
The Interplay between Autophagy and Redox Signaling in Cardiovascular Diseases


Abstract
:1. Introduction
2. Nrf-2 Redox Signaling in Cell Responses to Oxidative Stress
Nrf-2 Redox Signaling in Myocardium
3. Autophagy
3.1. Macro-Autophagy
3.2. Chaperon-Mediated Autophagy
3.3. Micro-Autophagy and Mitophagy
3.4. Autophagy in Myocardium
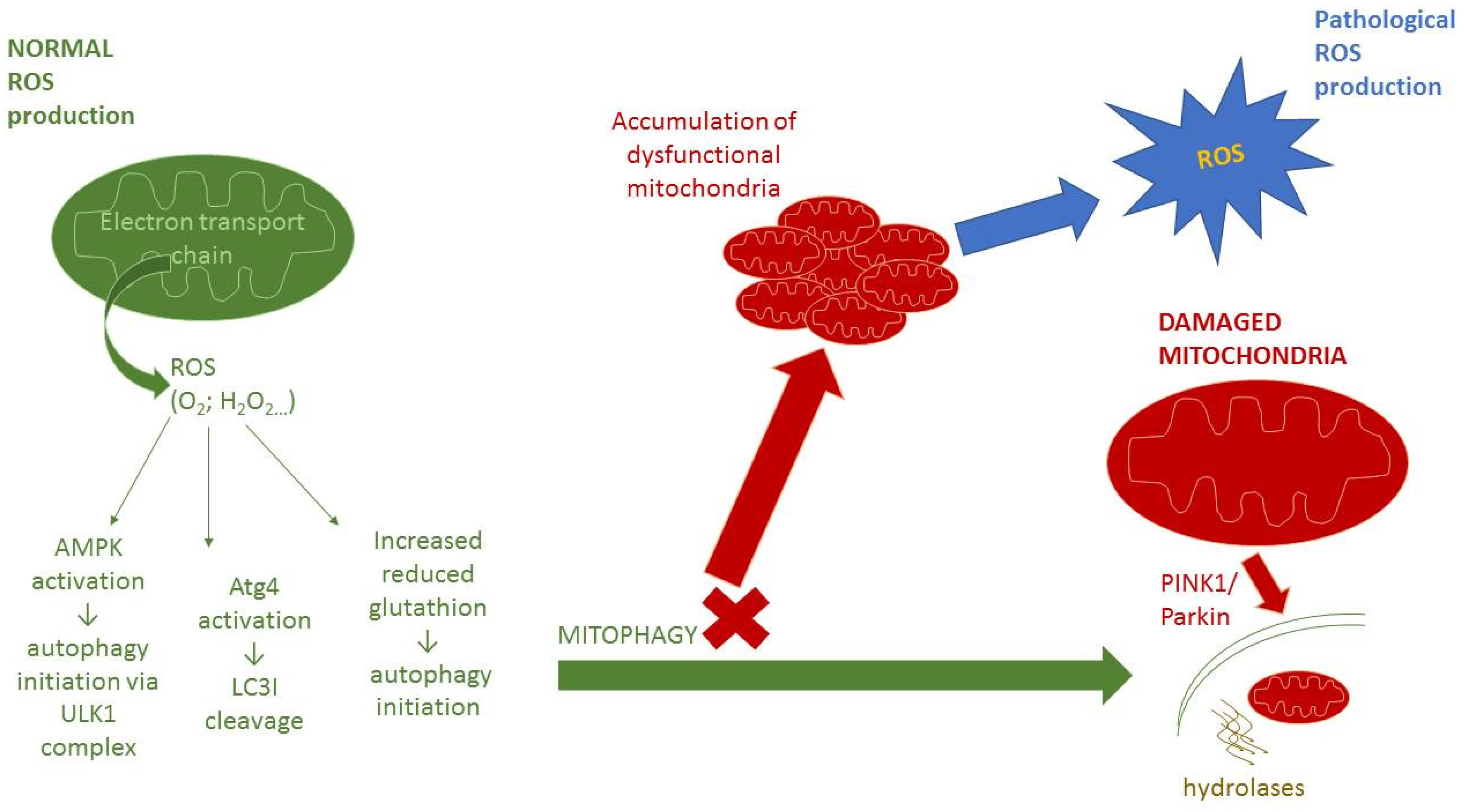
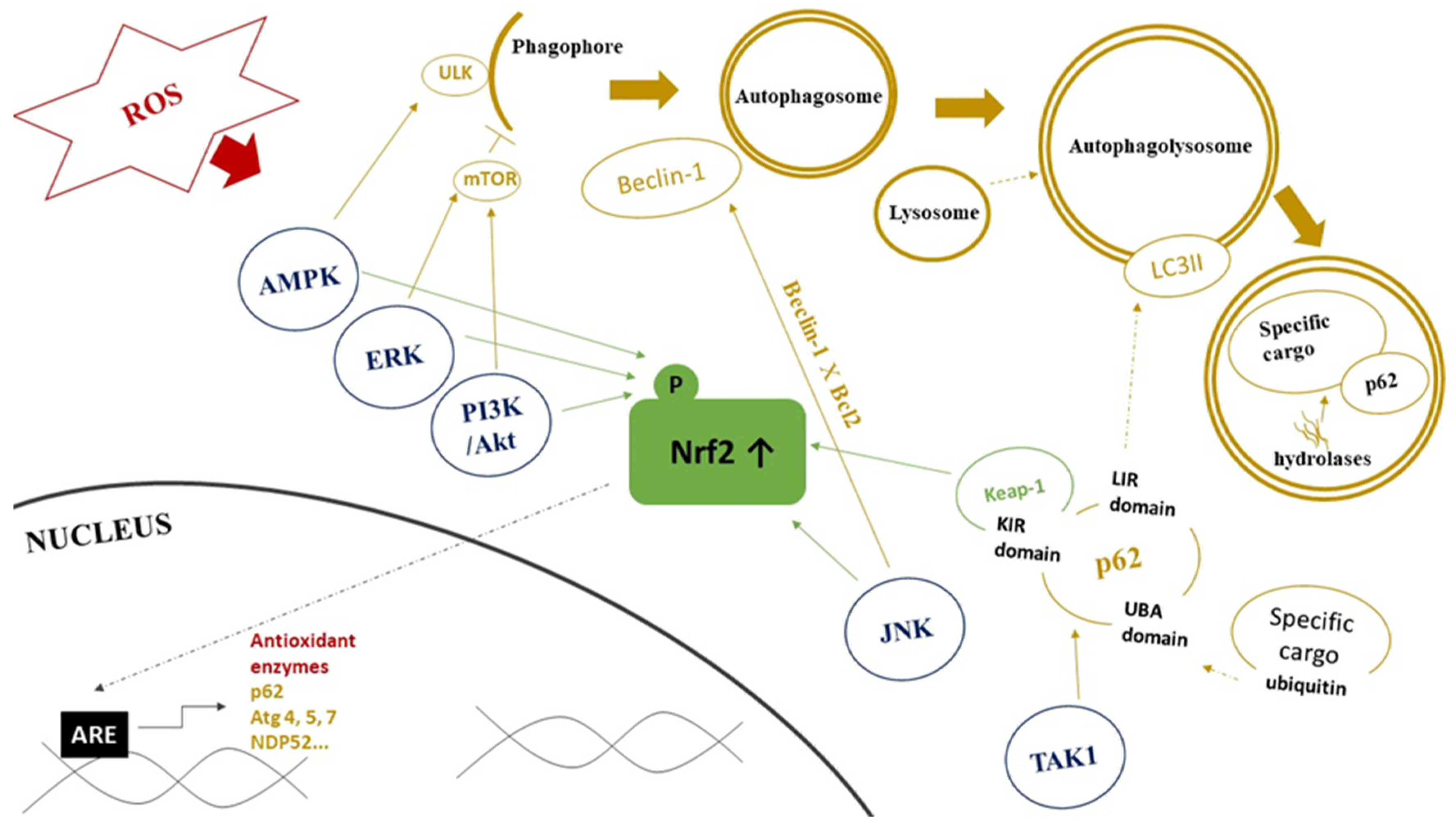
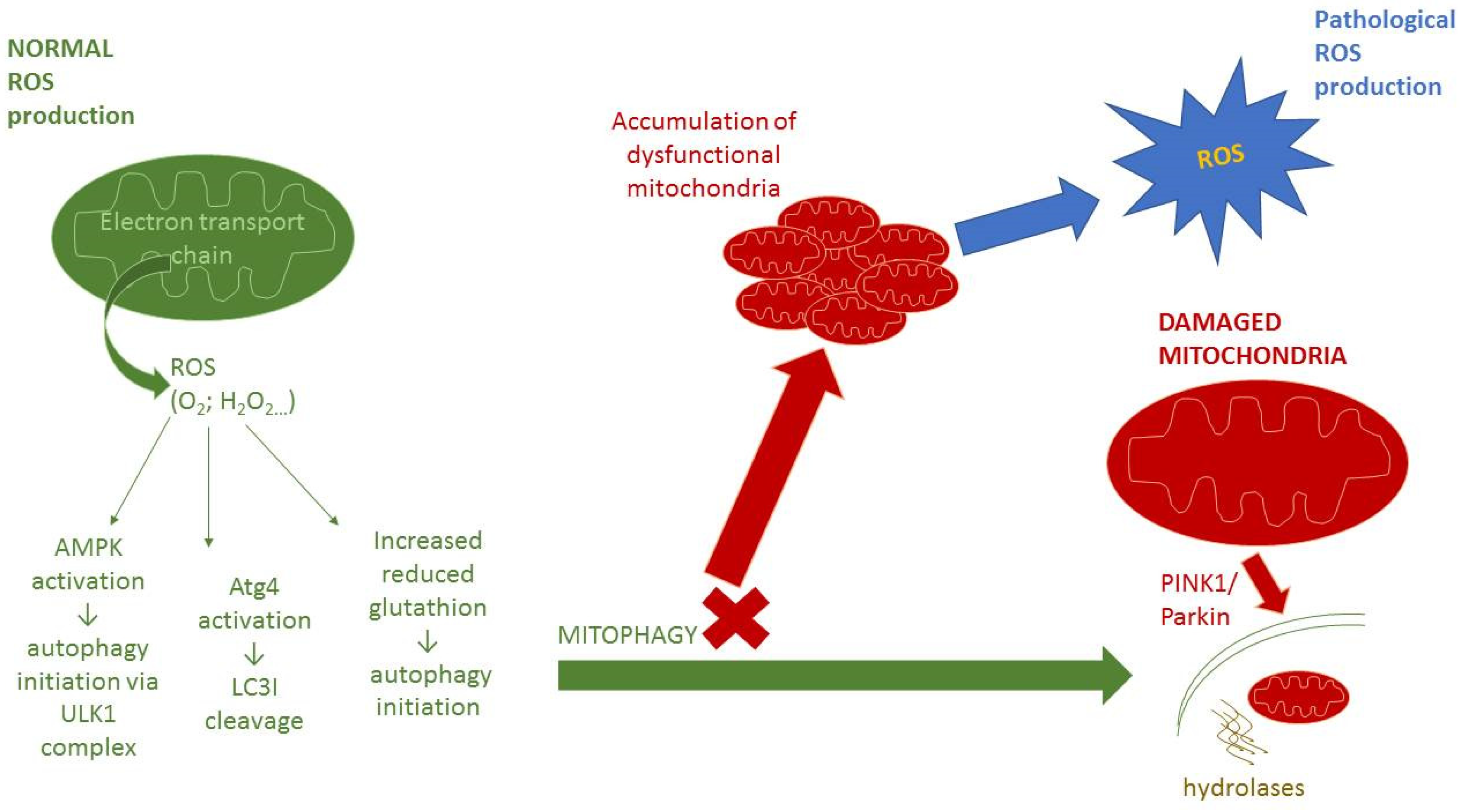
4. Interplay between Autophagy and Redox Signaling
5. Interplay between Autophagy and Redox Signaling in Cardiovascular Diseases
5.1. Interplay between Nrf2 Redox Signaling and Autophagy in Ischemic Heart Disease
5.2. Interplay between Nrf2 Redox Signaling and Autophagy in Cardiomyopathies
5.3. Interplay between Nrf2 Redox Signaling and Autophagy in Heart Failure
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Da Costa, R.M.; Fais, R.S.; Dechandt, C.R.P.; Louzada-Junior, P.; Alberici, L.C.; Lobato, N.S.; Tostes, R.C. Increased mitochondrial ROS generation mediates the loss of the anti-contractile effects of perivascular adipose tissue in high-fat diet obese mice. Br. J. Pharmacol. 2017, 174, 3527–3541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, G.; Din, J.U.; Zhao, F.; Liu, X. Effect of soybean peptides against hydrogen peroxide induced oxidative stress in HepG2 cells via Nrf2 signaling. Food Funct. 2020, 11, 2725–2737. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.-H.; Yan, X.-Y.; Zhou, L.; Xu, L.; Zhang, L.-C.; Yi, H.-W.; Su, J. p62 Suppressed VK3-induced Oxidative Damage Through Keap1/Nrf2 Pathway In Human Ovarian Cancer Cells. J. Cancer 2020, 11, 1299–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barančík, M.; Grešová, L.; Bartekova, M.; Dovinova, I. Nrf2 as a Key Player of Redox Regulation in Cardiovascular Diseases. Physiol. Res. 2016, S1–S10. [Google Scholar] [CrossRef]
- Papp, D.; Lenti, K.; Modos, D.; Fazekas, D.; Dúl, Z.; Turei, D.; Földvári-Nagy, L.; Nussinov, R.; Csermely, P.; Korcsmáros, T. The NRF2-related interactome and regulome contain multifunctional proteins and fine-tuned autoregulatory loops. FEBS Lett. 2012, 586, 1795–1802. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Li, W.; Su, Z.-Y.; Kong, A.-N.T. The complexity of the Nrf2 pathway: Beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413. [Google Scholar] [CrossRef]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef]
- Sun, B.; Xu, V.; Liu, Z.-Y.; Meng, W.-X.; Yang, H. Autophagy assuages myocardial infarction through Nrf2 signaling activa-tion-mediated reactive oxygen species clear. Eur. Rev. Med. Pharm. Sci. 2020, 24, 7381–7390. [Google Scholar]
- Yun, H.R.; Jo, Y.H.; Kim, J.; Shin, Y.; Kim, S.S.; Choi, T.G. Roles of Autophagy in Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 3289. [Google Scholar] [CrossRef]
- Pajares, M.; Cuadrado, A.; Engedal, N.; Jirsova, Z.; Cahova, M. The Role of Free Radicals in Autophagy Regulation: Implications for Ageing. Oxidative Med. Cell. Longev. 2018, 2018, 2450748. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell. Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönfeld, P.; Dymkowska, D.; Wojtczak, L. Acyl-CoA-induced generation of reactive oxygen species in mitochondrial preparations is due to the presence of peroxisomes. Free Radic. Biol. Med. 2009, 47, 503–509. [Google Scholar] [CrossRef]
- Liu, Q.; Berchner-Pfannschmidt, U.; Möller, U.; Brecht, M.; Wotzlaw, C.; Acker, H.; Jungermann, K.; Kietzmann, T. A Fenton reaction at the endoplasmic reticulum is involved in the redox control of hypoxia-inducible gene expression. Proc. Natl. Acad. Sci. USA 2004, 101, 4302–4307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, H.H.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.; Daiber, A.; Cuadrado, A. Antioxidants in Translational Medicine. Antioxid. Redox Signal. 2015, 23, 1130–1143. [Google Scholar] [CrossRef] [Green Version]
- Miersch, S.; Espey, M.G.; Chaube, R.; Akarca, A.; Tweten, R.; Ananvoranich, S.; Mutus, B. Plasma Membrane Cholesterol Content Affects Nitric Oxide Diffusion Dynamics and Signaling. J. Biol. Chem. 2008, 283, 18513–18521. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P. Redefining Oxidative Stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Elazar, Z. Oxidation as a post-translational modification that regulates autophagy. Autophagy 2007, 3, 371–373. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J. Keap1-dependent Proteasomal Degradation of Transcription Factor Nrf2 Contributes to the Negative Regulation of Antioxidant Response Element-driven Gene Expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [Green Version]
- Sajadimajd, S.; Khazaei, M. Oxidative Stress and Cancer: The Role of Nrf2. Curr. Cancer Drug Targets 2018, 18, 538–557. [Google Scholar] [CrossRef]
- Yuan, H.; Xu, Y.; Luo, Y.; Wang, N.-X.; Xiao, J.-H. Role of Nrf2 in cell senescence regulation. Mol. Cell. Biochem. 2020, 476, 247–259. [Google Scholar] [CrossRef] [PubMed]
- A Grimes, C.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2012, 85, 705–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen Synthase Kinase-3β Inhibits the Xenobiotic and Antioxidant Cell Response by Direct Phosphorylation and Nuclear Exclusion of the Transcription Factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo, A.I.; De Sagarra, M.R.; Cuadrado, A. GSK-3β down-regulates the transcription factor Nrf2 after oxidant damage: Relevance to exposure of neuronal cells to oxidative stress. J. Neurochem. 2008, 105, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Huang, Z.; Zhang, D.D. Phosphorylation of Nrf2 at Multiple Sites by MAP Kinases Has a Limited Contribution in Modulating the Nrf2-Dependent Antioxidant Response. PLoS ONE 2009, 4, e6588. [Google Scholar] [CrossRef] [Green Version]
- Keum, Y.-S.; Yu, S.; Chang, P.P.-J.; Yuan, X.; Kim, J.H.; Xu, C.; Han, J.; Agarwal, A.; Kong, A.-N.T. Mechanism of Action of Sulforaphane: Inhibition of p38 Mitogen-Activated Protein Kinase Isoforms Contributing to the Induction of Antioxidant Response Element–Mediated Heme Oxygenase-1 in Human Hepatoma HepG2 Cells. Cancer Res. 2006, 66, 8804–8813. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; Bai, Y.; Jiang, X.; Zhou, S.; Wang, Y.; Wintergerst, K.A.; Cui, T.; Ji, H.; Tan, Y.; Cai, L. Sulforaphane prevents angiotensin II-induced cardiomyopathy by activation of Nrf2 via stimulating the Akt/GSK-3ß/Fyn pathway. Redox Biol. 2018, 15, 405–417. [Google Scholar] [CrossRef]
- Li, S.; Wang, W.; Niu, T.; Wang, H.; Li, B.; Shao, L.; Lai, Y.; Li, H.; Janicki, J.S.; Wang, X.L.; et al. Nrf2 Deficiency Exaggerates Doxorubicin-Induced Cardiotoxicity and Cardiac Dysfunction. Oxidative Med. Cell. Longev. 2014, 2014, 748524. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Li, S.; Wang, H.; Li, B.; Shao, L.; Lai, Y.; Horvath, G.; Wang, Q.; Yamamoto, M.; Janicki, J.S.; et al. Nrf2 enhances myocardial clearance of toxic ubiquitinated proteins. J. Mol. Cell. Cardiol. 2014, 72, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.-F.; Yao, J.; Gao, S.-G.; Wang, X.-S.; Peng, X.-Q.; Yang, Y.-T.; Feng, X.-S. Nrf2 Overexpression Predicts Prognosis and 5-FU Resistance in Gastric Cancer. Asian Pac. J. Cancer Prev. 2013, 14, 5231–5235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.Z.A.; Zhao, D.; Hussain, T.; Sabir, N.; Mangi, M.H.; Yang, L. p62-Keap1-NRF2-ARE Pathway: A Contentious Player for Selective Targeting of Autophagy, Oxidative Stress and Mitochondrial Dysfunction in Prion Diseases. Front. Mol. Neurosci. 2018, 11, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, C.-H.; Zhang, J.-T.; Yang, G.-J.; Liu, H.; Han, Q.-B.; Ma, D.-L. Emerging Screening Approaches in the Development of Nrf2–Keap1 Protein–Protein Interaction Inhibitors. Int. J. Mol. Sci. 2019, 20, 4445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Ma, D.; Wang, P.; Pan, C.; Fang, Q.; Wang, J. Nrf2 overexpression increases risk of high tumor mutation burden in acute myeloid leukemia by inhibiting MSH2. Cell Death Dis. 2021, 12, 20. [Google Scholar] [CrossRef]
- Kapeta, S.; Chondrogianni, N.; Gonos, E.S. Nuclear Erythroid Factor 2-mediated Proteasome Activation Delays Senescence in Human Fibroblasts. J. Biol. Chem. 2010, 285, 8171–8184. [Google Scholar] [CrossRef] [Green Version]
- Wati, S.M.; Matsumaru, D.; Motohashi, H. NRF2 pathway activation by KEAP1 inhibition attenuates the manifestation of aging phenotypes in salivary glands. Redox Biol. 2020, 36, 101603. [Google Scholar] [CrossRef]
- Rajasekaran, N.S.; Varadharaj, S.; Khanderao, G.D.; Davidson, C.J.; Kannan, S.; Firpo, M.A.; Zweier, J.L.; Benjamin, I.J. Sustained Activation of Nuclear Erythroid 2-Related Factor 2/antioxidant Response Element Signaling Promotes Reductive Stress in the Human Mutant Protein Aggregation Cardiomyopathy in Mice. Antioxid. Redox Signal. 2011, 14, 957–971. [Google Scholar] [CrossRef] [Green Version]
- Satoh, H.; Moriguchi, T.; Saigusa, D.; Baird, L.; Yu, L.; Rokutan, H.; Igarashi, K.; Ebina, M.; Shibata, T.; Yamamoto, M. NRF2 Intensifies Host Defense Systems to Prevent Lung Carcinogenesis, but After Tumor Initiation Accelerates Malignant Cell Growth. Cancer Res. 2016, 76, 3088–3096. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Zhang, H.; Wu, F.; Liu, Z.; Cheng, Y.; Wang, C. Role of Nrf2 and Its Activators in Cardiocerebral Vascular Disease. Oxidative Med. Cell. Longev. 2020, 2020, 4683943. [Google Scholar] [CrossRef]
- Calvert, J.; Elston, M.; Nicholson, C.K.; Gundewar, S.; Jha, S.; Elrod, J.; Ramachandran, A.; Lefer, D.J. Genetic and Pharmacologic Hydrogen Sulfide Therapy Attenuates Ischemia-Induced Heart Failure in Mice. Circulation 2010, 122, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Strom, J.; Chen, Q.M. Loss of Nrf2 promotes rapid progression to heart failure following myocardial infarction. Toxicol. Appl. Pharmacol. 2017, 327, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Battelli, L.; Hubbs, A.F. Multiorgan Autoimmune Inflammation, Enhanced Lymphoproliferation, and Impaired Homeostasis of Reactive Oxygen Species in Mice Lacking the Antioxidant-Activated Transcription Factor Nrf2. Am. J. Pathol. 2006, 168, 1960–1974. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sano, M.; Shinmura, K.; Tamaki, K.; Katsumata, Y.; Matsuhashi, T.; Morizane, S.; Ito, H.; Hishiki, T.; Endo, J.; et al. 4-Hydroxy-2-nonenal protects against cardiac ischemia–reperfusion injury via the Nrf2-dependent pathway. J. Mol. Cell. Cardiol. 2010, 49, 576–586. [Google Scholar] [CrossRef]
- Li, J.; Ichikawa, T.; Villacorta, L.; Janicki, J.S.; Brower, G.; Yamamoto, M.; Cui, T. Nrf2 Protects Against Maladaptive Cardiac Responses to Hemodynamic Stress. Arter. Thromb. Vasc. Biol. 2009, 29, 1843–1850. [Google Scholar] [CrossRef] [PubMed]
- Sussan, T.E.; Rangasamy, T.; Blake, D.J.; Malhotra, D.; El-Haddad, H.; Bedja, D.; Yates, M.S.; Kombairaju, P.; Yamamoto, M.; Liby, K.T.; et al. Targeting Nrf2 with the triterpenoid CDDO- imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 250–255. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ichikawa, T.; Janicki, J.S.; Cui, T. Targeting the Nrf2 pathway against cardiovascular disease. Expert Opin. Ther. Targets 2009, 13, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, C.; Xing, Y.; Janicki, J.S.; Yamamoto, M.; Wang, X.L.; Tang, D.-Q.; Cui, T. Up-regulation of p27kip1 contributes to Nrf2-mediated protection against angiotensin II-induced cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 315–324. [Google Scholar] [CrossRef]
- Suzuki, H.; Osawa, T.; Fujioka, Y.; Noda, N.N. Structural biology of the core autophagy machinery. Curr. Opin. Struct. Biol. 2016, 43, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2011, 441, 523–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, W.E.; Beyer, A.M.; Gutterman, D.D. Vascular autophagy in health and disease. Basic Res. Cardiol. 2020, 115, 41. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hochstrasser, M. Microautophagy regulates proteasome homeostasis. Curr. Genet. 2020, 66, 683–687. [Google Scholar] [CrossRef]
- Dou, J.; Su, P.; Xu, C.; Wen, Z.; Mao, Z.; Li, W. Targeting Hsc70-based autophagy to eliminate amyloid β oligomers. Biochem. Biophys. Res. Commun. 2020, 524, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. USA 2011, 108, 9572–9577. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, S.R.; Simonsen, A. Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Fuhler, G.M.; Tyl, M.R.; Olthof, S.G.; Drayer, A.L.; Blom, N.; Vellenga, E. Distinct roles of the mTOR components Rictor and Raptor in MO7e megakaryocytic cells. Eur. J. Haematol. 2009, 83, 235–245. [Google Scholar] [CrossRef]
- Lin, X.; Zhang, Y.; Liu, L.; McKeehan, W.L.; Shen, Y.; Song, S.; Wang, F. FRS2α is Essential for the Fibroblast Growth Factor to Regulate the mTOR Pathway and Autophagy in Mouse Embryonic Fibroblasts. Int. J. Biol. Sci. 2011, 7, 1114–1121. [Google Scholar] [CrossRef] [Green Version]
- Noda, T. Regulation of Autophagy through TORC1 and mTORC1. Biomolecules 2017, 7, 52. [Google Scholar] [CrossRef]
- Wesselborg, S.; Stork, B. Autophagy signal transduction by ATG proteins: From hierarchies to networks. Cell. Mol. Life Sci. 2015, 72, 4721–4757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 2019, 63, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Sánchez, R.; Yakhine-Diop, S.M.; Arribas, M.R.; Pedro, J.M.B.-S.; Chacón, G.M.; Uribe-Carretero, E.; de Castro, D.C.P.; Pizarro-Estrella, E.; Fuentes, J.M.; González-Polo, R.A. mRNA and protein dataset of autophagy markers (LC3 and p62) in several cell lines. Data Brief 2016, 7, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Kirkin, V.; Lamark, T.; Sou, Y.-S.; Bjørkøy, G.; Nunn, J.L.; Bruun, J.-A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A Role for NBR1 in Autophagosomal Degradation of Ubiquitinated Substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Chiba, T.; Tatsumi, K.; Iemura, S.-I.; Tanida, I.; Okazaki, N.; Ueno, T.; Kominami, E.; Natsume, T.; Tanaka, K. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J. 2004, 23, 1977–1986. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.-S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.-I.; Ezaki, J.; Murata, S.; et al. Homeostatic Levels of p62 Control Cytoplasmic Inclusion Body Formation in Autophagy-Deficient Mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Gomes, A.V.; Barnes, J.A.; Dice, J.F. Selective Degradation of Annexins by Chaperone-mediated Autophagy. J. Biol. Chem. 2000, 275, 33329–33335. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Dice, J.F. Age-related Decline in Chaperone-mediated Autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef] [Green Version]
- Franch, H.A.; Sooparb, S.; Du, J.; Brown, N.S. A Mechanism Regulating Proteolysis of Specific Proteins during Renal Tubular Cell Growth. J. Biol. Chem. 2001, 276, 19126–19131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, V.; Dick, N.; Tawo, R.; Dreiseidler, M.; Wenzel, D.; Hesse, M.; Fürst, D.O.; Saftig, P.; Saint, R.; Fleischmann, B.K.; et al. Chaperone-Assisted Selective Autophagy Is Essential for Muscle Maintenance. Curr. Biol. 2010, 20, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Arias, E.; Koga, H.; Diaz, A.; Mocholi, E.; Patel, B.; Cuervo, A.M. Lysosomal mTORC2/PHLPP1/Akt Regulate Chaperone-Mediated Autophagy. Mol. Cell 2015, 59, 270–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endicott, S.J.; Ziemba, Z.J.; Beckmann, L.J.; Boynton, D.N.; Miller, R.A. Inhibition of class I PI3K enhances chaperone-mediated autophagy. J. Cell Biol. 2020, 219, e202001031. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, A.I.; Arias, E.; Díaz-Carretero, A.; Cuervo, A.M.; Cuadrado, A. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 2018, 14, 1310–1322. [Google Scholar] [CrossRef]
- Marzella, L.; Ahlberg, J.; Glaumann, H. Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1981, 36, 219–234. [Google Scholar] [CrossRef]
- Oku, M.; Sakai, Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. BioEssays 2018, 40, e1800008. [Google Scholar] [CrossRef]
- Sato, M.; Seki, T.; Konno, A.; Hirai, H.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H. Rapamycin activates mammalian microautophagy. J. Pharmacol. Sci. 2019, 140, 201–204. [Google Scholar] [CrossRef]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of Cytosolic Proteins by Late Endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Mejlvang, J.; Olsvik, H.; Svenning, S.; Bruun, J.-A.; Abudu, Y.P.; Larsen, K.B.; Brech, A.; Hansen, T.E.; Brenne, H.; Hansen, T.; et al. Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J. Cell Biol. 2018, 217, 3640–3655. [Google Scholar] [CrossRef] [Green Version]
- Gong, Z.; Pan, J.; Shen, Q.; Li, M.; Peng, Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflamm. 2018, 15, 242. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, P.C.; Ducret, A.; Langen, H.; Nogoceke, E.; Santos, R.H.B.; Nunes, J.P.S.; Benvenuti, L.; Levy, D.; Bydlowski, S.P.; Bocchi, E.A.; et al. Impairment of Multiple Mitochondrial Energy Metabolism Pathways in the Heart of Chagas Disease Cardiomyopathy Patients. Front. Immunol. 2021, 12, 755782. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmström, K.; Skujat, D.; Fiesel, F.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Han, E.; Bui, C.; Shin, W.; Lee, J.; Lee, S.; Choi, Y.; Lee, A.; Lee, K.; Park, C.; et al. Assurance of mitochondrial integrity and mammalian longevity by the p62–Keap1–Nrf2–Nqo1 cascade. EMBO Rep. 2012, 13, 150–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2009, 11, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK 1 translocates to mitochondria and phosphorylates FUNDC 1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Shirakabe, A.; Zhai, P.; Ikeda, Y.; Saito, T.; Maejima, Y.; Hsu, C.-P.; Nomura, M.; Egashira, K.; Levine, B.; Sadoshima, J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload–Induced Mitochondrial Dysfunction and Heart Failure. Circulation 2016, 133, 1249–1263. [Google Scholar] [CrossRef] [Green Version]
- Hein, S.; Arnon, E.; Kostin, S.; Schönburg, M.; Elsässer, A.; Polyakova, V.; Bauer, E.P.; Klövekorn, W.-P.; Schaper, J. Progression From Compensated Hypertrophy to Failure in the Pressure-Overloaded Human Heart: Structural deterioration and compensatory mechanisms. Circulation 2003, 107, 984–991. [Google Scholar] [CrossRef] [Green Version]
- Kanamori, H.; Takemura, G.; Goto, K.; Maruyama, R.; Tsujimoto, A.; Ogino, A.; Takeyama, T.; Kawaguchi, T.; Watanabe, T.; Fujiwara, T.; et al. The role of autophagy emerging in postinfarction cardiac remodelling. Cardiovasc. Res. 2011, 91, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Vatner, D.E.; Kim, S.-J.; Ge, H.; Masurekar, M.; Massover, W.H.; Yang, G.; Matsui, Y.; Sadoshima, J.; Vatner, S.F. Autophagy in chronically ischemic myocardium. Proc. Natl. Acad. Sci. USA 2005, 102, 13807–13812. [Google Scholar] [CrossRef] [Green Version]
- Takemura, G.; Miyata, S.; Kawase, Y.; Okada, H.; Maruyama, R.; Fujiwara, H. Autophagic degeneration and death of cardiomyocytes in heart failure. Autophagy 2006, 2, 212–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, A.B.; Gottlieb, R.A. Autophagy in Ischemic Heart Disease. Circ. Res. 2009, 104, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct Roles of Autophagy in the Heart During Ischemia and Reperfusion. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Asai, K.; Sato, S.; Hayashi, M.; Adachi, A.; Sasaki, Y.; Takano, H.; Mizuno, K.; Shimizu, W. Autophagic vacuoles in cardiomyocytes of dilated cardiomyopathy with initially decompensated heart failure predict improved prognosis. Autophagy 2016, 12, 579–587. [Google Scholar] [CrossRef]
- Schlossarek, S.; Mearini, G.; Carrier, L. Cardiac myosin-binding protein C in hypertrophic cardiomyopathy: Mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2011, 50, 613–620. [Google Scholar] [CrossRef]
- Takagi, H.; Matsui, Y.; Sadoshima, J. The Role of Autophagy in Mediating Cell Survival and Death During Ischemia and Reperfusion in the Heart. Antioxid. Redox Signal. 2007, 9, 1373–1382. [Google Scholar] [CrossRef]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 Mediates Mitochondrial Autophagy and Protects the Heart against Energy Stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef]
- Andres, A.M.; Hernandez, G.; Lee, P.; Huang, C.; Ratliff, E.P.; Sin, J.; Thornton, C.A.; Damasco, M.V.; Gottlieb, R.A. Mitophagy Is Required for Acute Cardioprotection by Simvastatin. Antioxid. Redox Signal. 2014, 21, 1960–1973. [Google Scholar] [CrossRef]
- Huang, C.; Andres, A.M.; Ratliff, E.P.; Hernandez, G.; Lee, P.; Gottlieb, R. Preconditioning Involves Selective Mitophagy Mediated by Parkin and p62/SQSTM1. PLoS ONE 2011, 6, e20975. [Google Scholar] [CrossRef]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef]
- Li, X.; Hu, X.; Wang, J.; Xu, W.; Yi, C.; Ma, R.; Jiang, H. Inhibition of autophagy via activation of PI3K/Akt/mTOR pathway contributes to the protection of hesperidin against myocardial ischemia/reperfusion injury. Int. J. Mol. Med. 2018, 42, 1917–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.J.; Wang, Z.V.; Battiprolu, P.K.; Jiang, N.; Morales, C.R.; Kong, Y.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl. Acad. Sci. USA 2011, 108, 4123–4128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, L.-Q.; Zhang, W.-B.; Ye, Y.; Yin, P.-P.; Yuan, J.; Wang, X.-X.; Kang, L.; Jiang, S.-S.; You, J.-Y.; Wu, J.; et al. Aliskiren ameliorates pressure overload-induced heart hypertrophy and fibrosis in mice. Acta Pharmacol. Sin. 2014, 35, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhao, G.; Hu, X.; Wang, M.; Li, H.; Ye, Y.; Du, Q.; Yao, J.; Bao, Z.; Hong, W.; et al. Aliskiren-attenuated myocardium apoptosis via regulation of autophagy and connexin-43 in aged spontaneously hypertensive rats. J. Cell. Mol. Med. 2014, 18, 1247–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanamori, H.; Takemura, G.; Maruyama, R.; Goto, K.; Tsujimoto, A.; Ogino, A.; Li, L.; Kawamura, I.; Takeyama, T.; Kawaguchi, T.; et al. Functional Significance and Morphological Characterization of Starvation-Induced Autophagy in the Adult Heart. Am. J. Pathol. 2009, 174, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Oyabu, J.; Yamaguchi, O.; Hikoso, S.; Takeda, T.; Oka, T.; Murakawa, T.; Yasui, H.; Ueda, H.; Nakayama, H.; Taneike, M.; et al. Autophagy-mediated degradation is necessary for regression of cardiac hypertrophy during ventricular unloading. Biochem. Biophys. Res. Commun. 2013, 441, 787–792. [Google Scholar] [CrossRef]
- Schreckenberger, Z.; Wenceslau, C.F.; Joe, B.; McCarthy, C.G. Mitophagy in Hypertension-Associated Premature Vascular Aging. Am. J. Hypertens. 2020, 33, 804–812. [Google Scholar] [CrossRef]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol. Ther. 2017, 178, 157–174. [Google Scholar] [CrossRef]
- Xiong, W.; Ma, Z.; An, D.; Liu, Z.; Cai, W.; Bai, Y.; Zhan, Q.; Lai, W.; Zeng, Q.; Ren, H.; et al. Mitofusin 2 Participates in Mitophagy and Mitochondrial Fusion against Angiotensin II-Induced Cardiomyocyte Injury. Front. Physiol. 2019, 10, 411. [Google Scholar] [CrossRef]
- Yang, M.; Linn, B.S.; Zhang, Y.; Ren, J. Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 2293–2302. [Google Scholar] [CrossRef]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to Hydrogen Peroxide Induces Oxidation and Activation of AMP-activated Protein Kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desideri, E.; Filomeni, G.; Ciriolo, M.R. Glutathione participates in the modulation of starvation-induced autophagy in carcinoma cells. Autophagy 2012, 8, 1769–1781. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Copple, I.; Lister, A.; Obeng, A.D.; Kitteringham, N.R.; Jenkins, R.E.; Layfield, R.; Foster, B.J.; Goldring, C.E.; Park, B.K. Physical and Functional Interaction of Sequestosome 1 with Keap1 Regulates the Keap1-Nrf2 Cell Defense Pathway. J. Biol. Chem. 2010, 285, 16782–16788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A Noncanonical Mechanism of Nrf2 Activation by Autophagy Deficiency: Direct Interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Tang, Z.; Chen, D.; Moughon, D.; Ding, X.; Chen, S.; Zhu, M.; Zhong, Q. Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy 2010, 6, 614–621. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Park, J.S.; Lee, Y.S.; Han, J.; Lee, D.-K.; Kwon, S.W.; Han, D.H.; Lee, Y.-H.; Bae, S.H. SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity. Autophagy 2020, 16, 1949–1973. [Google Scholar] [CrossRef]
- Tanji, K.; Miki, Y.; Ozaki, T.; Maruyama, A.; Yoshida, H.; Mimura, J.; Matsumiya, T.; Mori, F.; Imaizumi, T.; Itoh, K.; et al. Phosphorylation of serine 349 of p62 in Alzheimer’s disease brain. Acta Neuropathol. Commun. 2014, 2, 50. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, Y.; Waguri, S.; Sou, Y.-S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolini, D.; Dallaglio, K.; Torquato, P.; Piroddi, M.; Galli, F. Nrf2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma. Transl. Res. 2018, 193, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox Biol. 2017, 11, 543–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V.W. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496. [Google Scholar] [CrossRef] [PubMed]
- Herman-Antosiewicz, A.; Johnson, D.E.; Singh, S.V. Sulforaphane Causes Autophagy to Inhibit Release of Cytochrome c and Apoptosis in Human Prostate Cancer Cells. Cancer Res. 2006, 66, 5828–5835. [Google Scholar] [CrossRef] [Green Version]
- Mizunoe, Y.; Kobayashi, M.; Sudo, Y.; Watanabe, S.; Yasukawa, H.; Natori, D.; Hoshino, A.; Negishi, A.; Okita, N.; Komatsu, M.; et al. Trehalose protects against oxidative stress by regulating the Keap1–Nrf2 and autophagy pathways. Redox Biol. 2017, 15, 115–124. [Google Scholar] [CrossRef]
- Jena, K.K.; Kolapalli, S.P.; Mehto, S.; Nath, P.; Das, B.; Sahoo, P.K.; Ahad, A.; Syed, G.; Raghav, S.K.; Senapati, S.; et al. TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. EMBO J. 2018, 37, e98358. [Google Scholar] [CrossRef]
- Ding, Y.-W.; Zhao, G.-J.; Li, X.-L.; Hong, G.-L.; Li, M.-F.; Qiu, Q.-M.; Wu, B.; Lu, Z.-Q. SIRT1 exerts protective effects against paraquat-induced injury in mouse type II alveolar epithelial cells by deacetylating NRF2 in vitro. Int. J. Mol. Med. 2016, 37, 1049–1058. [Google Scholar] [CrossRef]
- Lee, I.H. Mechanisms and disease implications of sirtuin-mediated autophagic regulation. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; An, S.; Ro, S.-H.; Teixeira, F.; Park, G.J.; Kim, C.; Cho, C.-S.; Kim, J.-S.; Jakob, U.; Lee, J.H.; et al. Janus-faced Sestrin2 controls ROS and mTOR signalling through two separate functional domains. Nat. Commun. 2015, 6, 10025. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Joe, Y.; Kim, S.-K.; Park, S.-U.; Park, J.; Chen, Y.; Kim, J.; Ryu, J.; Cho, G.J.; Surh, Y.-J.; et al. Carbon monoxide protects against hepatic steatosis in mice by inducing sestrin-2 via the PERK-eIF2α-ATF4 pathway. Free Radic. Biol. Med. 2017, 110, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Pasha, M.; Eid, A.H.; Eid, A.A.; Gorin, Y.; Munusamy, S. Sestrin2 as a Novel Biomarker and Therapeutic Target for Various Diseases. Oxidative Med. Cell. Longev. 2017, 2017, 3296294. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Budanov, A.V.; Park, E.J.; Birse, R.; Kim, T.E.; Perkins, G.A.; Ocorr, K.; Ellisman, M.H.; Bodmer, R.; Bier, E.; et al. Sestrin as a Feedback Inhibitor of TOR That Prevents Age-Related Pathologies. Science 2010, 327, 1223–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Zhu, J.; Huang, H.; Xiang, D.; Li, Y.; Zhang, D.; Li, J.; Wang, Y.; Jin, H.; Jiang, G.; et al. SESN2/sestrin 2 induction-mediated autophagy and inhibitory effect of isorhapontigenin (ISO) on human bladder cancers. Autophagy 2016, 12, 1229–1239. [Google Scholar] [CrossRef] [Green Version]
- Ro, S.-H.; Semple, I.A.; Park, H.; Park, H.; Park, H.-W.; Kim, M.; Kim, J.S.; Lee, J.H. Sestrin2 promotes Unc-51-like kinase 1 mediated phosphorylation of p62/sequestosome-1. FEBS J. 2014, 281, 3816–3827. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, K.; Simmons, A.N.; Kajino-Sakamoto, R.; Tsuji, Y.; Ninomiya-Tsuji, J. TAK1 Regulates the Nrf2 Antioxidant System Through Modulating p62/SQSTM1. Antioxid. Redox Signal. 2016, 25, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.H.; Iskandar, K.B.; Yadav, S.K.; Hirpara, J.L.; Loh, T.; Pervaiz, S. Simultaneous Induction of Non-Canonical Autophagy and Apoptosis in Cancer Cells by ROS-Dependent ERK and JNK Activation. PLoS ONE 2010, 5, e9996. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-Mediated Phosphorylation of Bcl-2 Regulates Starvation-Induced Autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell. Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef] [Green Version]
- Kosztelnik, M.; Kurucz, A.; Papp, D.; Jones, E.; Sigmond, T.; Barna, J.; Traka, M.H.; Lorincz, T.; Szarka, A.; Banhegyi, G.; et al. Suppression of AMPK/aak-2 by NRF2/SKN-1 down-regulates autophagy during prolonged oxidative stress. FASEB J. 2018, 33, 2372–2387. [Google Scholar] [CrossRef] [Green Version]
- Rouschop, K.M.; Van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Wang, S.; Xu, H.; Li, X.; Guan, Y.; Yi, F.; Zhou, T.; Jiang, B.; Bai, N.; Ma, M.; et al. ATM-CHK 2-Beclin 1 axis promotes autophagy to maintain ROS homeostasis under oxidative stress. EMBO J. 2020, 39, e103111. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective Mitochondrial Autophagy, or Mitophagy, as a Targeted Defense against Oxidative Stress, Mitochondrial Dysfunction, and Aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Nieminen, A.-L.; Qian, T.; Trost, L.C.; Elmore, S.P.; Nishimura, Y.; Crowe, R.A.; Cascio, W.E.; Bradham, C.A.; Brenner, D.A.; et al. The mitochondrial permeability transition in cell death: A common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta 1998, 1366, 177–196. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Liu, Z.; Zhou, Y.; Hou, N.; Yan, W.; Qin, Y.; Ye, Q.; Cheng, X.; Xiao, Q.; Bao, Y.; et al. Urolithin B, a gut microbiota metabolite, protects against myocardial ischemia/reperfusion injury via p62/Keap1/Nrf2 signaling pathway. Pharmacol. Res. 2020, 153, 104655. [Google Scholar] [CrossRef]
- Hou, K.; Shen, J.; Yan, J.; Zhai, C.; Zhang, J.; Pan, J.-A.; Zhang, Y.; Jiang, Y.; Wang, Y.; Lin, R.Z.; et al. Loss of TRIM21 alleviates cardiotoxicity by suppressing ferroptosis induced by the chemotherapeutic agent doxorubicin. EBioMedicine 2021, 69, 103456. [Google Scholar] [CrossRef]
- Zang, H.; Wu, W.; Qi, L.; Tan, W.; Nagarkatti, P.; Nagarkatti, M.; Wang, X.; Cui, T. Autophagy Inhibition Enables Nrf2 to Exaggerate the Progression of Diabetic Cardiomyopathy in Mice. Diabetes 2020, 69, 2720–2734. [Google Scholar] [CrossRef]
- Bhide, S.; Trujillo, A.S.; O’Connor, M.T.; Young, G.H.; Cryderman, D.E.; Chandran, S.; Nikravesh, M.; Wallrath, L.L.; Melkani, G.C. Increasing autophagy and blocking Nrf2 suppress laminopathy-induced age-dependent cardiac dysfunction and shortened lifespan. Aging Cell 2018, 17, e12747. [Google Scholar] [CrossRef] [Green Version]
- Qin, Q.; Qu, C.; Niu, T.; Zang, H.; Qi, L.; Lyu, L.; Wang, X.; Nagarkatti, M.; Nagarkatti, P.; Janicki, J.S.; et al. Nrf2-Mediated Cardiac Maladaptive Remodeling and Dysfunction in a Setting of Autophagy Insufficiency. Hypertension 2016, 67, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, N.; Wang, H.; Chen, Y.; Li, H.; Viktor, K.; Huang, K.; Chen, X. Taurine attenuates OTA-promoted PCV2 replication through blocking ROS-dependent autophagy via inhibiting AMPK/mTOR signaling pathway. Chem. Interact. 2018, 296, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Qin, Q.; Ding, Y.; Zang, H.; Li, D.-S.; Nagarkatti, M.; Nagarkatti, P.; Wang, W.; Wang, X.; Cui, T. Autophagy Controls Nrf2-Mediated Dichotomy in Pressure Overloaded Hearts. Front. Physiol. 2021, 12, 673145. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.-H.; Chen, C.; Wang, D.W. AMPKα2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, J.; Xia, P.; Stratakis, C.A.; Cheng, Z. Loss of PKA regulatory subunit 1α aggravates cardiomyocyte necrosis and myocardial ischemia/reperfusion injury. J. Biol. Chem. 2021, 297, 100850. [Google Scholar] [CrossRef]
- Wang, Z.; Hill, J.A. Protein Quality Control and Metabolism: Bidirectional Control in the Heart. Cell Metab. 2015, 21, 215–226. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cui, T. Autophagy modulation: A potential therapeutic approach in cardiac hypertrophy. Am. J. Physiol. Circ. Physiol. 2017, 313, H304–H319. [Google Scholar] [CrossRef]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef]
- He, H.; Liu, X.; Lv, L.; Liang, H.; Leng, B.; Zhao, D.; Zhang, Y.; Du, Z.; Chen, X.; Li, S.; et al. Calcineurin suppresses AMPK-dependent cytoprotective autophagy in cardiomyocytes under oxidative stress. Cell Death Dis. 2014, 5, e997. [Google Scholar] [CrossRef]
- Lin, C.; Liu, Z.; Lu, Y.; Yao, Y.; Zhang, Y.; Ma, Z.; Kuai, M.; Sun, X.; Sun, S.; Jing, Y.; et al. Cardioprotective effect of Salvianolic acid B on acute myocardial infarction by promoting autophagy and neovascularization and inhibiting apoptosis. J. Pharm. Pharmacol. 2016, 68, 941–952. [Google Scholar] [CrossRef]
- Pan, H.; Niu, L.; Wu, Y.; Chen, L.; Zhou, X.; Zhao, Y. Lycium barbarum polysaccharide protects rats and cardiomyocytes against ischemia/reperfusion injury via Nrf2 activation through autophagy inhibition. Mol. Med. Rep. 2021, 24, 778. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.-G. Regulation of redox signalling and autophagy during cardiovascular diseases-role of resveratrol. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 1530–1536. [Google Scholar] [PubMed]
- Taneike, M.; Yamaguchi, O.; Nakai, A.; Hikoso, S.; Takeda, T.; Mizote, I.; Oka, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 2010, 6, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-L.; Yang, J.-J.; Zhang, H.-S. Carvedilol (CAR) combined with carnosic acid (CAA) attenuates doxorubicin-induced cardiotoxicity by suppressing excessive oxidative stress, inflammation, apoptosis and autophagy. Biomed. Pharmacother. 2018, 109, 71–83. [Google Scholar] [CrossRef]
- Ahmad, S.N.S.; Sanajou, D.; Kalantary-Charvadeh, A.; Hosseini, V.; Roshangar, L.; Khojastehfard, M.; Haiaty, S.; Mesgari-Abbasi, M. β-LAP achone ameliorates doxorubicin-induced cardiotoxicity via regulating autophagy and Nrf2 signalling pathways in mice. Basic Clin. Pharmacol. Toxicol. 2019, 126, 364–373. [Google Scholar] [CrossRef]
- Luo, J.; Yan, D.; Li, S.; Liu, S.; Zeng, F.; Cheung, C.W.; Liu, H.; Irwin, M.G.; Huang, H.; Xia, Z. Allopurinol reduces oxidative stress and activates Nrf2/p62 to attenuate diabetic cardiomyopathy in rats. J. Cell. Mol. Med. 2019, 24, 1760–1773. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wang, L.; Lu, J.; Hu, Y.; Wang, Q.; Li, Z.; Cai, S.; Liang, L.; Guo, K.; Xie, J.; et al. SESN2 protects against doxorubicin-induced cardiomyopathy via rescuing mitophagy and improving mitochondrial function. J. Mol. Cell. Cardiol. 2019, 133, 125–137. [Google Scholar] [CrossRef]
- Li, W.; Febbraio, M.; Reddy, S.P.; Yu, D.-Y.; Yamamoto, M.; Silverstein, R.L. CD36 participates in a signaling pathway that regulates ROS formation in murine VSMCs. J. Clin. Investig. 2010, 120, 3996–4006. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Disease | Model | Sample | Autophagy | Autophagy-Redox Signaling | Reference |
|---|---|---|---|---|---|
| Heart failure | TAC operation | Mice; Rat cardiac myocytes (H9c2) | Enhanced autophagosome formation and autophagic flux | Nrf2 increases autophagy-mediated clearance of ubiquitinated protein aggregates in cardiomyocytes | [30] |
| TAC operation | Mice | - | When autophagy is intact, Nrf2 is required for cardiac remodeling. When autophagy is impaired, Nrf2 nuclear export is decreased and Nrf2-driven angiotensinogen transcription is increased, which can lead to cardiac dysfunction | [151,153] | |
| TAC operation; patients with heart transplantation | Mice, human | Defective mitophagy | AMPK improves mitophagy via PINK-1 phosphorylation and decreases ROS formation | [154] | |
| Ischemic heart disease | LAD ligation | Mice | - | Autophagy increases Nrf2 signaling activation, which leads to MI damage improvement | [8] |
| I/R (LAD ligation) | Mice; NRCMs | Impaired mTORC-p62-Keap1-Nrf2 antioxidant defense system | Impaired mTORC-p62-Keap1-Nrf2 antioxidant defense system | [155] | |
| I/R (LAD ligation) Urolithin B treatment | Rats | Urolithin B decreases autophagy by Akt/mTOR/ULK1 pathway | Urolithin B increases p62/Keap1/Nrf2 signaling pathway activation | [147] | |
| Doxorubicin-induced cardiomyopathy | Doxorubicin treatment | Mice | Defective autophagy | P62-Keap1-Nrf2 activation leads to the positive regulation of oxidative stress and autophagy | [29,148] |
| Cardiac laminopathy | Lamin C mutant | Drosophila melanogaster | Increased autophagy genes expression | Increased Nrf2 levels lead to autophagy inhibition by mTOR activation | [150] |
| Diabetic cardiomyopathy | Type 1 diabetes | Mice | Defective autophagy | Autophagy inhibition leads to increased Nrf2 levels and thus to the progression of diabetic cardiomyopathy | [149] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boťanská, B.; Dovinová, I.; Barančík, M. The Interplay between Autophagy and Redox Signaling in Cardiovascular Diseases. Cells 2022, 11, 1203. https://doi.org/10.3390/cells11071203
Boťanská B, Dovinová I, Barančík M. The Interplay between Autophagy and Redox Signaling in Cardiovascular Diseases. Cells. 2022; 11(7):1203. https://doi.org/10.3390/cells11071203
Chicago/Turabian StyleBoťanská, Barbora, Ima Dovinová, and Miroslav Barančík. 2022. "The Interplay between Autophagy and Redox Signaling in Cardiovascular Diseases" Cells 11, no. 7: 1203. https://doi.org/10.3390/cells11071203
APA StyleBoťanská, B., Dovinová, I., & Barančík, M. (2022). The Interplay between Autophagy and Redox Signaling in Cardiovascular Diseases. Cells, 11(7), 1203. https://doi.org/10.3390/cells11071203