
Understanding Molecular Mechanisms of Phenotype Switching and Crosstalk with TME to Reveal New Vulnerabilities of Melanoma

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
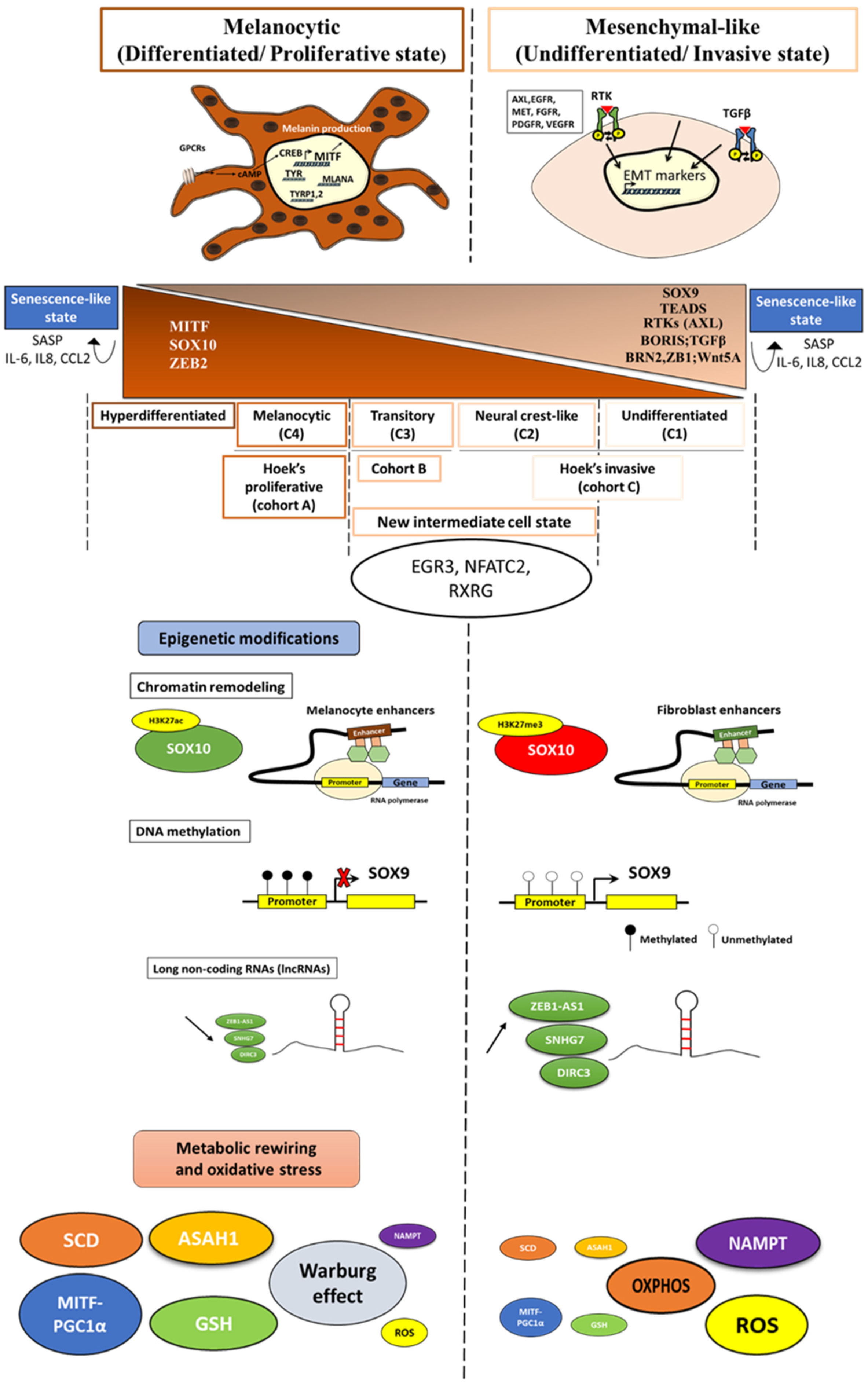
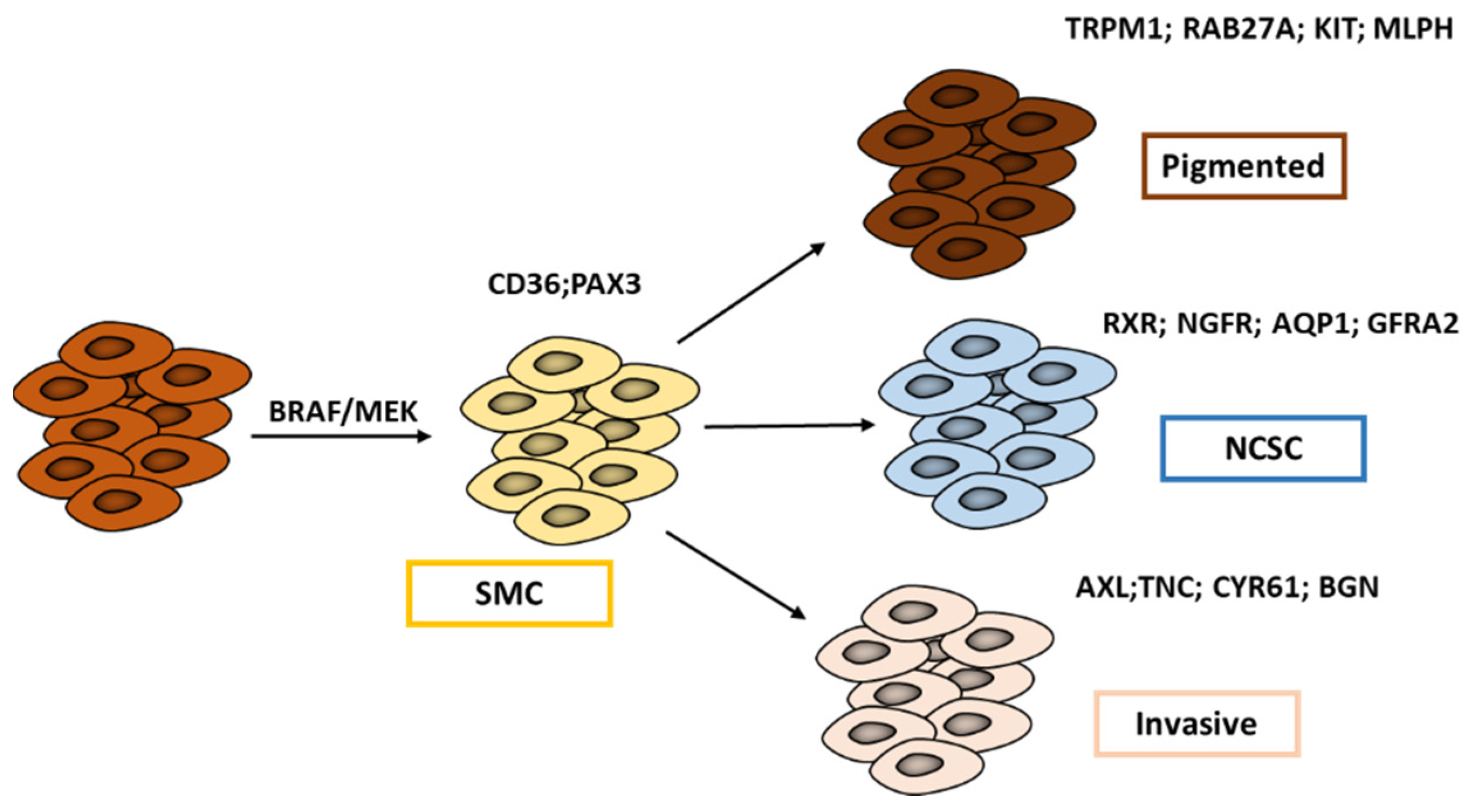
2. Phenotype Switching in Melanoma: Growing Ever More Complex
2.1. Key Regulator Factors of Phenotype Switching
2.1.1. The MITF “Rheostat Model”
2.1.2. ZEB2 and ZEB1 in Melanoma Plasticity
2.1.3. Opposing Roles of MITF and Wnt5A in Phenotype Switching
2.1.4. Role of MITF–BRN2 Axis in Phenotype Switching
2.1.5. Antagonistic Functions of SOX10 and SOX9 in the Regulation of Phenotype Switching
2.1.6. RTKs as Putative Drivers of Phenotype Switching
2.1.7. BORIS Mediates TGF-β-Driven Phenotype Switching
2.1.8. TEADs as Key Regulators of the Invasive Phenotype
2.1.9. Gene Regulatory Networks
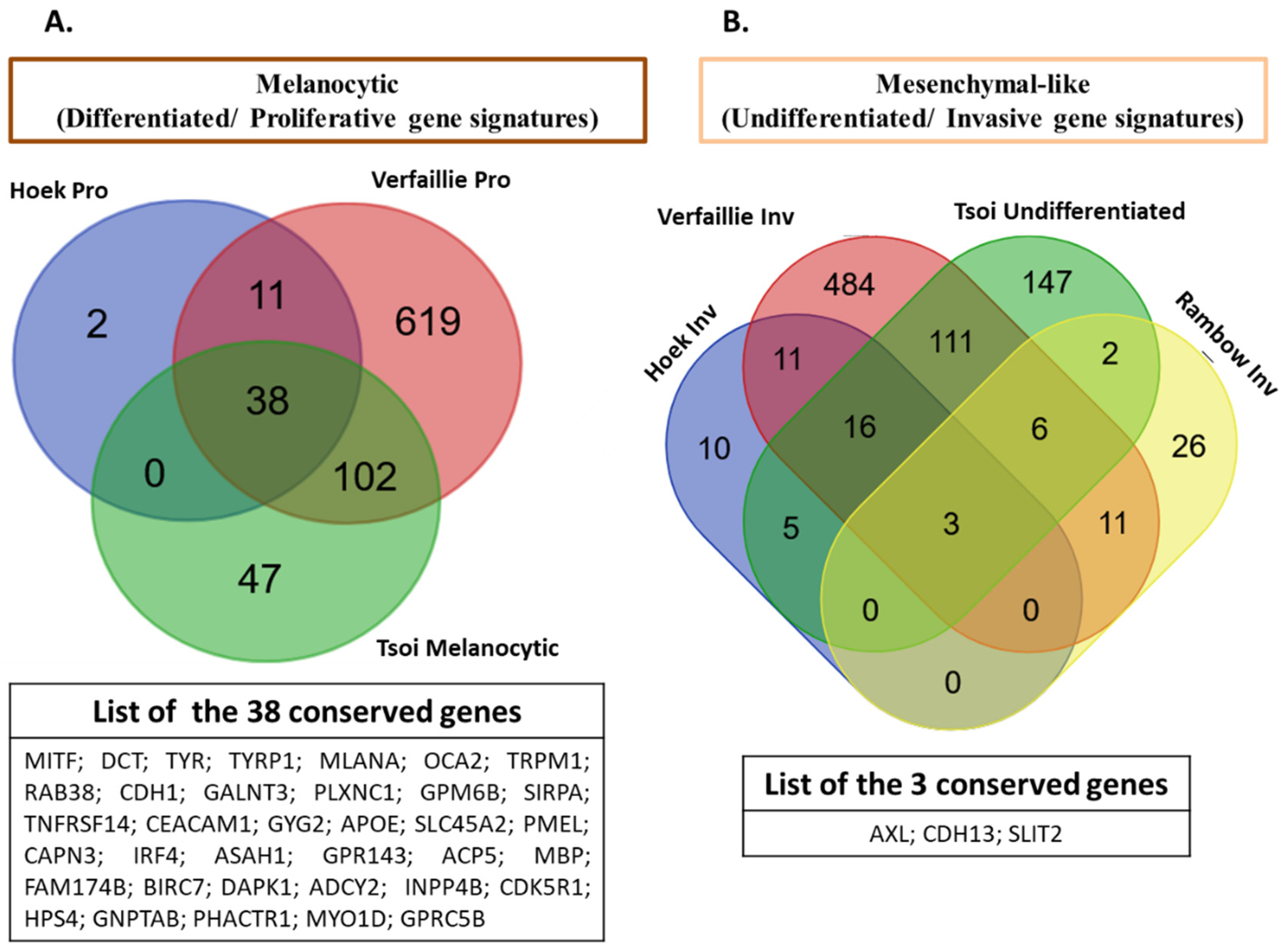
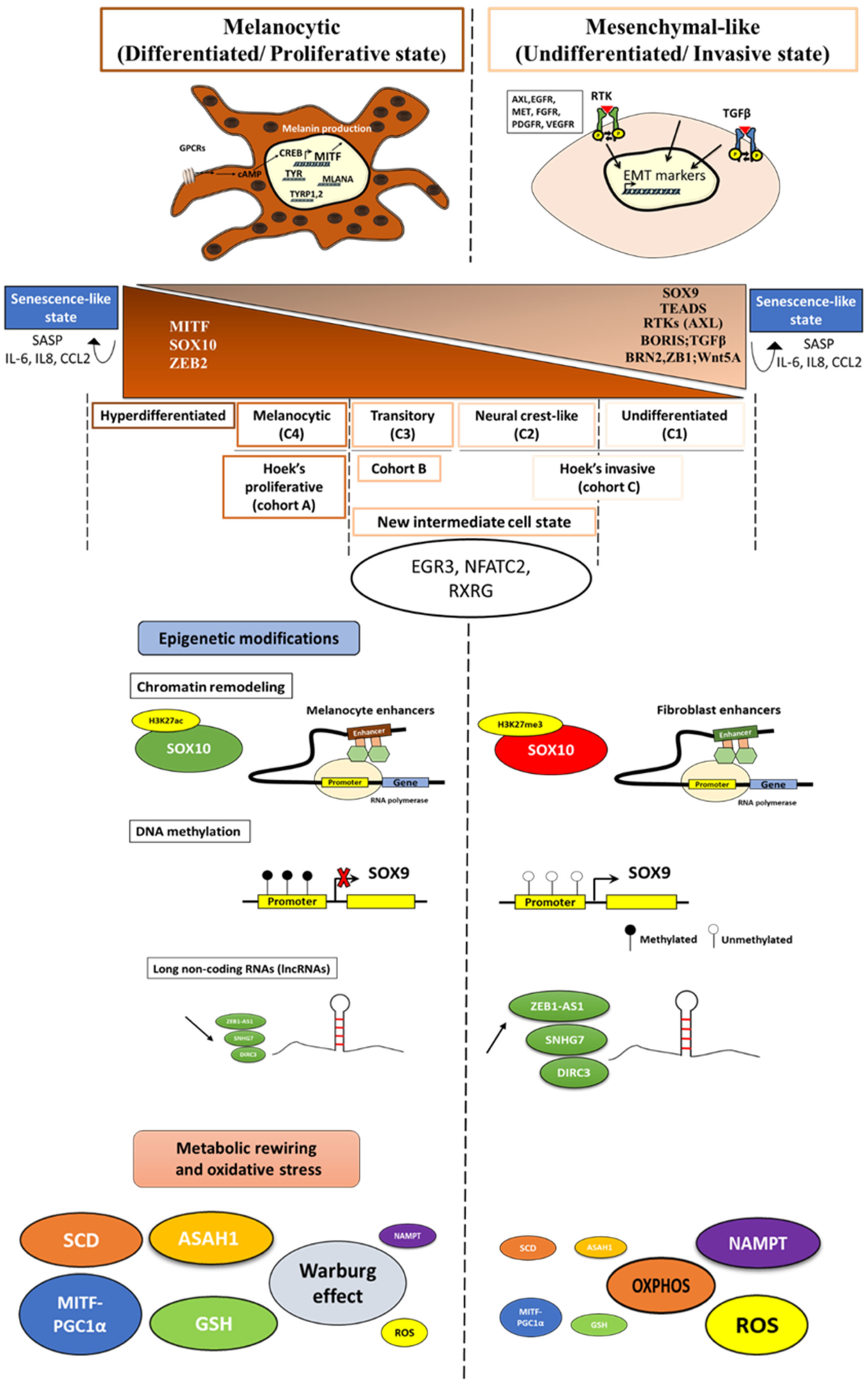
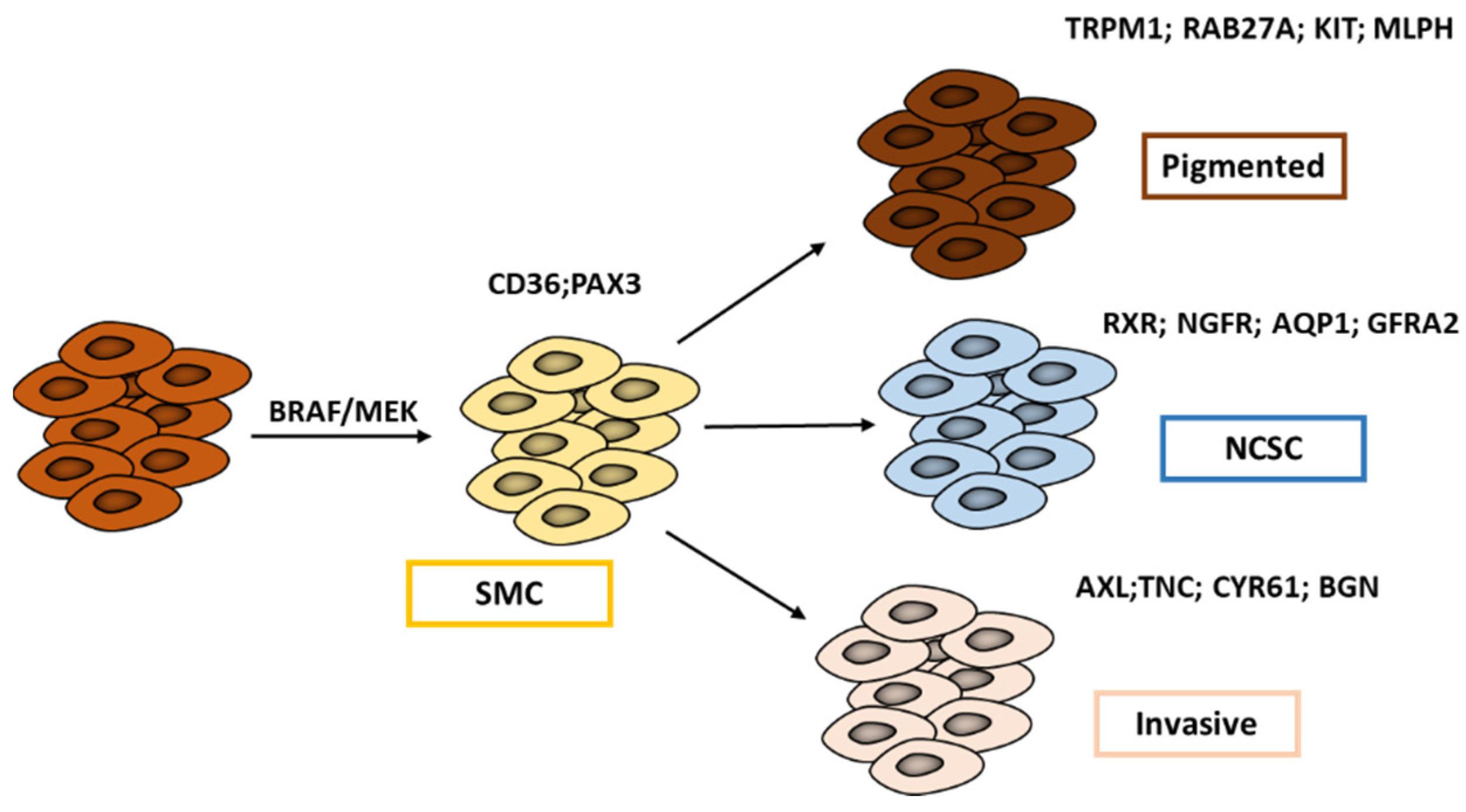
2.2. Refining Classification of Melanoma Phenotypes
2.3. New Intermediate Cell State and Major Regulators
2.4. Melanoma Cell States Associated with Minimal Residual Disease (MRD)
2.5. Senescence-like State
2.6. Hyperdifferentiated State
2.7. Deep Characterization of Melanoma Phenotypic Diversity: Lessons to Other Cancers
3. Role of Epigenetic Modifications in the Dynamic Phenotype Switch
3.1. Chromatin Remodeling
3.2. Methylation
3.3. Long Non-Coding RNAs (lncRNAs)
3.4. miRNAs (MicroRNAs)
4. Importance of the Metabolic Rewiring in the Phenotypic Switch
4.1. Glycolysis and Warburg Effect vs. OXPHOS
4.2. Glutamine Metabolism
4.3. Nicotinamide Phosphoribosyltransferase (NAMPT)
4.4. Lipid Metabolism
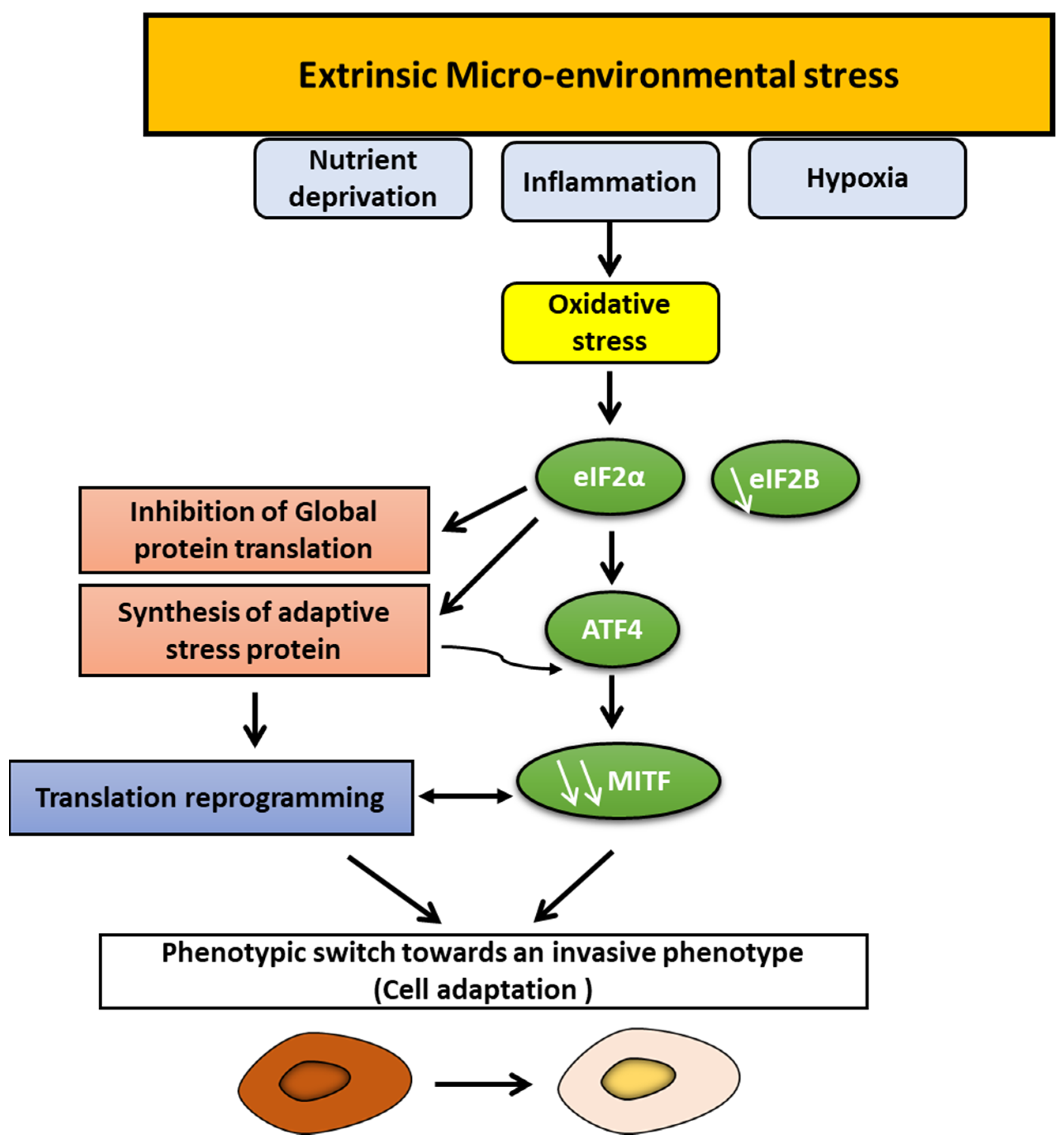
5. Prominent Role of Microenvironment Stress Signals in Phenotype Switching
5.1. Hypoxia
5.2. Inflammation
5.3. Nutrient Deprivation
5.4. Tumor Microenvironment (TME) Factors
5.4.1. Cancer-Associated Fibroblasts (CAFs)
5.4.2. CD73
5.4.3. Interleukin-like EMT Inducer (ILEI)/FAM3C
5.4.4. Endothelin-1 (ET-1)
6. Critical Role of Oxidative Stress in Regulating Phenotypic Plasticity
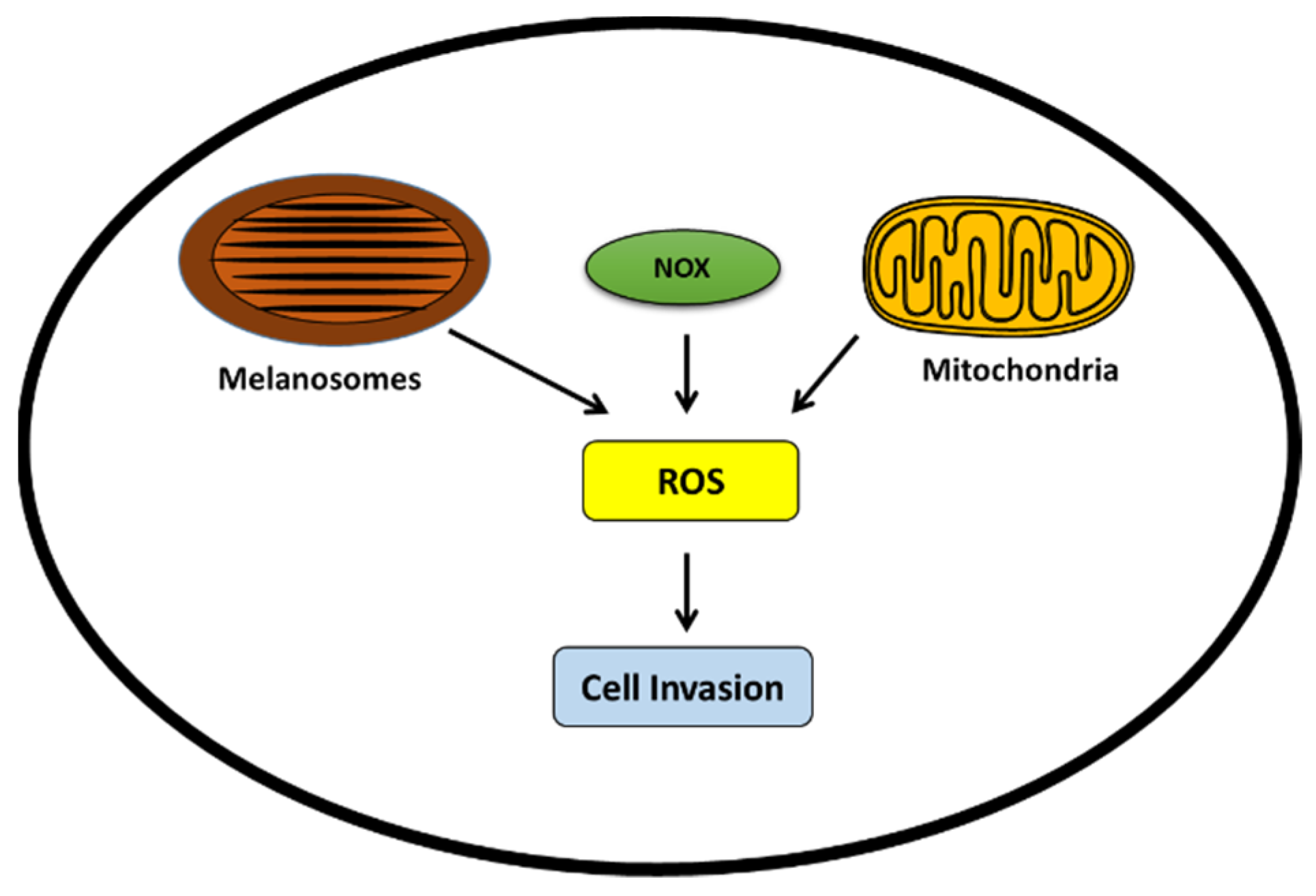
6.1. ROS Sources
6.1.1. Tyrosine-Induced Melanogenesis
6.1.2. NADPH Oxidases (NOX)
6.1.3. Mitochondria
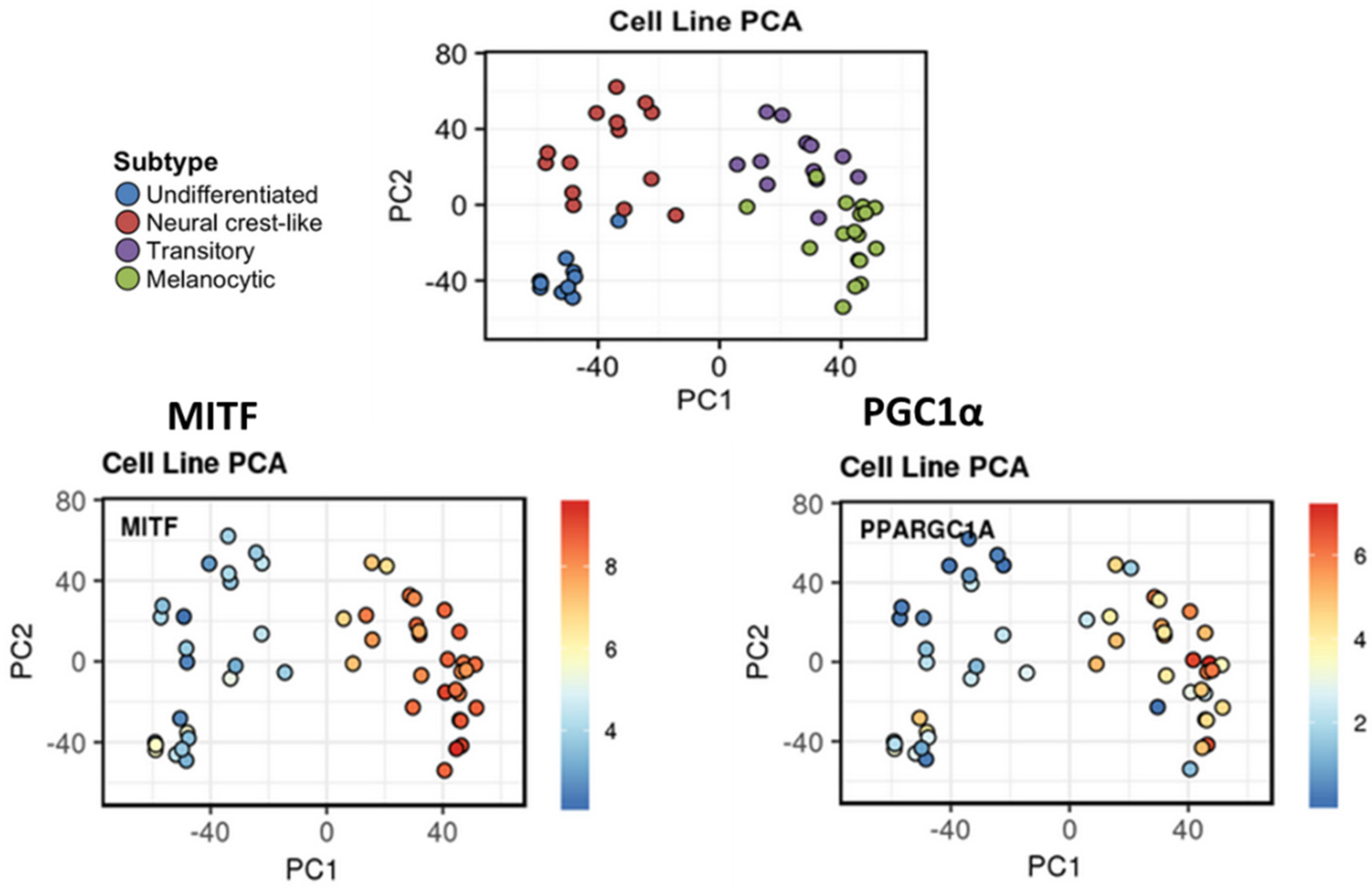
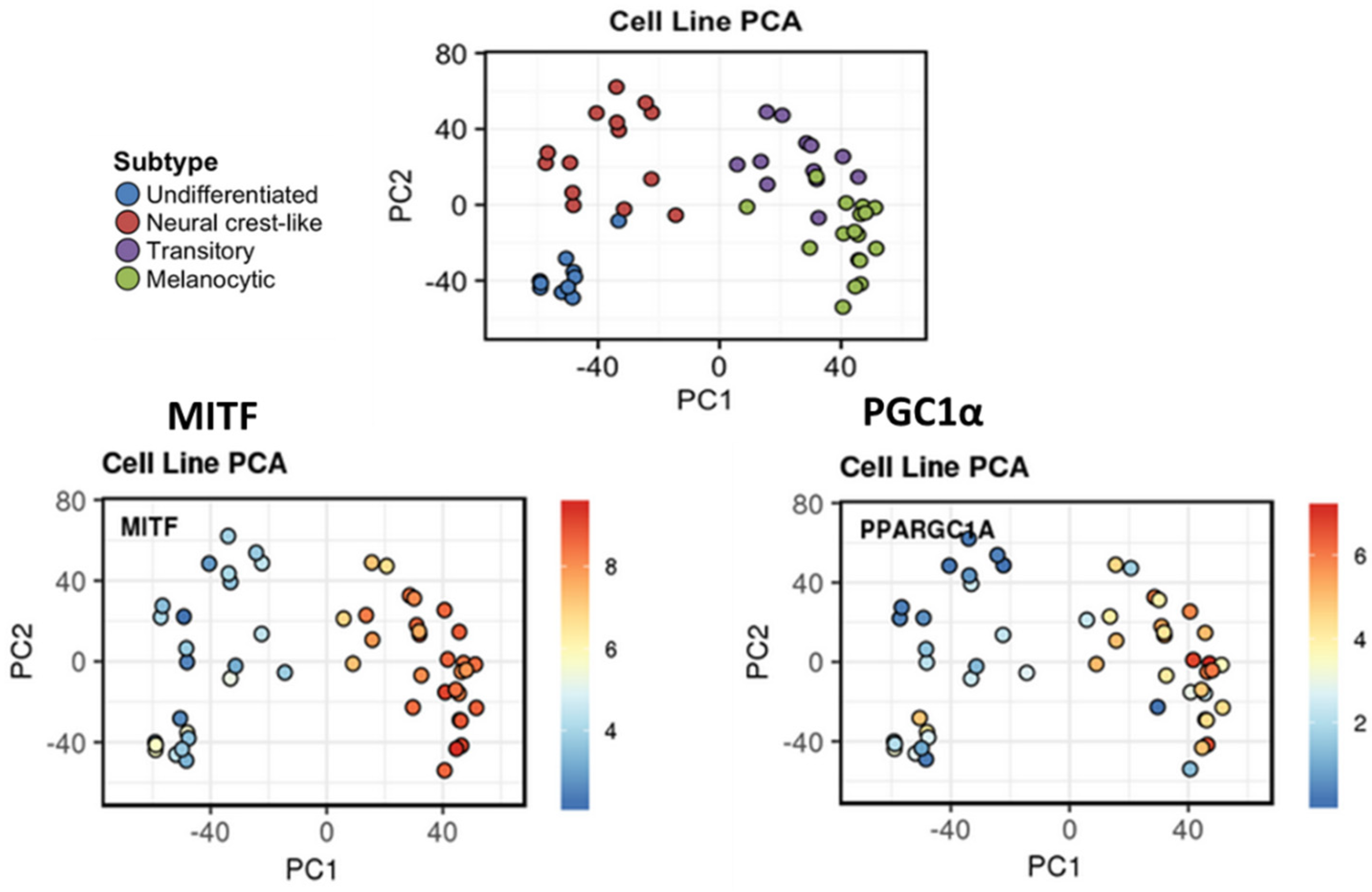
6.2. MITF-PGC1α Axis in the Regulation of Oxidative Stress and Phenotypic Plasticity
6.3. Oxidative Stress Defines the Invasive/Undifferentiated Phenotype
7. Targeting Phenotypic Plasticity: Identifying New Vulnerabilities of Melanoma Phenotypes and Future Challenges
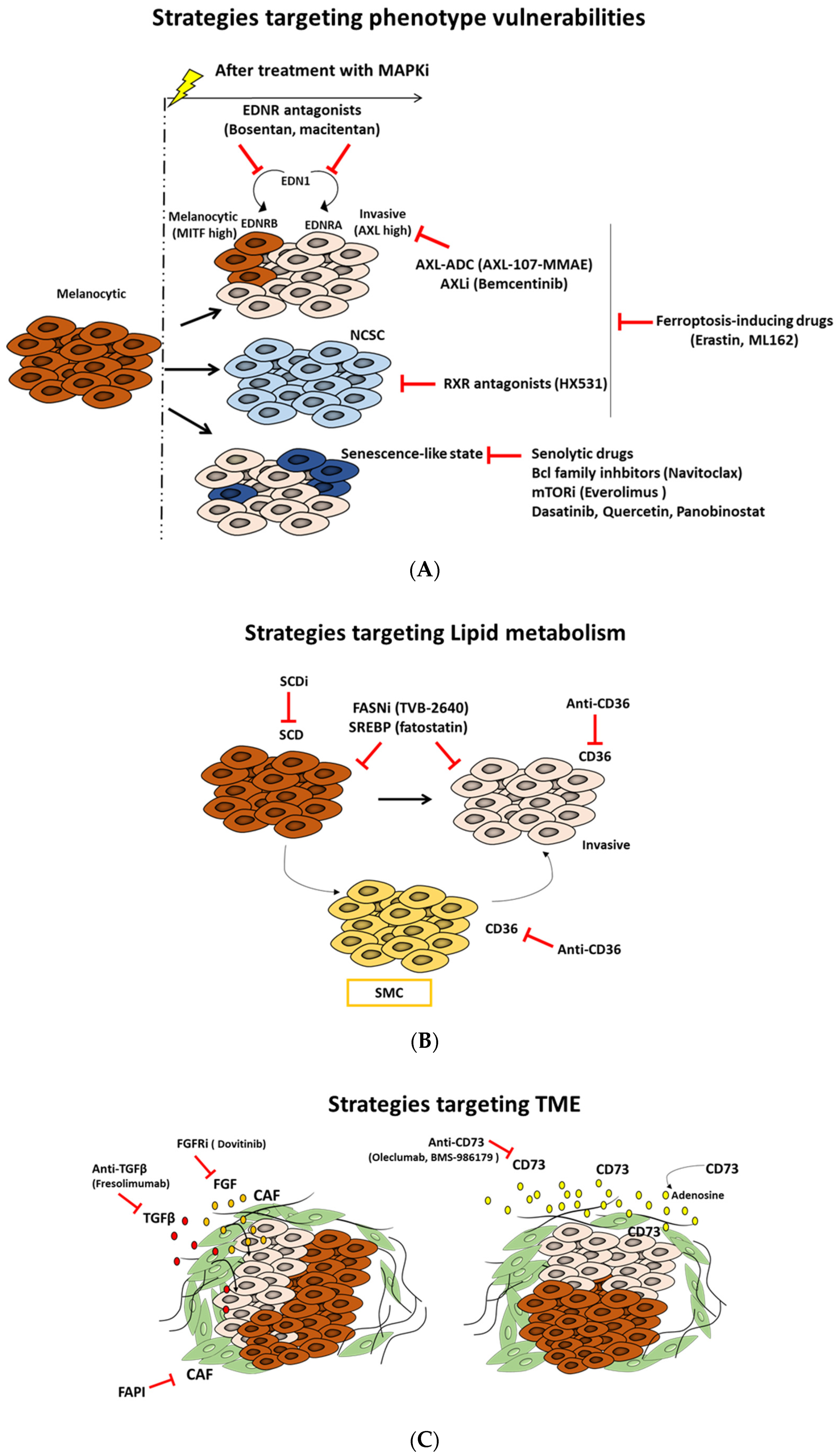
7.1. Targeting Phenotype-Specific Vulnerabilities
7.1.1. Targeting AXL Receptor Tyrosine Kinase as a New Promising Approach
7.1.2. Targeting The Neural Crest Stem Cell (NCSC) State and RXRG Signaling to Delay Melanoma Relapse
7.1.3. Targeting Ferroptosis to Block the Dedifferentiation Resistance Escape Route
7.1.4. Targeting the Senescence-like Phenotype
7.1.5. Targeting Endothelin Receptor Signaling as a Unique Approach to Overcome Phenotypic Heterogeneity
7.2. Targeting Lipid Metabolism as a New Strategy to Overcome Phenotypic Plasticity
7.2.1. Blocking Fatty Acid Translocase (FAT/CD36)
7.2.2. SCD Modulation
7.2.3. SREBPs Inhibition
7.2.4. Fatty Acid Synthase (FASN) Inhibition
7.3. Targeting Glutamine Metabolism to Reduce Melanoma Plasticity and Aggressiveness
7.3.1. Blocking Glutamine Import
7.3.2. Blocking Glutamine Use
7.4. Targeting TME Components and Signals
7.4.1. Targeting Cancer-Associated Fibroblasts (CAFs) to Disrupt Melanoma Plasticity
7.4.2. Targeting CD73 as a New Potential Therapeutic Opportunity
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Najem, A.; Krayem, M.; Perdrix, A.; Kerger, J.; Awada, A.; Journe, F.; Ghanem, G. New Drug Combination Strategies in Melanoma: Current Status and Future Directions. Anticancer Res. 2017, 37, 5941–5953. [Google Scholar] [PubMed] [Green Version]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef] [PubMed]
- Wouters, J.; Kalender-Atak, Z.; Minnoye, L.; Spanier, K.I.; De Waegeneer, M.; Bravo González-Blas, C.; Mauduit, D.; Davie, K.; Hulselmans, G.; Najem, A.; et al. Robust Gene Expression Programs Underlie Recurrent Cell States and Phenotype Switching in Melanoma. Nat. Cell Biol. 2020, 22, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the Regulatory Landscape of Melanoma Reveals TEADS as Regulators of the Invasive Cell State. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef] [Green Version]
- Rambow, F.; Marine, J.-C.; Goding, C.R. Melanoma Plasticity and Phenotypic Diversity: Therapeutic Barriers and Opportunities. Genes Dev. 2019, 33, 1295–1318. [Google Scholar] [CrossRef] [Green Version]
- Hoek, K.S.; Schlegel, N.C.; Brafford, P.; Sucker, A.; Ugurel, S.; Kumar, R.; Weber, B.L.; Nathanson, K.L.; Phillips, D.J.; Herlyn, M.; et al. Metastatic Potential of Melanomas Defined by Specific Gene Expression Profiles with No BRAF Signature. Pigment Cell Res. 2006, 19, 290–302. [Google Scholar] [CrossRef]
- Goding, C.R. Commentary. A Picture of Mitf in Melanoma Immortality. Oncogene 2011, 30, 2304–2306. [Google Scholar] [CrossRef]
- Hartman, M.L.; Czyz, M. MITF in Melanoma: Mechanisms behind Its Expression and Activity. Cell. Mol. Life Sci. 2015, 72, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Hoek, K.S.; Goding, C.R. Cancer Stem Cells versus Phenotype-Switching in Melanoma. Pigment Cell Melanoma Res. 2010, 23, 746–759. [Google Scholar] [CrossRef]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denecker, G.; Vandamme, N.; Akay, O.; Koludrovic, D.; Taminau, J.; Lemeire, K.; Gheldof, A.; De Craene, B.; Van Gele, M.; Brochez, L.; et al. Identification of a ZEB2-MITF-ZEB1 Transcriptional Network That Controls Melanogenesis and Melanoma Progression. Cell Death Differ. 2014, 21, 1250–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, G.; Dalle, S.; Monet, M.-A.; Ligier, M.; Boespflug, A.; Pommier, R.M.; de la Fouchardière, A.; Perier-Muzet, M.; Depaepe, L.; Barnault, R.; et al. ZEB1-Mediated Melanoma Cell Plasticity Enhances Resistance to MAPK Inhibitors. EMBO Mol. Med. 2016, 8, 1143–1161. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Arozarena, I. Microphthalmia-Associated Transcription Factor in Melanoma Development and MAP-Kinase Pathway Targeted Therapy. Pigment Cell Melanoma Res. 2015, 28, 390–406. [Google Scholar] [CrossRef] [Green Version]
- Dissanayake, S.K.; Olkhanud, P.B.; O’Connell, M.P.; Carter, A.; French, A.D.; Camilli, T.C.; Emeche, C.D.; Hewitt, K.J.; Rosenthal, D.T.; Leotlela, P.D.; et al. Wnt5A Regulates Expression of Tumor-Associated Antigens in Melanoma via Changes in Signal Transducers and Activators of Transcription 3 Phosphorylation. Cancer Res. 2008, 68, 10205–10214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fane, M.E.; Chhabra, Y.; Smith, A.G.; Sturm, R.A. BRN2, a POUerful Driver of Melanoma Phenotype Switching and Metastasis. Pigment Cell Melanoma Res. 2019, 32, 9–24. [Google Scholar] [CrossRef] [Green Version]
- Simmons, J.L.; Pierce, C.J.; Al-Ejeh, F.; Boyle, G.M. MITF and BRN2 Contribute to Metastatic Growth after Dissemination of Melanoma. Sci. Rep. 2017, 7, 10909. [Google Scholar] [CrossRef]
- Boyle, G.M.; Woods, S.L.; Bonazzi, V.F.; Stark, M.S.; Hacker, E.; Aoude, L.G.; Dutton-Regester, K.; Cook, A.L.; Sturm, R.A.; Hayward, N.K. Melanoma Cell Invasiveness Is Regulated by MiR-211 Suppression of the BRN2 Transcription Factor. Pigment Cell Melanoma Res. 2011, 24, 525–537. [Google Scholar] [CrossRef]
- Fane, M.E.; Chhabra, Y.; Hollingsworth, D.E.J.; Simmons, J.L.; Spoerri, L.; Oh, T.G.; Chauhan, J.; Chin, T.; Harris, L.; Harvey, T.J.; et al. NFIB Mediates BRN2 Driven Melanoma Cell Migration and Invasion Through Regulation of EZH2 and MITF. EBioMedicine 2017, 16, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Shakhova, O.; Cheng, P.; Mishra, P.J.; Zingg, D.; Schaefer, S.M.; Debbache, J.; Häusel, J.; Matter, C.; Guo, T.; Davis, S.; et al. Antagonistic Cross-Regulation between Sox9 and Sox10 Controls an Anti-Tumorigenic Program in Melanoma. PLoS Genet. 2015, 11, e1004877. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.M.; Foppen, M.H.G.; et al. Low MITF/AXL Ratio Predicts Early Resistance to Multiple Targeted Drugs in Melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef] [PubMed]
- Riesenberg, S.; Groetchen, A.; Siddaway, R.; Bald, T.; Reinhardt, J.; Smorra, D.; Kohlmeyer, J.; Renn, M.; Phung, B.; Aymans, P.; et al. MITF and C-Jun Antagonism Interconnects Melanoma Dedifferentiation with pro-Inflammatory Cytokine Responsiveness and Myeloid Cell Recruitment. Nat. Commun. 2015, 6, 8755. [Google Scholar] [CrossRef] [PubMed]
- Boshuizen, J.; Koopman, L.A.; Krijgsman, O.; Shahrabi, A.; van den Heuvel, E.G.-; Ligtenberg, M.A.; Vredevoogd, D.W.; Kemper, K.; Kuilman, T.; Song, J.-Y.; et al. Cooperative Targeting of Melanoma Heterogeneity with an AXL Antibody-Drug Conjugate and BRAF/MEK Inhibitors. Nat. Med. 2018, 24, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Erin Chen, Y.; Kumar, R.; Taylor, M.; Jenny Njauw, C.-N.; Miao, B.; Frederick, D.T.; Wargo, J.A.; Flaherty, K.T.; Jönsson, G.; et al. MITF Modulates Therapeutic Resistance through EGFR Signaling. J. Investig. Dermatol. 2015, 135, 1863–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-Stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, M.P.; Marchbank, K.; Webster, M.R.; Valiga, A.A.; Kaur, A.; Vultur, A.; Li, L.; Herlyn, M.; Villanueva, J.; Liu, Q.; et al. Hypoxia Induces Phenotypic Plasticity and Therapy Resistance in Melanoma via the Tyrosine Kinase Receptors ROR1 and ROR2. Cancer Discov. 2013, 3, 1378–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, M.P.; Weeraratna, A.T. Change Is in the Air: The Hypoxic Induction of Phenotype Switching in Melanoma. J. Investig. Dermatol. 2013, 133, 2316–2317. [Google Scholar] [CrossRef] [Green Version]
- Li, F.Z.; Dhillon, A.S.; Anderson, R.L.; McArthur, G.; Ferrao, P.T. Phenotype Switching in Melanoma: Implications for Progression and Therapy. Front. Oncol. 2015, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Janssen, S.M.; Moscona, R.; Elchebly, M.; Papadakis, A.I.; Redpath, M.; Wang, H.; Rubin, E.; van Kempen, L.C.; Spatz, A. BORIS/CTCFL Promotes a Switch from a Proliferative towards an Invasive Phenotype in Melanoma Cells. Cell Death Discov. 2020, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Pierrat, M.-J.; Marsaud, V.; Mauviel, A.; Javelaud, D. Expression of Microphthalmia-Associated Transcription Factor (MITF), Which Is Critical for Melanoma Progression, Is Inhibited by Both Transcription Factor GLI2 and Transforming Growth Factor-β. J. Biol. Chem. 2012, 287, 17996–18004. [Google Scholar] [CrossRef] [Green Version]
- Huh, H.D.; Kim, D.H.; Jeong, H.-S.; Park, H.W. Regulation of TEAD Transcription Factors in Cancer Biology. Cells 2019, 8, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent-Mistiaen, Z.; Elbediwy, A.; Vanyai, H.; Cotton, J.; Stamp, G.; Nye, E.; Spencer-Dene, B.; Thomas, G.J.; Mao, J.; Thompson, B. YAP Drives Cutaneous Squamous Cell Carcinoma Formation and Progression. eLife 2018, 7, e33304. [Google Scholar] [CrossRef] [PubMed]
- Hiemer, S.E.; Szymaniak, A.D.; Varelas, X. The Transcriptional Regulators TAZ and YAP Direct Transforming Growth Factor β-Induced Tumorigenic Phenotypes in Breast Cancer Cells. J. Biol. Chem. 2014, 289, 13461–13474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagai, L.; Peri-Naor, R.; Birk, R.Z. Docosahexaenoic Acid Significantly Stimulates Immediate Early Response Genes and Neurite Outgrowth. Neurochem. Res. 2009, 34, 867–875. [Google Scholar] [CrossRef]
- Baron, V.T.; Pio, R.; Jia, Z.; Mercola, D. Early Growth Response 3 Regulates Genes of Inflammation and Directly Activates IL6 and IL8 Expression in Prostate Cancer. Br. J. Cancer 2015, 112, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.V.; Birdsey, G.M.; Randi, A.M. Regulation of Endothelial Homeostasis, Vascular Development and Angiogenesis by the Transcription Factor ERG. Vasc. Pharmacol. 2016, 86, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Inoue, A.; Miki, Y.; Moriya, T.; Akahira, J.; Ishida, T.; Hirakawa, H.; Yamaguchi, Y.; Hayashi, S.; Sasano, H. Early Growth Responsive Gene 3 in Human Breast Carcinoma: A Regulator of Estrogen-Meditated Invasion and a Potent Prognostic Factor. Endocr. Relat. Cancer 2007, 14, 279–292. [Google Scholar] [CrossRef]
- Chien, M.-H.; Lee, W.-J.; Yang, Y.-C.; Li, Y.-L.; Chen, B.-R.; Cheng, T.-Y.; Yang, P.-W.; Wang, M.-Y.; Jan, Y.-H.; Lin, Y.-K.; et al. KSRP Suppresses Cell Invasion and Metastasis through MiR-23a-Mediated EGR3 MRNA Degradation in Non-Small Cell Lung Cancer. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 1013–1024. [Google Scholar] [CrossRef]
- Xiao, Z.-J.; Liu, J.; Wang, S.-Q.; Zhu, Y.; Gao, X.-Y.; Tin, V.P.-C.; Qin, J.; Wang, J.-W.; Wong, M.P. NFATc2 Enhances Tumor-Initiating Phenotypes through the NFATc2/SOX2/ALDH Axis in Lung Adenocarcinoma. eLife 2017, 6, e26733. [Google Scholar] [CrossRef]
- Perotti, V.; Baldassari, P.; Molla, A.; Vegetti, C.; Bersani, I.; Maurichi, A.; Santinami, M.; Anichini, A.; Mortarini, R. NFATc2 Is an Intrinsic Regulator of Melanoma Dedifferentiation. Oncogene 2016, 35, 2862–2872. [Google Scholar] [CrossRef]
- Kim, G.-C.; Kwon, H.-K.; Lee, C.-G.; Verma, R.; Rudra, D.; Kim, T.; Kang, K.; Nam, J.H.; Kim, Y.; Im, S.-H. Upregulation of Ets1 Expression by NFATc2 and NFKB1/RELA Promotes Breast Cancer Cell Invasiveness. Oncogenesis 2018, 7, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perotti, V.; Baldassari, P.; Bersani, I.; Molla, A.; Vegetti, C.; Tassi, E.; Col, J.D.; Dolcetti, R.; Anichini, A.; Mortarini, R. NFATc2 Is a Potential Therapeutic Target in Human Melanoma. J. Investig. Dermatol. 2012, 132, 2652–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eccles, M.R.; He, S.; Ahn, A.; Slobbe, L.J.; Jeffs, A.R.; Yoon, H.-S.; Baguley, B.C. MITF and PAX3 Play Distinct Roles in Melanoma Cell Migration; Outline of a “Genetic Switch” Theory Involving MITF and PAX3 in Proliferative and Invasive Phenotypes of Melanoma. Front. Oncol. 2013, 3, 229. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.P.; Rana, S.; Ferguson, J.; Rowling, E.J.; Flaherty, K.T.; Wargo, J.A.; Marais, R.; Wellbrock, C. A PAX3/BRN2 Rheostat Controls the Dynamics of BRAF Mediated MITF Regulation in MITFhigh/AXLlow Melanoma. Pigment Cell Melanoma Res. 2019, 32, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Aloia, A.; Müllhaupt, D.; Chabbert, C.D.; Eberhart, T.; Flückiger-Mangual, S.; Vukolic, A.; Eichhoff, O.; Irmisch, A.; Alexander, L.T.; Scibona, E.; et al. A Fatty Acid Oxidation-Dependent Metabolic Shift Regulates the Adaptation of BRAF-Mutated Melanoma to MAPK Inhibitors. Clin. Cancer Res. 2019, 25, 6852–6867. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Fisher, D.E.; Flaherty, K.T. Cell-State Dynamics and Therapeutic Resistance in Melanoma from the Perspective of MITF and IFNγ Pathways. Nat. Rev. Clin. Oncol. 2019, 16, 549–562. [Google Scholar] [CrossRef]
- Krayem, M.; Najem, A.; Journe, F.; Morandini, R.; Sales, F.; Awada, A.; Ghanem, G.E. Acquired Resistance to BRAFi Reverses Senescence-like Phenotype in Mutant BRAF Melanoma. Oncotarget 2018, 9, 31888–31903. [Google Scholar] [CrossRef]
- Najem, A.; Wouters, J.; Krayem, M.; Rambow, F.; Sabbah, M.; Sales, F.; Awada, A.; Aerts, S.; Journe, F.; Marine, J.-C.; et al. Tyrosine-Dependent Phenotype Switching Occurs Early in Many Primary Melanoma Cultures Limiting Their Translational Value. Front. Oncol. 2021, 11, 780654. [Google Scholar] [CrossRef]
- Strub, T.; Ballotti, R.; Bertolotto, C. The “ART” of Epigenetics in Melanoma: From Histone “Alterations, to Resistance and Therapies”. Theranostics 2020, 10, 1777–1797. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming Intrinsic Multidrug Resistance in Melanoma by Blocking the Mitochondrial Respiratory Chain of Slow-Cycling JARID1Bhigh Cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauvistré, H.; Daignault, S.M.; Shannan, B.; Ju, R.J.; Picard, D.; Vogel, F.C.E.; Egetemaier, S.; Krepler, C.; Rebecca, V.W.; Sechi, A.; et al. The Janus-Faced Role of KDM5B Heterogeneity in Melanoma: Differentiation as a Situational Driver of Both Growth Arrest and Drug-Resistance. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Kadoch, C.; Crabtree, G.R. Mammalian SWI/SNF Chromatin Remodeling Complexes and Cancer: Mechanistic Insights Gained from Human Genomics. Sci. Adv. 2015, 1, e1500447. [Google Scholar] [CrossRef] [Green Version]
- Laurette, P.; Strub, T.; Koludrovic, D.; Keime, C.; Le Gras, S.; Seberg, H.; Van Otterloo, E.; Imrichova, H.; Siddaway, R.; Aerts, S.; et al. Transcription Factor MITF and Remodeller BRG1 Define Chromatin Organisation at Regulatory Elements in Melanoma Cells. eLife 2015, 4, e06857. [Google Scholar] [CrossRef]
- Cheng, P.F.; Shakhova, O.; Widmer, D.S.; Eichhoff, O.M.; Zingg, D.; Frommel, S.C.; Belloni, B.; Raaijmakers, M.I.; Goldinger, S.M.; Santoro, R.; et al. Methylation-Dependent SOX9 Expression Mediates Invasion in Human Melanoma Cells and Is a Negative Prognostic Factor in Advanced Melanoma. Genome Biol. 2015, 16, 42. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Fullwood, M.J. Roles, Functions, and Mechanisms of Long Non-Coding RNAs in Cancer. Genom. Proteom. Bioinform. 2016, 14, 42–54. [Google Scholar] [CrossRef] [Green Version]
- Coe, E.A.; Tan, J.Y.; Shapiro, M.; Louphrasitthiphol, P.; Bassett, A.R.; Marques, A.C.; Goding, C.R.; Vance, K.W. The MITF-SOX10 Regulated Long Non-Coding RNA DIRC3 Is a Melanoma Tumour Suppressor. PLoS Genet. 2019, 15, e1008501. [Google Scholar] [CrossRef] [Green Version]
- Siena, Á.D.D.; Plaça, J.R.; Araújo, L.F.; de Barros, I.I.; Peronni, K.; Molfetta, G.; de Biagi, C.A.O.; Espreafico, E.M.; Sousa, J.F.; Silva, W.A. Whole Transcriptome Analysis Reveals Correlation of Long Noncoding RNA ZEB1-AS1 with Invasive Profile in Melanoma. Sci. Rep. 2019, 9, 11350. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhu, B.; Li, X.-B.; Cao, Y.-Q.; Yang, J.-C.; Li, X.; Liu, Y.-X.; Wang, Y.-B. Long Non-Coding RNA SNHG7 Promotes Migration and Invasion of Melanoma via Upregulating SOX4. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7553. [Google Scholar] [CrossRef]
- Vervoort, S.J.; Lourenço, A.R.; van Boxtel, R.; Coffer, P.J. SOX4 Mediates TGF-β-Induced Expression of Mesenchymal Markers during Mammary Cell Epithelial to Mesenchymal Transition. PLoS ONE 2013, 8, e53238. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, L.; Song, P.; Xu, J.; Li, G. Long Non-Coding RNA PANDAR Promotes Melanoma Cell Invasion through Regulating Epithelial-Mesenchymal Transition. Int. J. Clin. Exp. Pathol. 2018, 11, 2430–2439. [Google Scholar]
- Schmidt, K.; Joyce, C.E.; Buquicchio, F.; Brown, A.; Ritz, J.; Distel, R.J.; Yoon, C.H.; Novina, C.D. The LncRNA SLNCR1 Mediates Melanoma Invasion through a Conserved SRA1-like Region. Cell Rep. 2016, 15, 2025–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, K.; Carroll, J.S.; Yee, E.; Thomas, D.D.; Wert-Lamas, L.; Neier, S.C.; Sheynkman, G.; Ritz, J.; Novina, C.D. The LncRNA SLNCR Recruits the Androgen Receptor to EGR1-Bound Genes in Melanoma and Inhibits Expression of Tumor Suppressor P21. Cell Rep. 2019, 27, 2493–2507.e4. [Google Scholar] [CrossRef] [Green Version]
- Varrone, F.; Caputo, E. The MiRNAs Role in Melanoma and in Its Resistance to Therapy. Int. J. Mol. Sci. 2020, 21, 878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattore, L.; Costantini, S.; Malpicci, D.; Ruggiero, C.F.; Ascierto, P.A.; Croce, C.M.; Mancini, R.; Ciliberto, G. MicroRNAs in Melanoma Development and Resistance to Target Therapy. Oncotarget 2017, 8, 22262–22278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzywa, T.M.; Klicka, K.; Paskal, W.; Dudkiewicz, J.; Wejman, J.; Pyzlak, M.; Włodarski, P.K. MiR-410-3p Is Induced by Vemurafenib via ER Stress and Contributes to Resistance to BRAF Inhibitor in Melanoma. PLoS ONE 2020, 15, e0234707. [Google Scholar] [CrossRef] [PubMed]
- Segura, M.F.; Hanniford, D.; Menendez, S.; Reavie, L.; Zou, X.; Alvarez-Diaz, S.; Zakrzewski, J.; Blochin, E.; Rose, A.; Bogunovic, D.; et al. Aberrant MiR-182 Expression Promotes Melanoma Metastasis by Repressing FOXO3 and Microphthalmia-Associated Transcription Factor. Proc. Natl. Acad. Sci. USA 2009, 106, 1814–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg Effect: 80 Years On. Biochem Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Comparative Metabolic Flux Profiling of Melanoma Cell Lines: Beyond the Warburg Effect. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, J.; de Moura, M.B.; Lin, Y.; Vincent, G.; Thorne, S.; Duncan, L.M.; Hui-Min, L.; Kirkwood, J.M.; Becker, D.; Van Houten, B.; et al. Importance of Glycolysis and Oxidative Phosphorylation in Advanced Melanoma. Mol. Cancer 2012, 11, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feichtinger, R.G.; Lang, R.; Geilberger, R.; Rathje, F.; Mayr, J.A.; Sperl, W.; Bauer, J.W.; Hauser-Kronberger, C.; Kofler, B.; Emberger, M. Melanoma Tumors Exhibit a Variable but Distinct Metabolic Signature. Exp. Dermatol. 2018, 27, 204–207. [Google Scholar] [CrossRef] [Green Version]
- Vandyck, H.H.; Hillen, L.M.; Bosisio, F.M.; van den Oord, J.; Zur Hausen, A.; Winnepenninckx, V. Rethinking the Biology of Metastatic Melanoma: A Holistic Approach. Cancer Metastasis Rev. 2021, 40, 603–624. [Google Scholar] [CrossRef]
- Baenke, F.; Chaneton, B.; Smith, M.; Van Den Broek, N.; Hogan, K.; Tang, H.; Viros, A.; Martin, M.; Galbraith, L.; Girotti, M.R.; et al. Resistance to BRAF Inhibitors Induces Glutamine Dependency in Melanoma Cells. Mol. Oncol. 2016, 10, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Soumoy, L.; Schepkens, C.; Krayem, M.; Najem, A.; Tagliatti, V.; Ghanem, G.E.; Saussez, S.; Colet, J.-M.; Journe, F. Metabolic Reprogramming in Metastatic Melanoma with Acquired Resistance to Targeted Therapies: Integrative Metabolomic and Proteomic Analysis. Cancers 2020, 12, 1323. [Google Scholar] [CrossRef]
- Vara-Perez, M.; Maes, H.; Van Dingenen, S.; Agostinis, P. BNIP3 Contributes to the Glutamine-Driven Aggressive Behavior of Melanoma Cells. Biol. Chem. 2019, 400, 187–193. [Google Scholar] [CrossRef]
- Ohanna, M.; Cerezo, M.; Nottet, N.; Bille, K.; Didier, R.; Beranger, G.; Mograbi, B.; Rocchi, S.; Yvan-Charvet, L.; Ballotti, R.; et al. Pivotal Role of NAMPT in the Switch of Melanoma Cells toward an Invasive and Drug-Resistant Phenotype. Genes Dev. 2018, 32, 448–461. [Google Scholar] [CrossRef]
- Paulitschke, V.; Eichhoff, O.; Gerner, C.; Paulitschke, P.; Bileck, A.; Mohr, T.; Cheng, P.F.; Leitner, A.; Guenova, E.; Saulite, I.; et al. Proteomic Identification of a Marker Signature for MAPKi Resistance in Melanoma. EMBO J. 2019, 38, e95874. [Google Scholar] [CrossRef]
- Pellerin, L.; Carrié, L.; Dufau, C.; Nieto, L.; Ségui, B.; Levade, T.; Riond, J.; Andrieu-Abadie, N. Lipid Metabolic Reprogramming: Role in Melanoma Progression and Therapeutic Perspectives. Cancers 2020, 12, 3147. [Google Scholar] [CrossRef]
- Vivas-García, Y.; Falletta, P.; Liebing, J.; Louphrasitthiphol, P.; Feng, Y.; Chauhan, J.; Scott, D.A.; Glodde, N.; Chocarro-Calvo, A.; Bonham, S.; et al. Lineage-Restricted Regulation of SCD and Fatty Acid Saturation by MITF Controls Melanoma Phenotypic Plasticity. Mol. Cell 2020, 77, 120–137.e9. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, J.; Garandeau, D.; Pandiani, C.; Gaudel, C.; Bille, K.; Nottet, N.; Garcia, V.; Colosetti, P.; Pagnotta, S.; Bahadoran, P.; et al. Lysosomal Acid Ceramidase ASAH1 Controls the Transition between Invasive and Proliferative Phenotype in Melanoma Cells. Oncogene 2019, 38, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Louphrasitthiphol, P.; Ledaki, I.; Chauhan, J.; Falletta, P.; Siddaway, R.; Buffa, F.M.; Mole, D.R.; Soga, T.; Goding, C.R. MITF Controls the TCA Cycle to Modulate the Melanoma Hypoxia Response. Pigment Cell Melanoma Res. 2019, 32, 792–808. [Google Scholar] [CrossRef] [PubMed]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF Regulates Oxidative Metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [Green Version]
- Widmer, D.S.; Hoek, K.S.; Cheng, P.F.; Eichhoff, O.M.; Biedermann, T.; Raaijmakers, M.I.G.; Hemmi, S.; Dummer, R.; Levesque, M.P. Hypoxia Contributes to Melanoma Heterogeneity by Triggering HIF1α-Dependent Phenotype Switching. J. Investig. Dermatol. 2013, 133, 2436–2443. [Google Scholar] [CrossRef] [Green Version]
- Cheli, Y.; Giuliano, S.; Fenouille, N.; Allegra, M.; Hofman, V.; Hofman, P.; Bahadoran, P.; Lacour, J.-P.; Tartare-Deckert, S.; Bertolotto, C.; et al. Hypoxia and MITF Control Metastatic Behaviour in Mouse and Human Melanoma Cells. Oncogene 2012, 31, 2461–2470. [Google Scholar] [CrossRef] [Green Version]
- Hanna, S.C.; Krishnan, B.; Bailey, S.T.; Moschos, S.J.; Kuan, P.-F.; Shimamura, T.; Osborne, L.D.; Siegel, M.B.; Duncan, L.M.; O’Brien, E.T.; et al. HIF1α and HIF2α Independently Activate SRC to Promote Melanoma Metastases. J. Clin. Investig. 2013, 123, 2078–2093. [Google Scholar] [CrossRef] [Green Version]
- Hölzel, M.; Tüting, T. Inflammation-Induced Plasticity in Melanoma Therapy and Metastasis. Trends Immunol. 2016, 37, 364–374. [Google Scholar] [CrossRef]
- Falletta, P.; Sanchez-Del-Campo, L.; Chauhan, J.; Effern, M.; Kenyon, A.; Kershaw, C.J.; Siddaway, R.; Lisle, R.; Freter, R.; Daniels, M.J.; et al. Translation Reprogramming Is an Evolutionarily Conserved Driver of Phenotypic Plasticity and Therapeutic Resistance in Melanoma. Genes Dev. 2017, 31, 18–33. [Google Scholar] [CrossRef] [Green Version]
- Iurlaro, R.; Püschel, F.; León-Annicchiarico, C.L.; O’Connor, H.; Martin, S.J.; Palou-Gramón, D.; Lucendo, E.; Muñoz-Pinedo, C. Glucose Deprivation Induces ATF4-Mediated Apoptosis through TRAIL Death Receptors. Mol. Cell Biol. 2017, 37, e00479-16. [Google Scholar] [CrossRef] [Green Version]
- Gameiro, P.A.; Struhl, K. Nutrient Deprivation Elicits a Transcriptional and Translational Inflammatory Response Coupled to Decreased Protein Synthesis. Cell Rep. 2018, 24, 1415–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leucci, E.; Close, P.; Marine, J.-C. Translation Rewiring at the Heart of Phenotype Switching in Melanoma. Pigment Cell Melanoma Res. 2017, 30, 282–283. [Google Scholar] [CrossRef] [PubMed]
- Phung, B.; Cieśla, M.; Sanna, A.; Guzzi, N.; Beneventi, G.; Cao Thi Ngoc, P.; Lauss, M.; Cabrita, R.; Cordero, E.; Bosch, A.; et al. The X-Linked DDX3X RNA Helicase Dictates Translation Reprogramming and Metastasis in Melanoma. Cell Rep. 2019, 27, 3573–3586.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Kwa, M.Q.; Herum, K.M.; Brakebusch, C. Cancer-Associated Fibroblasts: How Do They Contribute to Metastasis? Clin. Exp. Metastasis 2019, 36, 71–86. [Google Scholar] [CrossRef]
- Passarelli, A.; Tucci, M.; Mannavola, F.; Felici, C.; Silvestris, F. The Metabolic Milieu in Melanoma: Role of Immune Suppression by CD73/Adenosine. Tumour Biol. 2019, 42, 1010428319837138. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, J.; Landsberg, J.; Schmid-Burgk, J.L.; Ramis, B.B.; Bald, T.; Glodde, N.; Lopez-Ramos, D.; Young, A.; Ngiow, S.F.; Nettersheim, D.; et al. MAPK Signaling and Inflammation Link Melanoma Phenotype Switching to Induction of CD73 during Immunotherapy. Cancer Res. 2017, 77, 4697–4709. [Google Scholar] [CrossRef] [Green Version]
- Chambers, A.M.; Matosevic, S. Immunometabolic Dysfunction of Natural Killer Cells Mediated by the Hypoxia-CD73 Axis in Solid Tumors. Front. Mol. Biosci. 2019, 6, 60. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Feng, B.; Meng, Y.; Wang, J.; Geng, B.; Cui, Q.; Zhang, H.; Yang, Y.; Yang, J. FAM3C-YY1 Axis Is Essential for TGFβ-promoted Proliferation and Migration of Human Breast Cancer MDA-MB-231 Cells via the Activation of HSF1. J. Cell Mol. Med. 2019, 23, 3464–3475. [Google Scholar] [CrossRef]
- Noguchi, K.; Dalton, A.C.; Howley, B.V.; McCall, B.J.; Yoshida, A.; Diehl, J.A.; Howe, P.H. Interleukin-like EMT Inducer Regulates Partial Phenotype Switching in MITF-Low Melanoma Cell Lines. PLoS ONE 2017, 12, e0177830. [Google Scholar] [CrossRef]
- Bagnato, A.; Spinella, F.; Rosanò, L. The Endothelin Axis in Cancer: The Promise and the Challenges of Molecularly Targeted Therapy. Can. J. Physiol. Pharm. 2008, 86, 473–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinella, F.; Caprara, V.; Cianfrocca, R.; Rosanò, L.; Di Castro, V.; Garrafa, E.; Natali, P.G.; Bagnato, A. The Interplay between Hypoxia, Endothelial and Melanoma Cells Regulates Vascularization and Cell Motility through Endothelin-1 and Vascular Endothelial Growth Factor. Carcinogenesis 2014, 35, 840–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.P.; Rowling, E.J.; Miskolczi, Z.; Ferguson, J.; Spoerri, L.; Haass, N.K.; Sloss, O.; McEntegart, S.; Arozarena, I.; von Kriegsheim, A.; et al. Targeting Endothelin Receptor Signalling Overcomes Heterogeneity Driven Therapy Failure. EMBO Mol. Med. 2017, 9, 1011–1029. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, S.A.N.; Finlay, G.J.; Baguley, B.C.; Askarian-Amiri, M.E. Signaling Pathways in Melanogenesis. Int. J. Mol. Sci 2016, 17, 1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulton, S.J.; Birch-Machin, M.A. Impact of Hyperpigmentation on Superoxide Flux and Melanoma Cell Metabolism at Mitochondrial Complex II. FASEB J. 2014, 29, 346–353. [Google Scholar] [CrossRef] [Green Version]
- Slominski, R.M.; Zmijewski, M.A.; Slominski, A.T. The Role of Melanin Pigment in Melanoma. Exp. Derm. 2015, 24, 258–259. [Google Scholar] [CrossRef] [Green Version]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF Inhibition Is Associated with Enhanced Melanoma Antigen Expression and a More Favorable Tumor Microenvironment in Patients with Metastatic Melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef] [Green Version]
- Funasaka, Y.; Sato, H.; Chakraborty, A.K.; Ohashi, A.; Chrousos, G.P.; Ichihashi, M. Expression of Proopiomelanocortin, Corticotropin-Releasing Hormone (CRH), and CRH Receptor in Melanoma Cells, Nevus Cells, and Normal Human Melanocytes. J. Investig. Dermatol. Symp. Proc. 1999, 4, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, M.; Adachi, Y.; Mitsushita, J.; Kuwabara, M.; Nagasawa, A.; Harada, S.; Furuta, S.; Zhang, Y.; Seheli, K.; Miyazaki, H.; et al. Reactive Oxygen Generated by NADPH Oxidase 1 (Nox1) Contributes to Cell Invasion by Regulating Matrix Metalloprotease-9 Production and Cell Migration. J. Biol. Chem. 2010, 285, 4481–4488. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Gomez Garcia, A.M.; Meyskens, F.L. NADPH Oxidase 1 Overexpression Enhances Invasion via Matrix Metalloproteinase-2 and Epithelial–Mesenchymal Transition in Melanoma Cells. J. Investig. Dermatol. 2012, 132, 2033–2041. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Peng, C.; Zhu, C.; Nie, S.; Qian, X.; Shi, Z.; Shi, M.; Liang, Y.; Ding, X.; Zhang, S.; et al. Hypoxia Promotes the Metastasis of Pancreatic Cancer through Regulating NOX4/KDM5A-Mediated Histone Methylation Modification Changes in a HIF1A-Independent Manner. Clin. Epigenetics 2021, 13, 18. [Google Scholar] [CrossRef] [PubMed]
- Abildgaard, C.; Guldberg, P. Molecular Drivers of Cellular Metabolic Reprogramming in Melanoma. Trends Mol. Med. 2015, 21, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Lim, J.-H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α Expression Defines a Subset of Human Melanoma Tumors with Increased Mitochondrial Capacity and Resistance to Oxidative Stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravel, S.-P. Deciphering the Dichotomous Effects of PGC-1α on Tumorigenesis and Metastasis. Front. Oncol. 2018, 8, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Journe, F.; Boufker, H.I.; Van Kempen, L.; Galibert, M.-D.; Wiedig, M.; Salès, F.; Theunis, A.; Nonclercq, D.; Frau, A.; Laurent, G.; et al. TYRP1 MRNA Expression in Melanoma Metastases Correlates with Clinical Outcome. Br. J. Cancer 2011, 105, 1726–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Lim, J.-H.; Lee, Y.; Granter, S.R.; Thomas, A.; Vazquez, F.; Widlund, H.R.; Puigserver, P. A PGC1α-Mediated Transcriptional Axis Suppresses Melanoma Metastasis. Nature 2016, 537, 422–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Balsa, E.; Perry, E.A.; Liang, J.; Tavares, C.D.; Vazquez, F.; Widlund, H.R.; Puigserver, P. H3K27me3-Mediated PGC1α Gene Silencing Promotes Melanoma Invasion through WNT5A and YAP. J. Clin. Investig. 2020, 130, 853–862. [Google Scholar] [CrossRef]
- Palomer, X.; Alvarez-Guardia, D.; Rodríguez-Calvo, R.; Coll, T.; Laguna, J.C.; Davidson, M.M.; Chan, T.O.; Feldman, A.M.; Vázquez-Carrera, M. TNF-α Reduces PGC-1α Expression through NF-κB and P38 MAPK Leading to Increased Glucose Oxidation in a Human Cardiac Cell Model. Cardiovasc. Res. 2009, 81, 703–712. [Google Scholar] [CrossRef]
- Lim, J.-H.; Luo, C.; Vazquez, F.; Puigserver, P. Targeting Mitochondrial Oxidative Metabolism in Melanoma Causes Metabolic Compensation through Glucose and Glutamine Utilization. Cancer Res. 2014, 74, 3535–3545. [Google Scholar] [CrossRef] [Green Version]
- Kaur, A.; Webster, M.R.; Marchbank, K.; Behera, R.; Ndoye, A.; Kugel, C.H.; Dang, V.M.; Appleton, J.; O’Connell, M.P.; Cheng, P.; et al. SFRP2 in the Aged Microenvironment Drives Melanoma Metastasis and Therapy Resistance. Nature 2016, 532, 250–254. [Google Scholar] [CrossRef] [Green Version]
- Santos Bernardes, S.; de Souza-Neto, F.P.; Pasqual Melo, G.; Guarnier, F.A.; Marinello, P.C.; Cecchini, R.; Cecchini, A.L. Correlation of TGF-Β1 and Oxidative Stress in the Blood of Patients with Melanoma: A Clue to Understanding Melanoma Progression? Tumour Biol. 2016, 37, 10753–10761. [Google Scholar] [CrossRef] [PubMed]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-Tolerant Persister Cancer Cells Are Vulnerable to GPX4 Inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Wei, Y.; Wei, X. AXL Receptor Tyrosine Kinase as a Promising Anti-Cancer Approach: Functions, Molecular Mechanisms and Clinical Applications. Mol. Cancer 2019, 18, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loges, S.; Heuser, M.; Chromik, J.; Vigil C., E.; Paschka, P.; Re, F.; Renzo N., D.; Lemoli R., M.; Mattei, D.G.; Ben-Batalla, I.; et al. The Combination of AXL Inhibitor Bemcentinib and Low Dose Cytarabine Is Well Tolerated and Efficacious in Elderly Relapsed AML Patients: Update from the Ongoing BGBC003 Phase II Trial (NCT02488408). Blood 2020, 136, 14. [Google Scholar] [CrossRef]
- Straume, O.; Lorens, J.B.; Gausdal, G.; Gjertsen, B.T.; Schuster, C. 1336TiP—A Randomized Phase Ib/II Study of the Selective Small Molecule Axl Inhibitor Bemcentinib (BGB324) in Combination with Either Dabrafenib/Trametinib (D/T) or Pembrolizumab in Patients with Metastatic Melanoma. Ann. Oncol. 2019, 30, v563. [Google Scholar] [CrossRef]
- Sabbah, M.; Najem, A.; Krayem, M.; Awada, A.; Journe, F.; Ghanem, G.E. RTK Inhibitors in Melanoma: From Bench to Bedside. Cancers 2021, 13, 1685. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Senolytic Drugs: From Discovery to Translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef]
- Zhu, M.; Meng, P.; Ling, X.; Zhou, L. Advancements in Therapeutic Drugs Targeting of Senescence. Adv. Chronic Dis. 2020, 11, 2040622320964125. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.-O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting Metastasis-Initiating Cells through the Fatty Acid Receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Yang, J.; Stack, M.S. Lipid Regulatory Proteins as Potential Therapeutic Targets for Ovarian Cancer in Obese Women. Cancers 2020, 12, 3469. [Google Scholar] [CrossRef]
- Fhu, C.W.; Ali, A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules 2020, 25, 3935. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of Fatty Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Davies, J.E.; Tran, T.Q.; Reid, M.A.; Rosales, K.R.; Lowman, X.H.; Pan, M.; Moriceau, G.; Yang, Y.; Wu, J.; Lo, R.S.; et al. Vemurafenib Resistance Reprograms Melanoma Cells towards Glutamine Dependence. J. Transl. Med. 2015, 13, 210. [Google Scholar] [CrossRef] [Green Version]
- Varghese, S.; Pramanik, S.; Williams, L.J.; Hodges, H.R.; Hudgens, C.W.; Fischer, G.M.; Luo, C.K.; Knighton, B.; Tan, L.; Lorenzi, P.L.; et al. The Glutaminase Inhibitor CB-839 (Telaglenastat) Enhances the Antimelanoma Activity of T-Cell-Mediated Immunotherapies. Mol. Cancer 2021, 20, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Lindner, T.; Loktev, A.; Giesel, F.; Kratochwil, C.; Altmann, A.; Haberkorn, U. Targeting of Activated Fibroblasts for Imaging and Therapy. EJNMMI Radiopharm. Chem. 2019, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Watabe, T.; Liu, Y.; Kaneda-Nakashima, K.; Shirakami, Y.; Lindner, T.; Ooe, K.; Toyoshima, A.; Nagata, K.; Shimosegawa, E.; Haberkorn, U.; et al. Theranostics Targeting Fibroblast Activation Protein in the Tumor Stroma: 64Cu and 225Ac Labelled FAPI-04 in Pancreatic Cancer Xenograft Mouse Models. J. Nucl. Med. 2020, 61, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, D.; Elez, E.; Tabernero, J.; Seoane, J. Clinical Development of Therapies Targeting TGFβ: Current Knowledge and Future Perspectives. Ann. Oncol. 2020, 31, 1336–1349. [Google Scholar] [CrossRef]
- Chandana, S.R.; Babiker, H.M.; Mahadevan, D. Clinical Complexity of Utilizing FGFR Inhibitors in Cancer Therapeutics. Expert Opin. Investig. Drugs 2020, 29, 1413–1429. [Google Scholar] [CrossRef]
- Harvey, J.B.; Phan, L.H.; Villarreal, O.E.; Bowser, J.L. CD73’s Potential as an Immunotherapy Target in Gastrointestinal Cancers. Front. Immunol. 2020, 11, 508. [Google Scholar] [CrossRef] [Green Version]
- Perrot, I.; Michaud, H.-A.; Giraudon-Paoli, M.; Augier, S.; Docquier, A.; Gros, L.; Courtois, R.; Déjou, C.; Jecko, D.; Becquart, O.; et al. Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep. 2019, 27, 2411–2425.e9. [Google Scholar] [CrossRef] [Green Version]
- Soleimani, A.; Farshchi, H.K.; Mirzavi, F.; Zamani, P.; Ghaderi, A.; Amini, Y.; Khorrami, S.; Mashayekhi, K.; Jaafari, M.R. The Therapeutic Potential of Targeting CD73 and CD73-Derived Adenosine in Melanoma. Biochimie 2020, 176, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Boshuizen, J.; Pencheva, N.; Krijgsman, O.; Altimari, D.D.; Castro, P.G.; de Bruijn, B.; Ligtenberg, M.A.; den Heuvel, E.G.; Vredevoogd, D.W.; Song, J.-Y.; et al. Cooperative Targeting of Immunotherapy-Resistant Melanoma and Lung Cancer by an AXL-Targeting Antibody-Drug Conjugate and Immune Checkpoint Blockade. Cancer Res. 2021, 81, 1775–1787. [Google Scholar] [CrossRef]
- Baggiolini, A.; Varum, S.; Mateos, J.M.; Bettosini, D.; John, N.; Bonalli, M.; Ziegler, U.; Dimou, L.; Clevers, H.; Furrer, R.; et al. Premigratory and Migratory Neural Crest Cells Are Multipotent in Vivo. Cell Stem Cell 2015, 16, 314–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Oatman, N.; Dasgupta, N.; Arora, P.; Choi, K.; Gawali, M.V.; Gupta, N.; Parameswaran, S.; Salomone, J.; Reisz, J.A.; Lawler, S.; et al. Mechanisms of Stearoyl CoA Desaturase Inhibitor Sensitivity and Acquired Resistance in Cancer. Sci. Adv. 2021, 7, eabd7459. [Google Scholar] [CrossRef]
- Talebi, A.; Dehairs, J.; Rambow, F.; Rogiers, A.; Nittner, D.; Derua, R.; Vanderhoydonc, F.; Duarte, J.A.G.; Bosisio, F.; Van den Eynde, K.; et al. Sustained SREBP-1-Dependent Lipogenesis as a Key Mediator of Resistance to BRAF-Targeted Therapy. Nat. Commun. 2018, 9, 2500. [Google Scholar] [CrossRef]
- Esslinger, C.S.; Cybulski, K.A.; Rhoderick, J.F. Ngamma-Aryl Glutamine Analogues as Probes of the ASCT2 Neutral Amino Acid Transporter Binding Site. Bioorg. Med. Chem. 2005, 13, 1111–1118. [Google Scholar] [CrossRef]
- Hassanein, M.; Qian, J.; Hoeksema, M.D.; Wang, J.; Jacobovitz, M.; Ji, X.; Harris, F.T.; Harris, B.K.; Boyd, K.L.; Chen, H.; et al. Targeting SLC1a5-Mediated Glutamine Dependence in Non-Small Cell Lung Cancer. Int. J. Cancer 2015, 137, 1587–1597. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Beaumont, K.A.; Otte, N.J.; Font, J.; Bailey, C.G.; van Geldermalsen, M.; Sharp, D.M.; Tiffen, J.C.; Ryan, R.M.; Jormakka, M.; et al. Targeting Glutamine Transport to Suppress Melanoma Cell Growth. Int. J. Cancer 2014, 135, 1060–1071. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.C. Targeting the Tumour Stroma to Improve Cancer Therapy. Nat. Rev. Clin. Oncol 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Grimm, J.; Hufnagel, A.; Wobser, M.; Borst, A.; Haferkamp, S.; Houben, R.; Meierjohann, S. BRAF Inhibition Causes Resilience of Melanoma Cell Lines by Inducing the Secretion of FGF1. Oncogenesis 2018, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Windisch, P.; Zwahlen, D.R.; Koerber, S.A.; Giesel, F.L.; Debus, J.; Haberkorn, U.; Adeberg, S. Clinical Results of Fibroblast Activation Protein (FAP) Specific PET and Implications for Radiotherapy Planning: Systematic Review. Cancers 2020, 12, 2629. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Yegutkin, G.G.; Pacher, P.; Blandizzi, C.; Haskó, G. Anti-CD73 in Cancer Immunotherapy: Awakening New Opportunities. Trends Cancer 2016, 2, 95–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Approach | Targets/Drugs | Stage of Development | References |
|---|---|---|---|
| Targeting phenotype-specific vulnerabilities | Invasive phenotype: AXL-(ADC): AXL-107-MMAE Enapotamab vedotin | Solid tumor phases I, II NCT02988817 | [23,123] |
| Invasive phenotype: Small molecule AXLi (bemcentinib, dubermatinib) | Solid tumor phases I, II Bemcentinib NCT03184571; NCT03649321 Dubermatinib: NCT02729298 | [124,125] | |
| Invasive phenotype: Multi-targeted TKI (sitravatinib) | Solid tumor phases I, II, III Sitravatinib NCT04518046; NCT03666143 NCT04123704;NCT03906071 | [126] | |
| Neural crest stem cell (NCSC) state: pan-RXR antagonist (HX531) | Preclinical | [43] | |
| Invasive/NCSC phenotypes: ferroptosis inducing drugs (erastin, ML162, and ML21s) | Preclinical | [25] | |
| Phenotypic heterogeneity/TME: EDNR antagonists | Bosentan NCT04158635 | [103] | |
| Senescence-like phenotype: Senolytics AKT/mTORi HDACi Bcl family inhibitor | Solid tumors phases I, II, III mTORi: everolimus (NCT00876395) HDACi: panobinostat (NCT04897880) Navitoclax (NCT03366103) | [127,128] | |
| Targeting lipid metabolism | Lipid uptake: Anti-CD36 | Preclinical | [129] |
| Lipid synthesis and uptake: SREBPi: fatostatin, botulin, and PF-429242 | Preclinical | [130] | |
| Lipogenesis: FASNi (TVB-2640) | Solid tumor phase II NCT03179904; NCT03808558 | [131,132] | |
| Targeting glutamine metabolism | Inhibition of GLS1: CB-839 | Phase I/II evaluation of CB-839 in combination with nivolumab in melanoma patients (NCT02771626) | [133,134] |
| Targeting TME components and signals | FAPI | Preclinical | [135,136] |
| Anti-TGF-beta | Solid tumor phase I NCT00356460 | [137] | |
| Small molecule FGFRi | Dovitinib NCT01831726 NCT01676714 | [138] | |
| Anti-CD73 | Solid tumor phases I, II Oleclumab: NCT03611556; NCT03381274; NCT04668300 CPI-006: NCT03454451 BMS-986179: NCT02754141 | [139,140,141] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Najem, A.; Soumoy, L.; Sabbah, M.; Krayem, M.; Awada, A.; Journe, F.; Ghanem, G.E. Understanding Molecular Mechanisms of Phenotype Switching and Crosstalk with TME to Reveal New Vulnerabilities of Melanoma. Cells 2022, 11, 1157. https://doi.org/10.3390/cells11071157
Najem A, Soumoy L, Sabbah M, Krayem M, Awada A, Journe F, Ghanem GE. Understanding Molecular Mechanisms of Phenotype Switching and Crosstalk with TME to Reveal New Vulnerabilities of Melanoma. Cells. 2022; 11(7):1157. https://doi.org/10.3390/cells11071157
Chicago/Turabian StyleNajem, Ahmad, Laura Soumoy, Malak Sabbah, Mohammad Krayem, Ahmad Awada, Fabrice Journe, and Ghanem E. Ghanem. 2022. "Understanding Molecular Mechanisms of Phenotype Switching and Crosstalk with TME to Reveal New Vulnerabilities of Melanoma" Cells 11, no. 7: 1157. https://doi.org/10.3390/cells11071157
APA StyleNajem, A., Soumoy, L., Sabbah, M., Krayem, M., Awada, A., Journe, F., & Ghanem, G. E. (2022). Understanding Molecular Mechanisms of Phenotype Switching and Crosstalk with TME to Reveal New Vulnerabilities of Melanoma. Cells, 11(7), 1157. https://doi.org/10.3390/cells11071157