting, T.; et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado–Joseph disease. Nature 2011, 480, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Paquola, A.C.; Ku, M.; Hatch, E.; Böhnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertens, J.; Stüber, K.; Poppe, D.; Doerr, J.; Ladewig, J.; Brüstle, O.; Koch, P. Embryonic Stem Cell–Based Modeling of Tau Pathology in Human Neurons. Am. J. Pathol. 2013, 182, 1769–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, S.T.; Siegel, G.J.; Albers, R.W.; Price, D.L. Basic Neurochemistry; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar] [CrossRef]
- De Santis, M.C.; Porporato, P.E.; Martini, M.; Morandi, A. Signaling Pathways Regulating Redox Balance in Cancer Metabolism. Front. Oncol. 2018, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Pollak, N.; Dölle, C.; Ziegler, M. The power to reduce: Pyridine nucleotides–small molecules with a multitude of functions. Biochem. J. 2007, 402, 205–218. [Google Scholar] [CrossRef]
- Hosios, A.M.; Heiden, M.G.V. The redox requirements of proliferating mammalian cells. J. Biol. Chem. 2018, 293, 7490–7498. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Luo, G.; Giannelli, S.G.; Szeto, H.H. Mitochondria-targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert-butyl hydroperoxide in neuronal cell lines. Biochem. Pharmacol. 2005, 70, 1796–1806. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Talalay, P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys. 2010, 501, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Nagarajan, N.; Oka, S.; Sadoshima, J. Modulation of signaling mechanisms in the heart by thioredoxin 1. Free Radic. Biol. Med. 2016, 109, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Zgheib, E.; Limonciel, A.; Jiang, X.; Wilmes, A.; Wink, S.; Van De Water, B.; Kopp-Schneider, A.; Bois, F.Y.; Jennings, P. Investigation of Nrf2, AhR and ATF4 Activation in Toxicogenomic Databases. Front. Genet. 2018, 9, 429. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1–Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, L.J.M.; Kuijper, I.A.; Schimming, J.P.; Wolters, L.; ter Braak, B.; Langenberg, J.P.; Noort, D.; Beltman, J.B.; van de Water, B. A systematic analysis of Nrf2 pathway activation dynamics during repeated xenobiotic exposure. Arch. Toxicol. 2018, 93, 435–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Wang, X.; Nisar, M.F.; Lin, M.; Zhong, J.L. Heme Oxygenases: Cellular Multifunctional and Protective Molecules against UV-Induced Oxidative Stress. Oxidative Med. Cell. Longev. 2019, 2019, 5416728. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W.; Cloonan, S.; Choi, A.M.K. Autophagy: A critical regulator of cellular metabolism and homeostasis. Mol. Cells 2013, 36, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Martín, P.; Saito, T.; Komatsu, M. p62/ SQSTM 1: ‘Jack of all trades’ in health and cancer. FEBS J. 2018, 286, 8–23. [Google Scholar] [CrossRef] [Green Version]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Jung, T.; Höhn, A.; Grune, T. The proteasome and the degradation of oxidized proteins: Part III—Redox regulation of the proteasomal system. Redox Biol. 2014, 2, 388–394. [Google Scholar] [CrossRef] [Green Version]
- Chondrogianni, N.; Georgila, K.; Kourtis, N.; Tavernarakis, N.; Gonos, E.S. 20S proteasome activation promotes life span extension and resistance to proteotoxicity in Caenorhabditis elegans. FASEB J. 2014, 29, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chondrogianni, N.; Tzavelas, C.; Pemberton, A.J.; Nezis, I.; Rivett, A.J.; Gonos, E.S. Overexpression of Proteasome β5 Assembled Subunit Increases the Amount of Proteasome and Confers Ameliorated Response to Oxidative Stress and Higher Survival Rates. J. Biol. Chem. 2005, 280, 11840–11850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkabetz, Y.; Panagiotakos, G.; Al Shamy, G.; Socci, N.D.; Tabar, V.; Studer, L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008, 22, 152–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.-Y.; Weick, J.P.; Yu, J.; Ma, L.-X.; Zhang, X.-Q.; Thomson, J.A.; Zhang, S.-C. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. USA 2010, 107, 4335–4340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertens, J.; Marchetto, M.C.; Bardy, C.; Gage, F.H. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat. Rev. Neurosci. 2016, 17, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Conti, L.; Cattaneo, E. Neural stem cell systems: Physiological players or in vitro entities? Nat. Rev. Neurosci. 2010, 11, 176–187. [Google Scholar] [CrossRef]
- Elkabetz, Y.; Studer, L. Human ESC-derived Neural Rosettes and Neural Stem Cell Progression. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Tailor, J.; Kittappa, R.; Leto, K.; Gates, M.; Borel, M.; Paulsen, O.; Spitzer, S.; Karadottir, R.T.; Rossi, F.; Falk, A.; et al. Stem Cells Expanded from the Human Embryonic Hindbrain Stably Retain Regional Specification and High Neurogenic Potency. J. Neurosci. 2013, 33, 12407–12422. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, K.E.; Joy, S.; Delhove, J.; Kotiadis, V.N.; Fernandez, E.; Fitzpatrick, L.M.; Whiteford, J.R.; King, P.J.; Bolanos, J.; Duchen, M.; et al. NRF2 Orchestrates the Metabolic Shift during Induced Pluripotent Stem Cell Reprogramming. Cell Rep. 2016, 14, 1883–1891. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.S.; Soares, N.L.; Sequeira, C.O.; Pereira, S.A.; Sonnewald, U.; Vieira, H.L. Improvement of neuronal differentiation by carbon monoxide: Role of pentose phosphate pathway. Redox Biol. 2018, 17, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Vargas, M. Redox Biology in Neurological Function, Dysfunction, and Aging. Antioxid. Redox Signal. 2018, 28, 1583–1586. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Joung, I.; Kim, H.J.; Kwon, Y.K. p62 modulates Akt activity via association with PKCζ in neuronal survival and differentiation. Biochem. Biophys. Res. Commun. 2005, 334, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Garrido, J.C.; Maffezzini, C.; Schober, F.A.; Clemente, P.; Uhlin, E.; Kele, M.; Stranneheim, H.; Lesko, N.; Bruhn, H.; Svenningsson, P.; et al. SQSTM1/p62-Directed Metabolic Reprogramming Is Essential for Normal Neurodifferentiation. Stem Cell Rep. 2019, 12, 696–711. [Google Scholar] [CrossRef] [Green Version]
- Copple, I.; Lister, A.; Obeng, A.D.; Kitteringham, N.R.; Jenkins, R.E.; Layfield, R.; Foster, B.J.; Goldring, C.E.; Park, B.K. Physical and Functional Interaction of Sequestosome 1 with Keap1 Regulates the Keap1-Nrf2 Cell Defense Pathway. J. Biol. Chem. 2010, 285, 16782–16788. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Matuzelski, E.; Bunt, J.; Harkins, D.; Lim, J.W.; Gronostajski, R.M.; Richards, L.J.; Harris, L.; Piper, M. Transcriptional regulation of Nfix by NFIB drives astrocytic maturation within the developing spinal cord. Dev. Biol. 2017, 432, 286–297. [Google Scholar] [CrossRef]
- Burke, E.E.; Chenoweth, J.G.; Shin, J.H.; Collado-Torres, L.; Kim, S.-K.; Micali, N.; Wang, Y.; Colantuoni, C.; Straub, R.E.; Hoeppner, D.J.; et al. Dissecting transcriptomic signatures of neuronal differentiation and maturation using iPSCs. Nat. Commun. 2020, 11, 462. [Google Scholar] [CrossRef] [Green Version]
- Silbereis, J.C.; Pochareddy, S.; Zhu, Y.; Li, M.; Sestan, N. The Cellular and Molecular Landscapes of the Developing Human Central Nervous System. Neuron 2016, 89, 248–268. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Moreno, N.; Stathakos, P.; Caldwell, M.A.; Lane, J.D. Induced Pluripotent Stem Cell Neuronal Models for the Study of Autophagy Pathways in Human Neurodegenerative Disease. Cells 2017, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yuan, X.; Sun, Q.; Ou, Y. Autophagy activator promotes neuronal differentiation of adult adipose-derived stromal cells. Neural Regen. Res. 2013, 8, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhou, P.; Guo, Y.; Zhou, B. The involvement of autophagy and cytoskeletal regulation in TDCIPP-induced SH-SY5Y cell differentiation. Neurotoxicology 2017, 62, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, J.; Cai, Z.; Xu, C. The effect of curcumin on the differentiation, apoptosis and cell cycle of neural stem cells is mediated through inhibiting autophagy by the modulation of Atg7 and p62. Int. J. Mol. Med. 2018, 42, 2481–2488. [Google Scholar] [CrossRef]
- Martorana, F.; Gaglio, D.; Bianco, M.R.; Aprea, F.; Virtuoso, A.; Bonanomi, M.; Alberghina, L.; Papa, M.; Colangelo, A.M. Differentiation by nerve growth factor (NGF) involves mechanisms of crosstalk between energy homeostasis and mitochondrial remodeling. Cell Death Dis. 2018, 9, 391. [Google Scholar] [CrossRef]
- Kulkarni, V.V.; Maday, S. Compartment-specific dynamics and functions of autophagy in neurons. Dev. Neurobiol. 2017, 78, 298–310. [Google Scholar] [CrossRef]
- Takei, N.; Nawa, H. mTOR signaling and its roles in normal and abnormal brain development. Front. Mol. Neurosci. 2014, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Ka, M.; Smith, A.L.; Kim, W.-Y. MTOR controls genesis and autophagy of GABAergic interneurons during brain development. Autophagy 2017, 13, 1348–1363. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L., Jr.; Iwata, J.-I.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 14489–14494. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.-C.; Wang, C.; Peng, X.; Gan, B.; Guan, J.-L. Neural-specific Deletion of FIP200 Leads to Cerebellar Degeneration Caused by Increased Neuronal Death and Axon Degeneration. J. Biol. Chem. 2010, 285, 3499–3509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.-I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Shibata, M.; Tadakoshi, M.; Gotoh, K.; Komatsu, M.; Waguri, S.; Kawahara, N.; Kuida, K.; Nagata, S.; Kominami, E.; et al. Inhibition of Autophagy Prevents Hippocampal Pyramidal Neuron Death after Hypoxic-Ischemic Injury. Am. J. Pathol. 2008, 172, 454–469. [Google Scholar] [CrossRef] [Green Version]
- Samara, C.; Syntichaki, P.; Tavernarakis, N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 2007, 15, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Uchiyama, Y.; Koike, M.; Shibata, M. Autophagic neuron death in neonatal brain ischemia/hypoxia. Autophagy 2008, 4, 404–408. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Fukui, K.; Koike, T.; Zheng, X. Induction of autophagy in neurite degeneration of mouse superior cervical ganglion neurons. Eur. J. Neurosci. 2007, 26, 2979–2988. [Google Scholar] [CrossRef]
- Li, Q.; Li, H.; Roughton, K.; Wang, X.; Kroemer, G.; Blomgren, K.; Zhu, C. Lithium reduces apoptosis and autophagy after neonatal hypoxia–ischemia. Cell Death Dis. 2010, 1, e56. [Google Scholar] [CrossRef]
- Kosztelnik, M.; Kurucz, A.; Papp, D.; Jones, E.; Sigmond, T.; Barna, J.; Traka, M.H.; Lorincz, T.; Szarka, A.; Banhegyi, G.; et al. Suppression of AMPK/aak-2 by NRF2/SKN-1 down-regulates autophagy during prolonged oxidative stress. FASEB J. 2018, 33, 2372–2387. [Google Scholar] [CrossRef] [Green Version]
- Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Koike, M.; Kuzumaki, N.; Hayakawa, H.; Nihira, T.; Kobayashi, T.; Ohyama, M.; Sato, S.; et al. Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain 2012, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Skibinski, G.; Hwang, V.; Ando, D.M.; Daub, A.; Lee, A.K.; Ravisankar, A.; Modan, S.; Finucane, M.M.; Shaby, B.A.; Finkbeiner, S. Nrf2 mitigates LRRK2- and α-synuclein–induced neurodegeneration by modulating proteostasis. Proc. Natl. Acad. Sci. USA 2017, 114, 1165–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, M.; Momiji, H.; Rabbani, N.; Bretschneider, T.; Rand, D.A.; Thornalley, P.J. Frequency modulated translocational oscillations of Nrf2, a transcription factor functioning like a wireless sensor. Biochem. Soc. Trans. 2015, 43, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Iso, T.; Suzuki, T.; Baird, L.; Yamamoto, M. Absolute Amounts and Status of the Nrf2-Keap1-Cul3 Complex within Cells. Mol. Cell. Biol. 2016, 36, 3100–3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T. Coordinate Regulation of Glutathione Biosynthesis and Release by Nrf2-Expressing Glia Potently Protects Neurons from Oxidative Stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef]
- Tang, X.; Zhou, L.; Wagner, A.M.; Marchetto, M.C.; Muotri, A.R.; Gage, F.H.; Chen, G. Astroglial cells regulate the developmental timeline of human neurons differentiated from induced pluripotent stem cells. Stem Cell Res. 2013, 11, 743–757. [Google Scholar] [CrossRef] [Green Version]
- Abe, K.; Saito, H. Characterization of t-Butyl Hydroperoxide Toxicity in Cultured Rat Cortical Neurones and Astrocytes. Pharmacol. Toxicol. 1998, 83, 40–46. [Google Scholar] [CrossRef]
- Dörrbaum, A.R.; Kochen, L.; Langer, J.D.; Schuman, E.M. Local and global influences on protein turnover in neurons and glia. eLife 2018, 7, e34202. [Google Scholar] [CrossRef]
- Aschner, M. Effects of Acrylamide on Primary Neonatal Rat Astrocyte Functions. Ann. N. Y. Acad. Sci. 2005, 1053, 444–454. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semkova, V.; Haupt, S.; Segschneider, M.; Bell, C.; Ingelman-Sundberg, M.; Hajo, M.; Weykopf, B.; Muthukottiappan, P.; Till, A.; Brüstle, O. Dynamics of Metabolic Pathways and Stress Response Patterns during Human Neural Stem Cell Proliferation and Differentiation. Cells 2022, 11, 1388. https://doi.org/10.3390/cells11091388
Semkova V, Haupt S, Segschneider M, Bell C, Ingelman-Sundberg M, Hajo M, Weykopf B, Muthukottiappan P, Till A, Brüstle O. Dynamics of Metabolic Pathways and Stress Response Patterns during Human Neural Stem Cell Proliferation and Differentiation. Cells. 2022; 11(9):1388. https://doi.org/10.3390/cells11091388
Chicago/Turabian StyleSemkova, Vesselina, Simone Haupt, Michaela Segschneider, Catherine Bell, Magnus Ingelman-Sundberg, Mohamad Hajo, Beatrice Weykopf, Pathma Muthukottiappan, Andreas Till, and Oliver Brüstle. 2022. "Dynamics of Metabolic Pathways and Stress Response Patterns during Human Neural Stem Cell Proliferation and Differentiation" Cells 11, no. 9: 1388. https://doi.org/10.3390/cells11091388
APA StyleSemkova, V., Haupt, S., Segschneider, M., Bell, C., Ingelman-Sundberg, M., Hajo, M., Weykopf, B., Muthukottiappan, P., Till, A., & Brüstle, O. (2022). Dynamics of Metabolic Pathways and Stress Response Patterns during Human Neural Stem Cell Proliferation and Differentiation. Cells, 11(9), 1388. https://doi.org/10.3390/cells11091388