
Involvement of Oxidative Stress in Protective Cardiac Functions of Calprotectin

 , and
, and 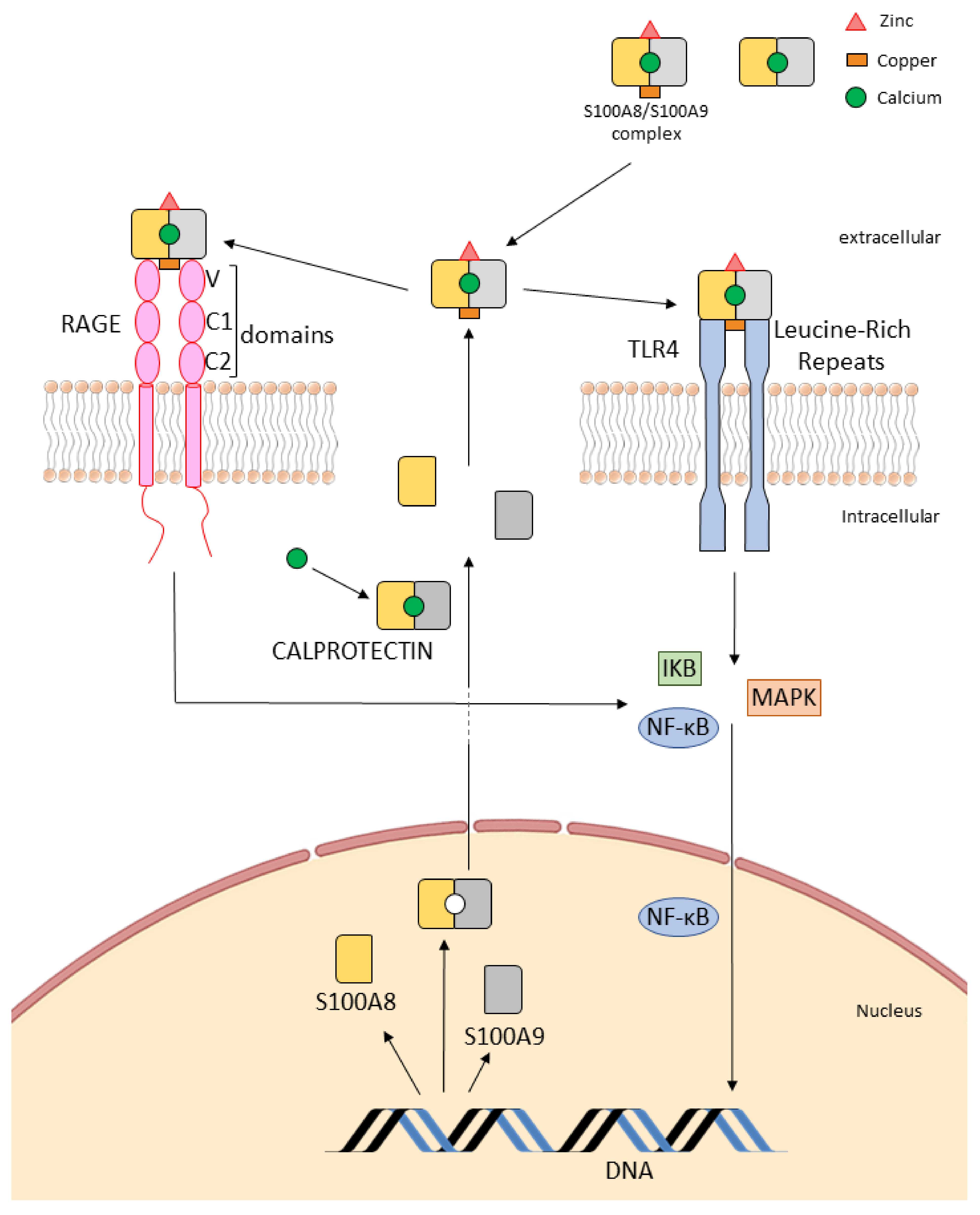{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Structure and Expression of Calprotectin
3. Calprotectin Receptors: Toll-Like Receptors (TLRs) and Receptor for Advanced Glycation End (RAGE)
3.1. Interaction with “Alarm” Receptors: Toll-Like Receptor (TLRs)
3.2. Binding to the Receptor RAGE
3.2.1. Extracellular Effects
3.2.2. Intracellular Mechanisms
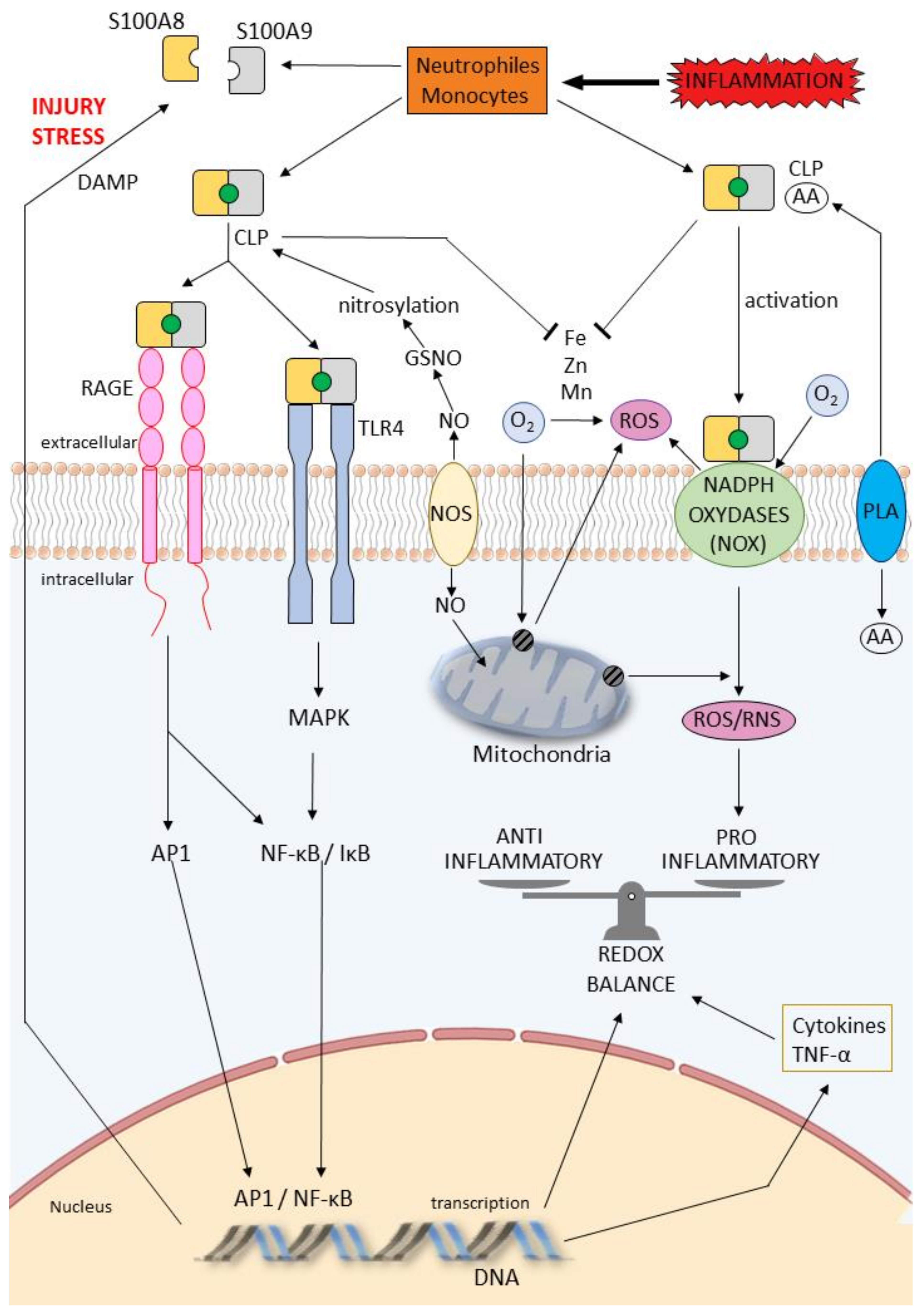
4. Calprotectin, Oxidative Stress and Redox Potential
4.1. Background on Oxidative Stress and Redox Potential
4.2. Effect of Calprotectin on Oxidative Stress and Redox Regulation
4.3. Calprotectin, Oxidative Stress and Viral Infection
4.4. S100 Proteins, Oxidative Stress and Transient Receptor Potential
5. Role of Calprotectin on Chemotaxis and Trans-Endothelial Migration
6. Calprotectin—A Pleiotropic Molecule in Acute and Chronic Inflammation
6.1. Anti-Inflammatory Properties of S100A8 and S100A9 Proteins
6.2. Modification of Proteins by S-Nitrosylation
6.3. Anti-Inflammatory Properties of Calprotectin
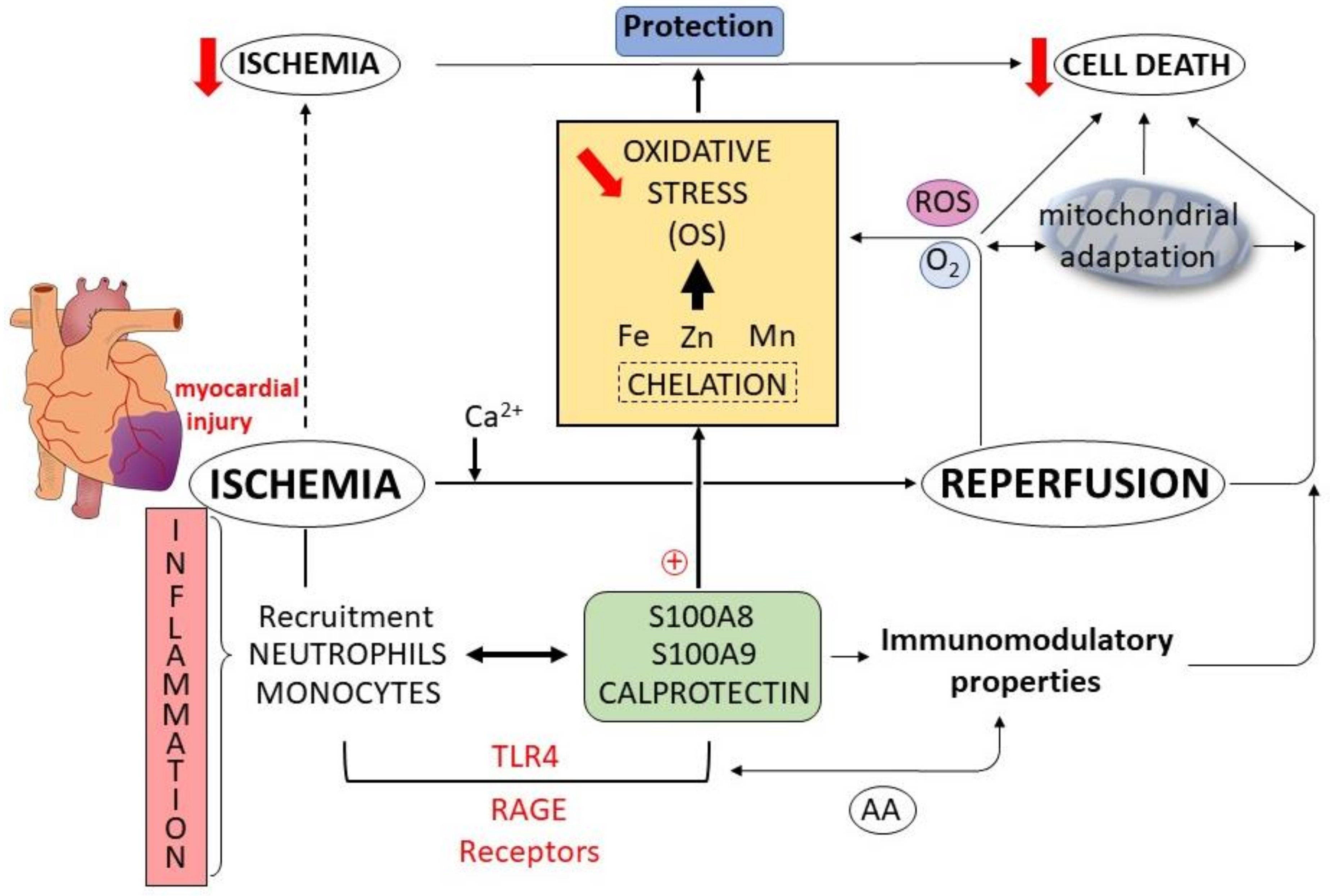
7. Calprotectin as Therapeutic Targets in Cardiac Disease
7.1. S100 Family Members and Cardioprotection
7.2. Interactions between S100 Proteins—Arachidonic Acid and Cardioprotection
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Austermann, J.; Zenker, S.; Roth, J. S100-alarmins: Potential therapeutic targets for arthritis. Expert Opin. Ther. Targets 2017, 21, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Stratis, A.; Wixler, V.; Völler, T.; Thurainayagam, S.; Jorch, S.K.; Zenker, S.; Dreiling, A.; Chakraborty, D.; Fröhling, M.; et al. Autoinhibitory regulation of S100A8/S100A9 alarmin activity locally restricts sterile inflammation. J. Clin. Investig. 2018, 128, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Cotoi, O.S.; Dunér, P.; Ko, N.; Hedblad, B.; Nilsson, J.; Björkbacka, H.; Schiopu, A. Plasma S100A8/A9 Correlates with Blood Neutrophil Counts, Traditional Risk Factors, and Cardiovascular Disease in Middle-Aged Healthy Individuals. Arter. Thromb. Vasc. Biol. 2014, 34, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, B.; Yang, X.; Zhang, C.; Jiao, Y.; Li, P.; Liu, Y.; Li, Z.; Qiao, B.; Lau, W.B.; et al. S100a8/a9 Signaling Causes Mitochondrial Dysfunction and Cardiomyocyte Death in Response to Ischemic/Reperfusion Injury. Circulation 2019, 140, 751–764. [Google Scholar] [CrossRef]
- Marinković, G.; Koenis, D.S.; De Camp, L.; Jablonowski, R.; Graber, N.; De Waard, V.; De Vries, C.J.; Goncalves, I.; Nilsson, J.; Jovinge, S.; et al. S100A9 Links Inflammation and Repair in Myocardial Infarction. Circ. Res. 2020, 127, 664–676. [Google Scholar] [CrossRef]
- Stríz, I.; Trebichavský, I. Calprotectin—A pleiotropic molecule in acute and chronic inflammation. Physiol. Res. 2004, 53, 245–253. [Google Scholar]
- Vogl, T.; Gharibyan, A.L.; Morozova-Roche, L.A. Pro-Inflammatory S100A8 and S100A9 Proteins: Self-Assembly into Multifunctional Native and Amyloid Complexes. Int. J. Mol. Sci. 2012, 13, 2893–2917. [Google Scholar] [CrossRef]
- Strupat, K.; Rogniaux, H.; Van Dorsselaer, A.; Roth, J.; Vogl, T. Calcium-induced noncovalently linked tetramers of MRP8 and MRP14 are confirmed by electrospray ionization-mass analysis. J. Am. Soc. Mass Spectrom. 2000, 11, 780–788. [Google Scholar] [CrossRef]
- Vogl, T.; Leukert, N.; Barczyk, K.; Strupat, K.; Roth, J. Biophysical characterization of S100A8 and S100A9 in the absence and presence of bivalent cations. Biochim. Biophys. Acta 2006, 1763, 1298–1306. [Google Scholar] [CrossRef]
- Nakashige, T.G.; Zhang, B.; Krebs, C.; Nolan, E.M. Human calprotectin is an iron-sequestering host-defense protein. Nat. Chem. Biol. 2015, 11, 765–771. [Google Scholar] [CrossRef]
- Freitas, M.; Porto, G.; Lima, J.L.F.C.; Fernandes, E. Zinc activates neutrophils’ oxidative burst. BioMetals 2010, 23, 31–41. [Google Scholar] [CrossRef]
- Nakashige, T.G.; Nolan, E.M. Human calprotectin affects the redox speciation of iron. Metallomics 2017, 9, 1086–1095. [Google Scholar] [CrossRef]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Redox Functions of Heme Oxygenase-1 and Biliverdin Reductase in Diabetes. Trends Endocrinol. Metab. 2018, 29, 74–85. [Google Scholar] [CrossRef]
- Steinckwich, N.; Schenten, V.; Melchior, C.; Bréchard, S.; Tschirhart, E.J. An Essential Role of STIM1, Orai1, and S100A8–A9 Proteins for Ca2+ Signaling and FcγR-Mediated Phagosomal Oxidative Activity. J. Immunol. 2011, 186, 2182–2191. [Google Scholar] [CrossRef]
- Heller, F.; Frischmann, S.; Grünbaum, M.; Zidek, W.; Westhoff, T.H. Urinary Calprotectin and the Distinction between Prerenal and Intrinsic Acute Kidney Injury. Clin. J. Am. Soc. Nephrol. 2011, 6, 2347–2355. [Google Scholar] [CrossRef]
- Jarlborg, M.; Courvoisier, D.S.; Lamacchia, C.; Prat, L.M.; Mahler, M.; Bentow, C.; Finckh, A.; Gabay, C.; Nissen, M.J.; on behalf of the Physicians of the Swiss Clinical Quality Management (SCQM) Registry. Serum calprotectin: A promising biomarker in rheumatoid arthritis and axial spondyloarthritis. Arthritis Res. Ther. 2020, 22, 105. [Google Scholar] [CrossRef]
- Yousefi, R.; Ardestani, S.K.; Saboury, A.A.; Kariminia, A.; Zeinali, M.; Amani, M. Investigation on the Surface Hydrophobicity and Aggregation Kinetics of Human Calprotectin in the Presence of Calcium. J. Biochem. Mol. Biol. 2005, 38, 407–413. [Google Scholar] [CrossRef]
- Schenten, V.; Bréchard, S.; Plançon, S.; Melchior, C.; Frippiat, J.-P.; Tschirhart, E.J. iPLA2, a novel determinant in Ca2+- and phosphorylation-dependent S100A8/A9 regulated NOX2 activity. Biochim. Biophys. Acta 2010, 1803, 840–847. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, M.X.; Meng, X.; Liu, F.Q.; Yu, G.S.; Zhang, C.; Sun, T.; Wang, X.P.; Li, L.; Wang, Y.Y.; et al. Atorvastatin suppresses LPS-induced rapid upregulation of Toll-like receptor 4 and its signaling pathway in endothelial cells. Am. J. Physiol. Circ. Physiol. 2011, 300, H1743–H1752. [Google Scholar] [CrossRef]
- Chan, J.K.; Roth, J.; Oppenheim, J.J.; Tracey, K.J.; Vogl, T.; Feldmann, M.; Horwood, N.; Nanchahal, J. Alarmins: Awaiting a clinical response. J. Clin. Investig. 2012, 122, 2711–2719. [Google Scholar] [CrossRef]
- Schattner, M. Platelet TLR4 at the crossroads of thrombosis and the innate immune response. J. Leukoc. Biol. 2018, 105, 873–880. [Google Scholar] [CrossRef]
- Hally, K.; Fauteux-Daniel, S.; Hamzeh-Cognasse, H.; Larsen, P.; Cognasse, F. Revisiting Platelets and Toll-Like Receptors (TLRs): At the Interface of Vascular Immunity and Thrombosis. Int. J. Mol. Sci. 2020, 21, 6150. [Google Scholar] [CrossRef]
- Lecour, S.; Chevet, D.; Maupoil, V.; Moisant, M.; Bernard, C.; Zahnd, J.P.; Touchard, G.; Briot, F.; Rochette, L. Intrarenal Detection of Nitric Oxide Using Electron Spin Resonance Spectroscopy in Hypertensive Lipopolysaccharide-Treated Rats. J. Cardiovasc. Pharmacol. 2002, 40, 9–17. [Google Scholar] [CrossRef]
- Rochette, L.; Lorin, J.; Zeller, M.; Guilland, J.-C.; Lorgis, L.; Cottin, Y.; Vergely, C. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: Possible therapeutic targets? Pharmacol. Ther. 2013, 140, 239–257. [Google Scholar] [CrossRef]
- Grylls, A.; Seidler, K.; Neil, J. Link between microbiota and hypertension: Focus on LPS/TLR4 pathway in endothelial dysfunction and vascular inflammation, and therapeutic implication of probiotics. Biomed. Pharmacother. 2021, 137, 111334. [Google Scholar] [CrossRef]
- Yan, S.F.; Barile, G.R.; D'Agati, V.; Du Yan, S.; Ramasamy, R.; Schmidt, A.M. The biology of RAGE and its ligands: Uncovering mechanisms at the heart of diabetes and its complications. Curr. Diabetes Rep. 2007, 7, 146–153. [Google Scholar] [CrossRef]
- Guarneri, F.; Custurone, P.; Papaianni, V.; Gangemi, S. Involvement of RAGE and Oxidative Stress in Inflammatory and Infectious Skin Diseases. Antioxidants 2021, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Moskalev, A.; Stambler, I.; Caruso, C. Innate and Adaptive Immunity in Aging and Longevity: The Foundation of Resilience. Aging Dis. 2020, 11, 1363–1373. [Google Scholar] [CrossRef]
- Donato, R. Intracellular and extracellular roles of S100 proteins. Microsc. Res. Tech. 2003, 60, 540–551. [Google Scholar] [CrossRef]
- Schenten, V.; Plançon, S.; Jung, N.; Hann, J.; Bueb, J.-L.; Bréchard, S.; Tschirhart, E.J.; Tolle, F. Secretion of the Phosphorylated Form of S100A9 from Neutrophils Is Essential for the Proinflammatory Functions of Extracellular S100A8/A9. Front. Immunol. 2018, 9, 447. [Google Scholar] [CrossRef]
- Jung, N.; Schenten, V.; Bueb, J.-L.; Tolle, F.; Bréchard, S. miRNAs Regulate Cytokine Secretion Induced by Phosphorylated S100A8/A9 in Neutrophils. Int. J. Mol. Sci. 2019, 20, 5699. [Google Scholar] [CrossRef] [PubMed]
- Gudjoncik, A.; Guenancia, C.; Zeller, M.; Cottin, Y.; Vergely, C.; Rochette, L. Iron, oxidative stress, and redox signaling in the cardiovascular system. Mol. Nutr. Food Res. 2014, 58, 1721–1738. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Diabetes, oxidative stress and therapeutic strategies. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 2709–2729. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Gudjoncik, A.; Guenancia, C.; Zeller, M.; Cottin, Y.; Vergely, C. The iron-regulatory hormone hepcidin: A possible therapeutic target? Pharmacol. Ther. 2015, 146, 35–52. [Google Scholar] [CrossRef]
- Diaz-Ochoa, V.E.; Ejellbauer, S.; Eklaus, S.; Eraffatellu, M. Transition metal ions at the crossroads of mucosal immunity and microbial pathogenesis. Front. Cell. Infect. Microbiol. 2014, 4, 2. [Google Scholar] [CrossRef]
- Weiss, G. Metals: Calprotectin and iron match up. Nat. Chem. Biol. 2015, 11, 756–757. [Google Scholar] [CrossRef]
- Gagnon, D.M.; Brophy, M.B.; Bowman, S.E.J.; Stich, T.A.; Drennan, C.L.; Britt, R.D.; Nolan, E.M. Manganese Binding Properties of Human Calprotectin under Conditions of High and Low Calcium: X-ray Crystallographic and Advanced Electron Paramagnetic Resonance Spectroscopic Analysis. J. Am. Chem. Soc. 2015, 137, 3004–3016. [Google Scholar] [CrossRef]
- Hoskin, T.S.; Crowther, J.; Cheung, J.; Epton, M.; Sly, P.D.; Elder, P.A.; Dobson, R.C.; Kettle, T.; Dickerhof, N. Oxidative cross-linking of calprotectin occurs in vivo, altering its structure and susceptibility to proteolysis. Redox Biol. 2019, 24, 101202. [Google Scholar] [CrossRef]
- Zygiel, E.M.; Nolan, E.M. Transition Metal Sequestration by the Host-Defense Protein Calprotectin. Annu. Rev. Biochem. 2018, 87, 621–643. [Google Scholar] [CrossRef]
- Babior, B.M. NADPH oxidase. Curr. Opin. Immunol. 2004, 16, 42–47. [Google Scholar] [CrossRef]
- Monteith, A.J.; Skaar, E.P. The impact of metal availability on immune function during infection. Trends Endocrinol. Metab. 2021, 32, 916–928. [Google Scholar] [CrossRef]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. GDF15: An emerging modulator of immunity and a strategy in COVID-19 in association with iron metabolism. Trends Endocrinol. Metab. 2021, 32, 875–889. [Google Scholar] [CrossRef]
- Sharif-Askari, N.S.; Sharif-Askari, F.S.; Mdkhana, B.; Alsayed, H.A.H.; Alsafar, H.; Alrais, Z.F.; Hamid, Q.; Halwani, R. Upregulation of oxidative stress gene markers during SARS-COV-2 viral infection. Free Radic. Biol. Med. 2021, 172, 688–698. [Google Scholar] [CrossRef]
- Zielińska, W.; Zabrzyński, J.; Gagat, M.; Grzanka, A. The Role of TRPM2 in Endothelial Function and Dysfunction. Int. J. Mol. Sci. 2021, 22, 7635. [Google Scholar] [CrossRef]
- Kozai, D.; Ogawa, N.; Mori, Y. Redox Regulation of Transient Receptor Potential Channels. Antioxid. Redox Signal. 2014, 21, 971–986. [Google Scholar] [CrossRef]
- Pires, P.W.; Earley, S. Redox regulation of transient receptor potential channels in the endothelium. Microcirculation 2017, 24, e12329. [Google Scholar] [CrossRef]
- Zouharova, M.; Herman, P.; Hofbauerová, K.; Vondrasek, J.; Bousova, K. TRPM6 N-Terminal CaM- and S100A1-Binding Domains. Int. J. Mol. Sci. 2019, 20, 4430. [Google Scholar] [CrossRef]
- Kannan, S. Inflammation: A novel mechanism for the transport of extracellular nucleotide-induced arachidonic acid by S100A8/A9 for transcellular metabolism. Cell Biol. Int. 2003, 27, 593–595. [Google Scholar] [CrossRef]
- Vandal, K.; Rouleau, P.; Boivin, A.; Ryckman, C.; Talbot, M.; Tessier, P. Blockade of S100A8 and S100A9 Suppresses Neutrophil Migration in Response to Lipopolysaccharide. J. Immunol. 2003, 171, 2602–2609. [Google Scholar] [CrossRef]
- Viemann, D.; Strey, A.; Janning, A.; Jurk, K.; Klimmek, K.; Vogl, T.; Hirono, K.; Ichida, F.; Foell, D.; Kehrel, B.; et al. Myeloid-related proteins 8 and 14 induce a specific inflammatory response in human microvascular endothelial cells. Blood 2005, 105, 2955–2962. [Google Scholar] [CrossRef]
- Frangogiannis, N.; Smith, C.; Entman, M.L. The inflammatory response in myocardial infarction. Cardiovasc. Res. 2002, 53, 31–47. [Google Scholar] [CrossRef]
- Ryckman, C.; Vandal, K.; Rouleau, P.; Talbot, M.; Tessier, P. Proinflammatory Activities of S100: Proteins S100A8, S100A9, and S100A8/A9 Induce Neutrophil Chemotaxis and Adhesion. J. Immunol. 2003, 170, 3233–3242. [Google Scholar] [CrossRef]
- Basta, G. Receptor for advanced glycation endproducts and atherosclerosis: From basic mechanisms to clinical implications. Atherosclerosis 2008, 196, 9–21. [Google Scholar] [CrossRef]
- Pruenster, M.; Kurz, A.; Chung, K.-J.; Cao-Ehlker, X.; Bieber, S.; Nussbaum, C.F.; Bierschenk, S.; Eggersmann, T.K.; Rohwedder, I.; Heinig, K.; et al. Extracellular MRP8/14 is a regulator of β2 integrin-dependent neutrophil slow rolling and adhesion. Nat. Commun. 2015, 6, 6915. [Google Scholar] [CrossRef]
- Sabapathy, V.; Venkatadri, R.; Dogan, M.; Sharma, R. The Yin and Yang of Alarmins in Regulation of Acute Kidney Injury. Front. Med. 2020, 7, 441. [Google Scholar] [CrossRef] [PubMed]
- Sreejit, G.; Flynn, M.C.; Patil, M.; Krishnamurthy, P.; Murphy, A.J.; Nagareddy, P.R. S100 family proteins in inflammation and beyond. Adv. Clin. Chem. 2020, 98, 173–231. [Google Scholar] [CrossRef]
- Harman, J.L.; Loes, A.N.; Warren, G.D.; Heaphy, M.C.; Lampi, K.J.; Harms, M.J. Evolution of multifunctionality through a pleiotropic substitution in the innate immune protein S100A9. eLife 2020, 9, e54100. [Google Scholar] [CrossRef]
- Lim, S.Y.; Raftery, M.J.; Goyette, J.; Hsu, K.; Geczy, C.L. Oxidative modifications of S100 proteins: Functional regulation by redox. J. Leukoc. Biol. 2009, 86, 577–587. [Google Scholar] [CrossRef]
- Jiang, L.; Shao, Y.; Tian, Y.; Ouyang, C.; Wang, X. Nuclear Alarmin Cytokines in Inflammation. J. Immunol. Res. 2020, 2020, 7206451. [Google Scholar] [CrossRef]
- Lim, S.Y.; Raftery, M.; Cai, H.; Hsu, K.; Yan, W.X.; Hseih, H.-L.; Watts, R.N.; Richardson, D.; Thomas, S.; Perry, M.; et al. S-Nitrosylated S100A8: Novel Anti-Inflammatory Properties. J. Immunol. 2008, 181, 5627–5636. [Google Scholar] [CrossRef]
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404. [Google Scholar] [CrossRef] [PubMed]
- Goyette, J.; Geczy, C.L. Inflammation-associated S100 proteins: New mechanisms that regulate function. Amino Acids 2010, 41, 821–842. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Yang, C.; Qu, S.-L.; Shao, Y.-D.; Zhou, C.-Y.; Chao, R.; Huang, L.; Zhang, C. S100 proteins in atherosclerosis. Clin. Chim. Acta 2020, 502, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Ikemoto, M.; Murayama, H.; Itoh, H.; Totani, M.; Fujita, M. Intrinsic function of S100A8/A9 complex as an anti-inflammatory protein in liver injury induced by lipopolysaccharide in rats. Clin. Chim. Acta 2006, 376, 197–204. [Google Scholar] [CrossRef]
- Otsuka, K.; Terasaki, F.; Ikemoto, M.; Fujita, S.; Tsukada, B.; Katashima, T.; Kanzaki, Y.; Sohmiya, K.; Kono, T.; Toko, H.; et al. Suppression of inflammation in rat autoimmune myocarditis by S100A8/A9 through modulation of the proinflammatory cytokine network. Eur. J. Heart Fail. 2009, 11, 229–237. [Google Scholar] [CrossRef]
- De Jong, H.K.; Achouiti, A.; Koh, G.C.K.W.; Parry, C.M.; Baker, S.; Faiz, M.A.; van Dissel, J.T.; Vollaard, A.M.; van Leeuwen, E.M.M.; Roelofs, J.J.T.H.; et al. Expression and Function of S100A8/A9 (Calprotectin) in Human Typhoid Fever and the Murine Salmonella Model. PLoS Negl. Trop. Dis. 2015, 9, e0003663. [Google Scholar] [CrossRef]
- Zhang, W.; Lavine, K.J.; Epelman, S.; Evans, S.A.; Weinheimer, C.J.; Barger, P.M.; Mann, D.L. Necrotic Myocardial Cells Release Damage-Associated Molecular Patterns That Provoke Fibroblast Activation In Vitro and Trigger Myocardial Inflammation and Fibrosis In Vivo. J. Am. Heart Assoc. 2015, 4, e001993. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef]
- Cai, X.Y.; Lu, L.; Wang, Y.N.; Jin, C.; Zhang, R.Y.; Zhang, Q.; Chen, Q.J.; Shen, W.F. Association of increased S100B, S100A6 and S100P in serum levels with acute coronary syndrome and also with the severity of myocardial infarction in cardiac tissue of rat models with ischemia–reperfusion injury. Atherosclerosis 2011, 217, 536–542. [Google Scholar] [CrossRef]
- Gebhardt, C.; Riehl, A.; Durchdewald, M.; Németh, J.; Fürstenberger, G.; Müller-Decker, K.; Enk, A.; Arnold, B.; Bierhaus, A.; Nawroth, P.P.; et al. RAGE signaling sustains inflammation and promotes tumor development. J. Exp. Med. 2008, 205, 275–285. [Google Scholar] [CrossRef]
- Riva, M.; Källberg, E.; Björk, P.; Hancz, D.; Vogl, T.; Roth, J.; Ivars, F.; Leanderson, T. Induction of nuclear factor-κB responses by the S100A9 protein is Toll-like receptor-4-dependent. Immunology 2012, 137, 172–182. [Google Scholar] [CrossRef]
- Zhong, A.; Xu, W.; Zhao, J.; Xie, P.; Jia, S.; Sun, J.; Galiano, R.D.; Mustoe, T.A.; Hong, S.J. S100A8 and S100A9 Are Induced by Decreased Hydration in the Epidermis and Promote Fibroblast Activation and Fibrosis in the Dermis. Am. J. Pathol. 2016, 186, 109–122. [Google Scholar] [CrossRef]
- Boyd, J.H.; Kan, B.; Roberts, H.; Wang, Y.; Walley, K.R. S100A8 and S100A9 Mediate Endotoxin-Induced Cardiomyocyte Dysfunction via the Receptor for Advanced Glycation End Products. Circ. Res. 2008, 102, 1239–1246. [Google Scholar] [CrossRef]
- Imbalzano, E.; Mandraffino, G.; Casciaro, M.; Quartuccio, S.; Saitta, A.; Gangemi, S. Pathophysiological mechanism and therapeutic role of S100 proteins in cardiac failure: A systematic review. Heart Fail. Rev. 2016, 21, 463–473. [Google Scholar] [CrossRef]
- Ma, L.-P.; Haugen, E.; Ikemoto, M.; Fujita, M.; Terasaki, F.; Fu, M. S100A8/A9 complex as a new biomarker in prediction of mortality in elderly patients with severe heart failure. Int. J. Cardiol. 2011, 155, 26–32. [Google Scholar] [CrossRef]
- Wang, W.; Asp, M.L.; Guerrero-Serna, G.; Metzger, J.M. Differential effects of S100 proteins A2 and A6 on cardiac Ca2+ cycling and contractile performance. J. Mol. Cell. Cardiol. 2014, 72, 117–125. [Google Scholar] [CrossRef][Green Version]
- Baumann, M.; Sollinger, D.; Roos, M.; Lutz, J.; Heemann, U. Prehypertensive Preconditioning Improves Adult Antihypertensive and Cardioprotective Treatment. J. Pharmacol. Exp. Ther. 2010, 332, 1121–1126. [Google Scholar] [CrossRef]
- Yang, N.K.; Choi, B.Y.; Lee, Y.-H.; Kim, Y.-G.; Cho, M.-C.; Hong, S.-E.; Kim, H.; Hajjar, R.J.; Park, W.J. Gene profiling during regression of pressure overload-induced cardiac hypertrophy. Physiol. Genom. 2007, 30, 1–7. [Google Scholar] [CrossRef]
- Wei, X.; Wu, B.; Zhao, J.; Zeng, Z.; Xuan, W.; Cao, S.; Huang, X.; Asakura, M.; Xu, D.; Bin, J.; et al. Myocardial Hypertrophic Preconditioning Attenuates Cardiomyocyte Hypertrophy and Slows Progression to Heart Failure Through Upregulation of S100A8/A9. Circulation 2015, 131, 1506–1517. [Google Scholar] [CrossRef]
- Takano, A.P.C.; Munhoz, C.; Moriscot, A.S.; Gupta, S.; Barreto-Chaves, M.L.M. S100A8/MYD88/NF-κB: A novel pathway involved in cardiomyocyte hypertrophy driven by thyroid hormone. Klin. Wochenschr. 2017, 95, 671–682. [Google Scholar] [CrossRef]
- Fujiu, K.; Shibata, M.; Nakayama, Y.; Ogata, F.; Matsumoto, S.; Noshita, K.; Iwami, S.; Nakae, S.; Komuro, I.; Nagai, R.; et al. A heart–brain–kidney network controls adaptation to cardiac stress through tissue macrophage activation. Nat. Med. 2017, 23, 611–622. [Google Scholar] [CrossRef]
- Sugita, J.; Fujiu, K.; Nakayama, Y.; Matsubara, T.; Matsuda, J.; Oshima, T.; Liu, Y.; Maru, Y.; Hasumi, E.; Kojima, T.; et al. Cardiac macrophages prevent sudden death during heart stress. Nat. Commun. 2021, 12, 1910. [Google Scholar] [CrossRef]
- Trostchansky, A.; Bonilla, L.; González-Perilli, L.; Rubbo, H. Nitro-Fatty Acids: Formation, Redox Signaling, and Therapeutic Potential. Antioxid. Redox Signal. 2013, 19, 1257–1265. [Google Scholar] [CrossRef]
- Kerkhoff, C.; Vogl, T.; Nacken, W.; Sopalla, C.; Sorg, C. Zinc binding reverses the calcium-induced arachidonic acid-binding capacity of the S100A8/A9 protein complex. FEBS Lett. 1999, 460, 134–138. [Google Scholar] [CrossRef]
- De Caterina, R.; Liao, J.K.; Libby, P. Fatty acid modulation of endothelial activation. Am. J. Clin. Nutr. 2000, 71, 213S–223S. [Google Scholar] [CrossRef]
- Baker, E.J.; Yusof, H.M.; Yaqoob, P.; Miles, E.; Calder, P.C. Omega-3 fatty acids and leukocyte-endothelium adhesion: Novel anti-atherosclerotic actions. Mol. Asp. Med. 2018, 64, 169–181. [Google Scholar] [CrossRef]
- Kerkhoff, C.; Nacken, W.; Benedyk, M.; Dagher, M.C.; Sopalla, C.; Doussiere, J. The arachidonic acid-binding protein S100A8/A9 promotes NADPH oxidase activation by interaction with p67phoxand Rac-2. FASEB J. 2004, 19, 467–469. [Google Scholar] [CrossRef]
- Sreejit, G.; Abdel-Latif, A.; Athmanathan, B.; Annabathula, R.; Dhyani, A.; Noothi, S.K.; Quaife-Ryan, G.; Al-Sharea, A.; Pernes, G.; Dragoljevic, D.; et al. Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction. Circulation 2020, 141, 1080–1094. [Google Scholar] [CrossRef]
- Gao, S.; Yang, Y.; Fu, Y.; Guo, W.; Liu, G. Diagnostic and prognostic value of myeloid-related protein complex 8/14 for sepsis. Am. J. Emerg. Med. 2015, 33, 1278–1282. [Google Scholar] [CrossRef]
- Bouma, G.; Lam-Tse, W.K.; Wierenga-Wolf, A.F.; Drexhage, H.A.; Versnel, M.A. Increased Serum Levels of MRP-8/14 in Type 1 Diabetes Induce an Increased Expression of CD11b and an Enhanced Adhesion of Circulating Monocytes to Fibronectin. Diabetes 2004, 53, 1979–1986. [Google Scholar] [CrossRef][Green Version]
- Hirata, A.; Kishida, K.; Nakatsuji, H.; Hiuge-Shimizu, A.; Funahashi, T.; Shimomura, I. High serum S100A8/A9 levels and high cardiovascular complication rate in type 2 diabetics with ultrasonographic low carotid plaque density. Diabetes Res. Clin. Pract. 2012, 97, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Padilla, L.; Dakhel, S.; Adan, J.; Masa, M.; Martinez, J.M.; Roque, L.; Coll, T.; Hervas, R.; Calvis, C.; Llinas, L.; et al. S100A7: From mechanism to cancer therapy. Oncogene 2017, 36, 6749–6761. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rochette, L.; Dogon, G.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. Involvement of Oxidative Stress in Protective Cardiac Functions of Calprotectin. Cells 2022, 11, 1226. https://doi.org/10.3390/cells11071226
Rochette L, Dogon G, Rigal E, Zeller M, Cottin Y, Vergely C. Involvement of Oxidative Stress in Protective Cardiac Functions of Calprotectin. Cells. 2022; 11(7):1226. https://doi.org/10.3390/cells11071226
Chicago/Turabian StyleRochette, Luc, Geoffrey Dogon, Eve Rigal, Marianne Zeller, Yves Cottin, and Catherine Vergely. 2022. "Involvement of Oxidative Stress in Protective Cardiac Functions of Calprotectin" Cells 11, no. 7: 1226. https://doi.org/10.3390/cells11071226
APA StyleRochette, L., Dogon, G., Rigal, E., Zeller, M., Cottin, Y., & Vergely, C. (2022). Involvement of Oxidative Stress in Protective Cardiac Functions of Calprotectin. Cells, 11(7), 1226. https://doi.org/10.3390/cells11071226