Abstract
Epithelial cells that line tissues such as the intestine serve as the primary barrier to the outside world. Epithelia provide selective permeability in the presence of a large constellation of microbes, termed the microbiota. Recent studies have revealed that the symbiotic relationship between the healthy host and the microbiota includes the regulation of cell–cell interactions at the level of epithelial tight junctions. The most recent findings have identified multiple microbial-derived metabolites that influence intracellular signaling pathways which elicit activities at the epithelial apical junction complex. Here, we review recent findings that place microbiota-derived metabolites as primary regulators of epithelial cell–cell interactions and ultimately mucosal permeability in health and disease.
1. Introduction
The gastrointestinal (GI) tract is lined by a monolayer of epithelial cells that form a critical barrier separating our body from the external environment. In this, the intestinal epithelium plays an essential role in the regulation of the immune system and maintenance of health [1]. The GI tract performs two key tasks: it acts as a highly selective filter, allowing movement of nutrients and water into circulation and the internal milieu in general. As a barrier, it prevents exposure to harmful entities and restricts infiltration of pathogenic organisms [2]. This seemingly conflicting task is finely regulated by an interplay of structural components and molecular interactions at the intestinal mucosa to maintain intestinal integrity and immune homeostasis [3]. The function of the mucosal barrier can be affected through severe structural damage or more subtle changes in its regulating components [4]. Although the exact mechanisms are still unclear, loss of barrier function is tightly linked to chronic inflammatory diseases, such as inflammatory bowel disease (IBD) [5].
The intestine is home to the largest constellation of microorganisms in our body including bacteria, viruses and fungi termed the gut microbiota (GM), mostly present in the colon. The fact that the intestine is not easily overtaken by infection is a testament to the monumental task accomplished by the mucosal barrier. A recently revised study revealed that in the body, the ratio of human:bacterial cells is close to 1:1 [6]. Interestingly, the human genome consists of approximately 23,000 genes, whereas the microbiome encodes over 3 million genes [7]. Therefore, the “human superorganism” is actually approximately 1% human in this regard. These microbial passengers in return offer many symbiotic benefits to the host in the form of thousands of metabolites, acting upon a range of physiological functions including strengthening gut integrity, modulating cellular energetics and shaping the intestinal epithelium [8]. There is a wide variety of metabolites necessary for maintaining and restoring barrier function including short-chain fatty acids (SCFAs), indoles, purines, bile acids, and polyamines. This review will focus on the role of microbial-derived metabolites in the regulation of intestinal barrier function and homeostasis.
2. Intestinal Barrier Composition
The intestinal barrier structure is composed of several elements that, in conjunction, provide this complex physical and immunological defense mechanism. The first line of defense is an outer mucus barrier, where the commensal bacteria, antimicrobial peptides (AMPs), and secretory immunoglobulin A (sIgA) reside; next, a central monolayer of specialized intestinal epithelial cells (IECs) are tightly held together by tight junctions (TJs), adherens junctions (AJs), and desmosomes; lastly, the inner lamina propria, where the last defense resort of innate and adaptive immune cells reside, such as T cells, B cells, macrophages, and dendritic cells [1,9].
Bacteria arriving at the gut lumen are immediately met by the mucus barrier, which inhibits their direct contact with the intestinal epithelium [9]. This layer of protection is composed of a hydrated gel of heavily glycosylated proteins termed mucins. In the intestinal mucosa, mucin 2 (MUC2) is the most abundant mucus protein secreted by goblet cells [1]. MUC2 is the primary component of the mucus barrier in the colon and is critical for protection against disease, as demonstrated in a classical model of MUC2 knockout mice spontaneously developing colitis [10]. Furthermore, transmembrane mucins expressed by IECs remain attached to their apical surface and, together with glycolipids, form the glycocalyx [11]. Composed of various receptors for bacterial adhesion, a critical function of the glycocalyx is to act as a landing hub for normal microbiota and limit colonization by pathogens [12,13,14,15]. Remarkably, the mucus in the colon is organized in two layers: an inner layer that is “firmly” adherent to epithelial cells and an outer non-attached “soluble” layer. While the inner layer is dense and does not allow bacteria to penetrate, thus keeping the epithelial cell surface free of bacteria, the outer layer plays a crucial role as a proliferating habitat of the microbiota in the colon [16]. Lastly, there is the presence of immune regulators such as AMPs and sIgA molecules as final reinforcement in the physical separation of the epithelium to the bacteria-invaded lumen [17]. AMPs have the capacity to rapidly kill [18] or arrest [19] a wide variety of microorganisms. An important class of AMPs are defensins, which are broadly classified as α and β defensins. Alpha-enteric defensins (HD5 and HD6) are secreted by Paneth cells and act as antimicrobial agents in the small intestine [20,21]. Secretion of these peptides is upregulated in the colonic mucosa of IBD patients through metaplastic Paneth cells [22]. Additionally, β-defensins secreted by the colonic epithelium play a major role in innate host defenses to maintain a healthy microbiota. Accordingly, their important role is reflected in IBD patients, where the defective expression and function of β-defensins lead to altered microbiota, infection and inflammation as reviewed here [23]. To briefly elucidate their antimicrobial mechanisms, defensins are cationic and arginine-rich peptides that bind to negatively charged microbial membranes, causing membrane disruption and cell death [24]. An interesting alternative mechanism was reported in HD6, where the peptide spontaneously self assembles into multi-peptide nanonets in the presence of bacteria [19]. In other words, rather than killing bacteria, HD6 function by aggregating and sequestering bacteria. Notably, the mucosal layer and the gut microbiota are interdependent: a shift in the microbiota will affect the composition of the mucosa and vice versa [25].
Epithelial cells provide the most selective component of the intestinal physical barrier. A pool of pluripotent stem cells residing in the crypt produce a distinct type of cells including absorptive enterocytes, goblet cells, enteroendocrine cells, Paneth cells, and microfold cells [26]. These cells, in conjunction, form a continuous and polarized monolayer barrier that separates the lamina propria from the lumen. The epithelial monolayer allows for selective passage of water, nutrients, and electrolytes while excluding harmful microbial pathogens, toxins, and other foreign agents [27]. This selective filter is regulated by the presence of junctional complexes. The three main complexes are the TJs, AJs, and desmosomes [28]. TJs are located in the apical side of the epithelial layer and form a continuous belt-like ring between the apical and lateral membrane [29]. TJs consist of transmembrane proteins (e.g., occludin and claudin), peripheral membrane proteins (e.g., zonula occludens ZO-1 and ZO-2), and regulatory proteins. TJ proteins regulate molecule passage through the epithelial layer based on their size and charge. They are essential to maintain a strong epithelial barrier and gut health. AJs and desmosomes are found below TJs and provide strong mechanical attachments between neighboring cells [29]. The structures, properties, and functions of these complexes have been extensively reviewed elsewhere [30,31]. The disruption of these complexes leads to decreased barrier and immediate invasion of inflammatory agents. Continuous inflammation perpetuates the physical impairment of their function and can lead to further aggravation of permeability and chronic inflammation.
Finally, following their production by B cell lineage cells in the lamina propria, dimeric immunoglobulin A (IgA) complexes bind to the immunoglobulin receptor (pIgR) on the basolateral membrane of IECs and are transported to the lumen [32]. This collaboration between immune cells and IECs provides an immune component to regulate commensal bacteria populations and contributes to intestinal homeostasis [33,34]. Indeed, several studies have shown that IgA binds colitogenic members of the microbiota [35], and that mice deficient in IgA or the receptor pIgR develop more severe colitis [36].
Intestinal barrier dysfunction has been directly linked to inflammatory disorders including IBD [5]. Marin et al. demonstrated that the tight junctions of Crohn’s patients are misaligned, fragmented, and severely disorganized [37]. Later, Gassler et al. reported the downregulation of junctional proteins (E-cadherin and α-catenin) and their mRNA in actively inflamed IBD patients [38]. In fact, intestinal barrier permeability is used as a prognostic indicator of relapse in patients with quiescent IBD [39,40]. In 2020, two novel studies demonstrated that intestinal barrier permeability can start years before the clinical diagnosis of IBD, suggesting the possibility that IBD can be prevented by early intervention; a brief review of these studies can be found here [41]. Immunologically, IBD results in impaired innate and adaptive immune responses. Specifically, high levels of T helper 17 cells lead to perpetuating disease. Furthermore, a decrease in Treg cells, a specialized subpopulation of T cells that act to suppress immune response, leads to defective anti-inflammatory mechanisms [42]. The role of GM metabolites in maintaining and synchronizing these main intestinal elements for continuous homeostasis and their downregulation in disease will be discussed here.
3. Oxygen Gradient Environment of the Mammalian Intestine
The mammalian intestine has evolved to function as a uniquely suited environment for the growth and survival of anaerobic microbes [43]. The GI tract harbors a distinct oxygenation profile [44] and is characterized by a high rate of metabolite circulation. Even at baseline, barrier epithelial cells that line the mucosa exist at a low-oxygen tension environment, defined as ‘physiologic hypoxia’ [45] (see Figure 1). Original studies revealed that the countercurrent oxygen exchange mechanisms of the GI tract provide for arterial blood supply diffusion to adjacent venules, along the crypt villus axis, resulting in graded hypoxia [46]. This steep oxygen gradient has been well documented in the distal colon of the GI tract, spanning from the anaerobic lumen, across the epithelium to the richly vascularized sub-epithelial mucosa [47]. Given the high energy requirement of the gut and the integral role of the epithelium in maintaining intestinal homeostasis, it is not surprising that these cells have evolved a number of mechanisms to cope with this austere metabolic environment [48]. During active inflammation, the combination of recruited leukocytes, edema, and vasculitis enhances the hypoxic gradient to become “inflammatory hypoxia” [45]. The microbiome plays a key role in maintaining this oxygen gradient, which is critical for nutrient absorption, barrier function, and immune responses in the intestine [49].
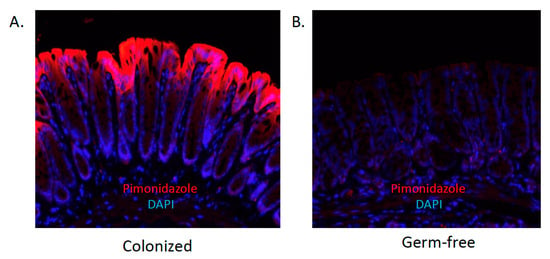
Figure 1.
The contribution of the microbiota to “physiologic hypoxia”. Shown here are histologic sections of healthy colon derived from colonized (A) or germ-free (B) mice documenting low-oxygen regions (red) visualized by staining with pimonidazole, a dye that stains only in low-oxygen tensions. Nuclear counter-stain with DAPI is shown in blue. Note the near total lack of pimonidazole localization in the absence of microbial colonization.
Tissue oxygenation has been observed using 2-nitroimidazole dyes (e.g., pimonidazole), a class of compounds known to undergo intracellular metabolism dependent on the level of tissue oxygenation [50] (Figure 1). These dyes were developed to image the low-oxygen environment of growing tumors [51] and have subsequently been used as tools to monitor levels of tissue oxygenation ex vivo. Nitroimidazoles can form covalent bonds with thiol groups of various tissue macromolecules only at a pO2 < 10 mmHg. Antibodies specific for these conjugated adducts provide a histochemical approach to estimation of tissue pO2. It is notable that this approach has not been established to titer tissue pO2; rather it is a qualitative estimate of tissue hypoxia (e.g., above or below pO2 of ~10 mmHg). In addition, studies in germ-free mice have revealed that such physiologic hypoxia results in large part to contributions from the microbiota [52] (see Figure 1).
Given the rather unique environment of the intestine, particularly the colon, a number of studies have shown that stabilization of the transcription factor hypoxia-inducible factor (HIF) in low-oxygen environments triggers the expression of genes that are essential to epithelial barrier function [53,54,55,56]. Additionally, HIF is one of the central regulators of overall tissue metabolism [57] and has profound influences on the inflammatory response [48]. HIF function is dependent on stabilization of an O2-dependent degradation domain (ODD) expressed on the α-subunit and subsequent nuclear translocation to form a functional complex with HIF-1β [58]. In normally oxygenated tissues, ferrous iron, 2-oxoglutarate (2-OG), and O2-dependent hydroxylation of two prolines (Pro564 and Pro402 of HIF-1α in humans) carried out by prolyl hydroxylase enzymes (PHD1-3) within the ODD of the alpha subunit initiates the association with the von Hippel-Lindau tumor suppressor protein (pVHL) and the recruitment of a ubiquitin-E3 ligase for degradation via proteasomal targeting [59,60]. Alternatively, in low-oxygen environments or chemically induced PHD inhibition, HIFα is stabilized. Recently, it was discovered that some metabolites play an indirect role in gut health through HIF stabilization. An in-depth review of the adaptive role of HIF in the context of barrier function has been reviewed in detail elsewhere [43].
4. Microbial Metabolites, Oxygen Metabolism and Barrier Regulation
The GM is a vital and permanent passenger represented in vast richness of microbial life inhabiting the gastrointestinal tract. The GM is composed of several species of microorganisms, including bacteria, yeasts, and viruses. Classified by phylum, Firmicutes and Bacteroidetes represent 90% of the gut bacterial composition, along other subdominant phyla such as Proteobacteria, Actinobacteria, and Verrucomicrobia [61]. Lactobacillus, Bacillus, Clostridium, Enterococcus, and Ruminicoccus are examples of the more than 200 different genera representing the Firmicutes phyla in the GM where Clostridium constitutes 95% of its composition. Bacteroidetes consists predominantly of Bacteroides and Prevotella. Less abundantly, the Actinobacteria phylum is mostly represented by the Bifidobacterium genus [62]. Similar to a fingerprint, everyone is born with a unique GM profile that will play a lifetime role in nutrient metabolism, maintenance of structural integrity of the gut barrier, energy, and immunomodulation. Established at birth and early life, GM composition can be directly impacted by factors including gestational age, delivery method, milk feeding, and weaning [63] as well as antibiotic use [62]. Likewise, the complex GM–host interaction begins at birth where the GM initiates immune development, while at the same time the host orchestrates GM composition [64]. The first years of life represent a critical timepoint where the establishment of a healthy GM and a respectful GM–host balance will regulate metabolism, immunity, and prevention of disease for life. A comprehensive report of the microbiota composition and individual variations thereof can be found here [62].
In healthy mammals, the host–microbiota interaction is symbiotic with regard to host immunity, energy metabolism, and cellular communication [29,65,66,67,68]. The GM is key in harvesting nutrients from the diet. GM metabolizes undigested food that safely reaches the large intestine (e.g., fiber and some starches, sugars). The metabolism of these dietary components yields many active microbial metabolites. Furthermore, the GM interacts with the epithelium in the local environment, which initiates the production of additional metabolites [69], facilitating a metabolite-mediated communication or “crosstalk” essential for gut health [70]. It is notable that shifts in GM composition and metabolite production may contribute to the development of mucosal diseases such as IBD [71,72]. These microbiota-derived metabolites include short-chain fatty acids, tryptophan catabolites, purines, secondary bile acids, and polyamines. The following section will discuss the role of selected metabolites in cell communication, energy balance, immunomodulatory activities, and gut barrier regulation.
4.1. Short-Chain Fatty Acids (SCFAs)
Dietary fibers (DF) are important carbohydrates that the human body is unable to digest. These fibers pass through the small intestine into the colon, where they are fermented by the GM. Pectin, inulin, α-glucans, oligosaccharides, and guar gum are fermentable fibers that constitute the main energy source for GM [61]. The distribution of consumed DF and resultant fermentation can shape the diversity and function of the GM [73], influencing the production of microbiota-derived metabolites. High DF intake correlates with reduced disease incidence and death [74,75,76]. It has been shown that DF are important for homeostatic gut barrier function [77,78], whereas the lack of DF intake is associated with autoimmune disease and IBD development [79,80]. These positive effects are attributed to microbiota-derived metabolites, notably SCFAs, interacting with intestinal cells through various mechanisms. Briefly, SCFAs are a requisite waste product to balance redox equivalent production in the anaerobic gut lumen. In the body, SCFAs are classified as saturated aliphatic carboxylate salts between one and six carbons in length—formate, acetate, propionate, butyrate, valerate, and hexanoate. Acetate, propionate, and butyrate are the most abundant, comprising > 95% of SCFAs, and exist in a molar colonic ratio of approximately 60:20:20, with total SCFAs reaching 140 millimolar (mM) in the proximal colon and 70 mM in the distal colon [81]. The majority of SCFAs are rapidly absorbed by colonocytes, with only 5–10% secreted in feces. These SCFAs have a significant impact on host physiology as energy substrates, gene expression regulators, and signaling molecules for specific receptors [82,83,84,85].
4.1.1. SCFAs, Transporters, and Anti-Inflammation
Carrier-mediated transport represents the most important route of entry of SCFAs in their anionic form into the colonic epithelium [86]. Several transport systems including monocarboxylic transporter 1 (MCT1) and 4 (MCT4) operate in cellular uptake of SCFAs. Specifically, sequestration of butyrate to the colon is due in large part to the different affinities of the apical (Km = 1.5 mM) and basolateral (Km = 17.5 mM) SCFA–HCO3- exchange transporters, which confine butyrate to colonocytes [87,88] (Figure 2). Similarly, the affinity of the apical MCT1 transporter for butyrate is also higher than the basolateral transporter MCT4, and the higher intracellular pH renders all SCFAs in the dissociated form, which limits passive diffusion across the basolateral membrane. In line with butyrate production, MCT1 is mostly observed in the colon and it is considered to be the primary transporter of butyrate [89]. Peripheral systemic availability of microbiota-derived butyrate has been shown to be less than 2%, as the vast majority of butyrate is utilized by colonocytes [90].
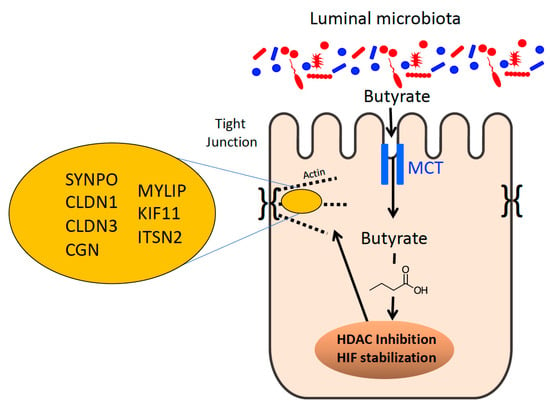
Figure 2.
Microbial-derived butyrate regulates epithelial tight junction expression and function. Shown here is the influence of butyrate on tight junction protein expression. Through the actions of butyrate on HDACs and HIF, butyrate influences the expression of multiple TJ-associated proteins, including those indicated here. See text for further clarification.
SCFAs exert anti-inflammatory influences in the intestinal mucosa in part by activation of G protein-coupled receptors (GPCRs) present in IECs and immune cells. GPCR41 and GPCR43 play a role in the immune surveillance of the colonic mucosa providing communication between SCFAs and mast cells [91]. Butyrate reduces lipopolysaccharide-induced NF-κB activation via GPR109A acting as a tumor suppressor [92]. Additionally, GPR43-bound acetate promotes potassium efflux and hyperpolarization in colonic cells, leading to NLRP3 inflammasome activation [93]. NLRP3 inflammasome activation leads to the release of proinflammatory cytokines (e.g., IL-1β and IL-18). The current body of knowledge in the relationship of these receptors and SCFAs has been reviewed elsewhere [94]. Furthermore, butyrate directly regulates immune cells, such as macrophages [95], dendritic cells [96], lymphocytes [95], and inhibits cytokines (e.g., IL12-p70 and IL-23) [97]. Lastly, as it is well known, butyrate is a potent histone deacetylase (HDAC) inhibitor [98] (Figure 2), affecting many processes including the inflammatory response. HDACs regulate innate immunity pathways, such as myeloid cell differentiation, TLR and IFN-mediated inflammatory response [99]. This demonstrates the importance of the localization of SCFAs in the colon and all the interactions with receptors, immune cells, and enzymes to regulate and coordinate proper immune function and intestinal health.
4.1.2. SCFAs as the Primary Colonocyte Energy Source
Another aspect of butyrate sequestration is that it is the preferred energy source of the colonic epithelium, with oxidation of this SCFA accounting for over 70% of cellular oxygen consumption in the distal colon [100]. Colonocytes preferentially utilize butyrate over acetate and propionate, where it is oxidized to ketone bodies and CO2. Greater than 95% of microbiota-derived butyrate is utilized by colonocytes for energy. An energy deprived state (monitored by a decrease in enzymes involved in the tricarboxylic acid cycle) results in lower ATP levels, and ultimately increased autophagic flux, in colonocytes from germ-free (GF) mice. Recolonization of GF mice with butyrate-producing bacteria (mostly of the Firmicutes phylum) or butyrate treatment of GF colonocytes increases oxidative phosphorylation and returns autophagy to baseline [89,101]. As an energy substrate, butyrate undergoes β-oxidation to form acetyl-CoA, which enters into the tricarboxylic acid cycle (TCA) to produce the reducing factors that drives the electron transport chain (ETC) and oxygen consumption to ultimately regenerate ATP. Maintenance of the mucosal barrier requires cytoskeleton stability, requiring substantial energy pools, and the rapid utilization of butyrate not only prevents butyrate from escaping into systemic circulation but also provides the requisite energy for IECs to rapidly polarize and form strong AJCs [102].
4.1.3. Butyrate and the Epithelial Barrier
Multiple studies have shown that the selective sequestration of butyrate in the colon influences intestinal barrier function. For instance, an mRNA-based screen of intestinal epithelial cells exposed to physiologic concentrations of butyrate revealed the repression of claudin-2 (CLDN2), a “leaky” claudin that increases permeability. The mechanisms of butyrate activity were subsequently traced to induction of the IL-10 receptor (IL-10Rα) and IL-10-enhanced protein expression on IECs through HDAC inhibition [103]. Other studies have shown butyrate to induce the expression of “sealing” TJ proteins such as CLDN1, also through HDAC inhibition [104]. More recently, a newly characterized gene of interest to barrier function, synaptopodin (SYNPO) was identified via a single-cell RNA sequencing approach (scRNAseq). SYNPO is an important intestinal epithelial tight junction protein. Like claudins, the mechanisms of butyrate regulation of SYNPO were via HDAC inhibition (Figure 2). SYNPO was additionally shown to promote wound healing as an important component of the butyrate-derived wound healing response in vivo [105]. Altogether, butyrate can coordinate the repression of a “leaky” TJ protein, the induction of “sealing” TJ proteins, and coordination of wound healing responses through HDAC inhibition. While HDAC inhibition impacts the expression of ~2% of mammalian genes [98], butyrate regulates barrier function by targeting multiple genes through HDAC inhibition. Additionally, SYNPO was induced at the protein level after 6 h of butyrate treatment, while other studies showed that CLDN2 levels were reduced by butyrate after 24 h, which corresponded with peak IL-10Rα induction at 24 h, followed by a CLDN1 increase after 36 h [103,104]. The observations suggest that butyrate may temporally orchestrate the induction and repression of specific TJ proteins to ultimately promote barrier function. Wang et al. showed in their scRNAseq that multiple TJ and actin-associated genes were upregulated by butyrate including cingulin (CGN), CLDN1, CLDN3, as well as genes related to cellular motility including MYLIP and KIF11 [105] (Figure 2). This suggests that butyrate simultaneously and purposefully influences multiple genes related to these processes as, naturally, the AJC is comprised of many components and active epithelial restitution to begin wound healing requires many proteins working in concert.
4.1.4. Butyrate-HIF Regulation of Epithelial Barrier
Butyrate-producing bacteria are anaerobic, taking advantage of the unique oxygen environment in the intestine with a low O2 concentration in the lumen. Furthermore, the metabolism of butyrate in the epithelium further reduces the already low levels of O2 to the point of HIF stabilization (a transcription factor involved in barrier protection) [106]. Accordingly, mice lacking microbiota-derived butyrate (e.g., GF mice) show diminished hypoxia localization (Figure 1) and reduced HIF stabilization at baseline [52]. Recently, an alternative butyrate-HIF stabilization mechanism independent of β-oxidation was reported [107]. Analysis using a combination of recombinant HIF prolyl hydroxylase (PHD) enzyme and 1D-NMR experiments found that butyrate stabilizes HIF by acting as a direct, non-competitive inhibitor of HIF PHD in vitro and in vivo. Interestingly, shorter or longer SCFAs (e.g., acetate, propionate, and valerate) exhibited a much higher PHD IC50 and no binding according to NMR. This implicates butyrate as a significant and dynamic endogenous regulator of HIF in IECs [107].
HIF contributes to epithelial barrier function in a number of ways. Butyrate induces barrier function in vitro (measured by FITC-dextran flux) but not in IEC monolayers lacking HIF1α, indicating a fundamental role for HIF in maintaining barrier [52]. Selective knockdown of HIF-1α in murine IECs demonstrated major defects in mucosal barrier integrity. This could partly be due to HIF-1α directly regulating the expression of CLDN1, a crucial TJ protein [108]. HIF-1α also upregulates MUC2, the major component of the mucus layer, as well as human β-defensin 1 (HBD-1), which is the only constitutively secreted antimicrobial peptide in the intestine [109,110]. Likewise, HIF is a transcriptional regulator of intestinal trefoil factor (i.e., TTF3), a 40 amino acid lectin protein that binds and crosslinks mucins [53]. HIF-2α regulates creatine kinases and the creatine transporter that co-localize with AJs and supply energy at junctional sites for tasks such as tight junction assembly, maintenance, and restitution [111,112]. Additionally, HIF-1α regulates the expression of ectonucleoside triphosphate diophosphohydrolase 1 (CD39) and 5′-nucleotidase (CD73), which enzymatically convert adenosine triphosphate (ATP)/adenosine diphosphate (ADP) to adenosine monophosphate (AMP) and AMP to adenosine, respectively. Adenosine signaling plays a key role in the perfusion of the intestinal mucosa and promotes intestinal barrier function through activating the adenosine 2B receptor (A2BR), which is highly expressed in the intestinal mucosa and is transcriptionally regulated by HIF-1α [113].
4.1.5. SCFAs in Disease
Dysbiosis in IBD patients is characterized by loss of GM diversity, with higher abundance of Proteobacteria and loss of SCFAs/butyrate-producing bacteria mainly of the Firmicutes phylum. Specific studies show that a decrease in F. prausnitzii, a butyrate-producing bacteria is a hallmark of active IBD patients [114,115], ultimately resulting in lower luminal concentrations of butyrate. Multiple studies, for example, have revealed lower concentrations of luminal butyrate levels and reduced overall abundance of butyrate-producing organisms in patients with IBD (e.g., certain Roseburia and Faecalibacterium genera) [116,117,118]. While not conclusive, this correlative observation is supported by data demonstrating that specifically increasing fecal butyrate levels diminishes intestinal inflammation and some symptoms in patients with ulcerative colitis [119]. A recent meta-analysis of 25 studies of patients with ulcerative colitis suggested a nearly 65% clinical response rate to fecal microbiota transplant (FMT) that corresponded with a broad increase in microbial diversity including butyrate producers (e.g., Firmicutes and Clostridium clusters IV, XIVa, XVIII) [120].
Furthermore, among the abnormalities observed in IBD patients, it has been found that MCT1 protein expression (butyrate transporter) and the transcript of its encoding gene SLC16A1 are reduced in inflamed mucosa [121,122,123]. In fact, an important inverse correlation is observed between butyrate uptake/oxidation and the Mayo endoscopic subscore and Geboes histological score [121]. Specifically, genes encoding enzymes responsible in butyrate oxidation/uptake are downregulated in inflamed mucosa of IBD patients [121,122,123]. Additionally, inflammatory cytokines inhibit butyrate uptake, oxidation (e.g., TNF-α), and MCT1/SLC16A1 expression [124,125,126]. These findings suggest that inflammation is directly related to SCFAs (specifically butyrate) synthesis, uptake, and metabolism, suggesting that SCFAs supplementation alone may not be sufficient to regain homeostasis. Promising combined clinical approaches utilizing butyrate and butyrate-producing bacteria have resulted in prevention of relapse in IBD patients [127,128]. Collectively, these findings demonstrate the importance SCFAs in gut health and that a combination of approaches to regain butyrate uptake and metabolism may be a promising treatment in the clinical management of IBD.
4.2. Indoles and Indole Derivatives
4.2.1. Biosynthesis of Indoles
1H-indole (hereafter, “indole”) and indole derivatives are gut bacterial metabolites resulting from degradation of dietary tryptophan [129]. Such derivatives, for example indole-3-propionic acid (IPA), indole-3-pyruvate (IPy), and indole-3-acetic acid (IAA), resemble indole except for substitution at the 3 position with a variety of diverse moieties. Generation of indole from tryptophan occurs through a single-step deamination reaction (EC 4.1.99.1), which generates pyruvate and ammonia as additional products [130]. The enzyme responsible for this reaction, TnaA (tryptophanase), has been described in diverse bacteria including both Gram-positive and Gram-negative species with varying degrees of conservation to the prototypic E. coli K-12 sequence [131]. Generation of substituted indoles from tryptophan occurs through a more complicated process often unique to the synthesizing organism; for example, the obligate anaerobe Clostridium sporogenes generates IPA through a multi-step pathway involving the production of IPy, indole-3-lactic acid (ILA), and indole-3-acrylate (IA) as immediate precursors along with synthesis of IAA under certain circumstances [132]. Similar multi-step biosynthetic pathways have been described for other bacteria, highlighting the diversity of microbial tryptophan metabolism (reviewed in [133]). Notably, indole and indole derivatives have been shown to be exclusively derived from the gut microbiota: studies using germ-free mice and mice treated with broad-spectrum antibiotics have shown a loss of indoles in the absence of an intact GM, indicating an essential role for intestinal microbiota in the metabolism of dietary tryptophan [132,134,135]. Indoles are abundant in the gut, and indole has been consistently measured in the millimolar range in human fecal extracts [134,135]. In addition to the influences on the mammalian intestinal tract (discussed in further detail below), indoles have been characterized as intercellular signaling molecules, controlling a variety of cellular processes including virulence gene expression, sporulation, plasmid stability, cell cycle control, and biofilm formation [136,137,138,139,140]. The ability of indoles to modulate eukaryotic cell function implies that these molecules act as interkingdom signaling molecules and suggests a co-evolution between metazoans and their gut symbionts [141].
4.2.2. Protective Actions of Indoles
The protective influence of indoles on the mammalian gastrointestinal tract has been known since at least 2010, when Bansal et al. described the induction of tight junction genes by indole treatment in the colorectal adenocarcinoma cell line HCT-8 [142]. Bansal et al. further demonstrated that acute treatment with indole enhances HCT-8 barrier function, as measured by transepithelial electrical resistance (TEER), and is protective during TNF-α-induced inflammation by suppressing nF-κB signaling and IL-8 secretion, while inducing expression of the anti-inflammatory cytokine IL-10. These results were corroborated by subsequent studies that demonstrated a positive influence of indoles on barrier function in Caco-2 and T84 colorectal adenocarcinoma cell lines, including protection against TNF-α-induced barrier dysfunction, as measured by both TEER and flux of the fluorescent tracer FITC-dextran across the cellular monolayer [143,144,145]. IPA was further shown to be protective in the dextran sodium sulfate (DSS) animal model of colitis, reducing tissue pro-inflammatory cytokines and ameliorating intestinal histopathology [145]. The protective benefits of indole derivatives have been associated with stimulation of aryl hydrocarbon receptor (AhR) signaling (Figure 3), as studies have indicated that the protective benefits of indoles are lost when AhR is knocked out, pharmacologically inhibited, or its signaling obviated due to loss of its nuclear binding partner ARNT [144,145]. AhR is a ligand-dependent transcription factor expressed in diverse cell types that binds a wide variety of both xenobiotic factors (e.g., dioxins) and endogenous ligands, resulting in activation of an anti-inflammatory/pro-barrier transcription program (reviewed in [146]). Activation of AhR by endogenous ligands, such as the host-derived tryptophan metabolite kynurenine, has been shown to induce expression of IL-10R1 and promote wound healing following intestinal insult [147]. Similar observations have been made for indoles: treatment of intestinal epithelial cells with indole derivatives induces IL-10R1 in vitro, and mice treated with IPA show enhanced colonic IL-10R1 expression during DSS colitis [145]. Additionally, indole-dependent AhR signaling was found to prevent inflammation-induced activation of the actin-regulatory protein ezrin, a protein implicated in loss of apical junction complex integrity, as well as activation of myosin light-chain kinase, which was similarly involved in regulation of epithelial barrier integrity [144,148] (Figure 3).
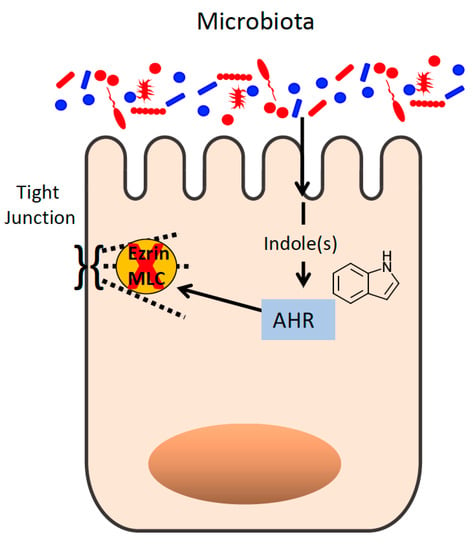
Figure 3.
Microbe-derived indole regulates epithelial tight junction function. Indoles act through the aryl hydrocarbon receptor (AHR) to inhibit ezrin and myosin light-chain (MLC) kinase activity.
Although the importance of AhR signaling in mediating the pro-barrier influences of indoles has been demonstrated by several studies, evidence suggests that the pleiotropic influences of indoles maintain epithelial homeostasis through diverse mechanisms. One study found that indoles, namely indole and IPA, act synergistically upon the human pregnane X receptor (PXR) to mitigate tissue histopathology and pro-inflammatory cytokine expression in the indomethacin model of small intestine inflammation [149]. PXR is a transcription factor that can bind a diverse array of ligands and has been implicated in attenuating intestinal inflammation through suppressing nF-KB target gene expression [150]. The importance of PXR in maintaining epithelial homeostasis was found to be dependent on Toll-like receptor (TLR) signaling: mice deficient in both PXR and TLR4 (which recognizes lipopolysaccharide) did not show the increased susceptibility to indomethacin evidenced by mice deficient in PXR alone. These findings suggest that the intestinal epithelium exists in a finely tuned state between pro- and anti-inflammatory responses, and disruption of one pathway may elicit deleterious influences by disturbing this balance.
Indoles have also been shown to be protective in the context of intestinal inflammation by acting on the innate immune system. For example, indoles, including indole, IPA, and IAA, have been demonstrated to inhibit neutrophil myeloperoxidase (MPO) in assays using both purified, recombinant human MPO and primary human neutrophils [151,152]. MPO, through its generation of hypochlorous acid from hydrogen peroxide, is essential for neutrophil anti-microbial activity but can cause damage to “bystander” tissues during uncontrolled inflammation [153]. Indole and IPA have been found to bind MPO directly to block the generation of HOCl, suggesting a role for regulation of innate immune processes by gut microbial metabolites [151].
4.2.3. Indole-Derived Gut Microbiota Shifts
Indoles may regulate intestinal homeostasis through modulating the identity of the gut microbiota. One study demonstrated that exogenous supplementation of indole significantly reduced gut microbiota perturbation induced by indomethacin, correlating with less severe tissue histopathology and lower fecal calprotectin (a metric of intestinal inflammation) [154]. Other studies have observed loss of intestinal indole metabolites during active inflammation, including in samples from IBD patients, suggesting a link between gut indoles and disease progression [145]. Similarly, treatment of mice with indole-3-carbinol (IC) during 2,4,6-trinitrobenzenesulfonic acid-induced (TNBS-induced) colitis prevented a pathologic shift in the gut microbiota and ameliorated disease in a manner dependent on IL-22, a member of the IL-10 cytokine family [155]. Indole has also been implicated in the pathogenesis of Clostridioides difficile and has been determined to be a predictor of C. difficile disease relapse [156,157]. In this way, future studies should investigate the potential of indole and indole derivatives as potential therapies for intestinal inflammatory disorders through their ability to directly modulate barrier function, inflammatory responses, and the identity of the gut microbiome.
4.3. Hypoxanthine and Other Purines
Intestinal healing and barrier repair require sufficient nucleotide generation to provide RNA for protein transcription and DNA for proliferation. Furthermore, high levels of ATP are needed to maintain the energy balance necessary to drive cytoskeletal function for wound restitution and endoplasmic reticulum function for mucin synthesis and secretion [158]. Since 1998, it has been known that adenine nucleotides (specifically ADP and ATP) significantly stimulate IEC migration and enhance structural and functional regeneration in vivo [159], indicative of the fundamental role nucleotides and associating metabolism play in eukaryotic cellular function.
4.3.1. Hypoxanthine as a Biological Marker of Intestinal Barrier
Pursuant to understanding intestinal barrier-related adenylate energy flux, Lee et al. developed an HPLC-based profiling method to monitor changes in high-energy phosphates and adenylate metabolites [160]. In this study, they elucidated the role of hypoxanthine (Hpx, a naturally occurring purine) as a checkpoint metabolite in IEC function, with Hpx promoting cellular energetics to the benefit of cytoskeletal and barrier function. A “calcium switch” experiment [161] disrupts epithelial tight junctions and results in a >90% loss of barrier that recovers over time as monitored by TEER. Surprisingly, Hpx levels had an inverse correlation with barrier integrity, with a 3.5-fold increase in Hpx at the lowest barrier function. This suggests that Hpx is a marker of the rapid adenylate metabolite pool regulation by IECs during stress. Hpx supplementation studies demonstrated an enhanced barrier formation rate and an epithelial monolayer with significantly higher resistance in Hpx-treated cells. Hpx significantly accelerated wound closure rate, demonstrating a role in improving cellular migration. Additionally, it was discovered that Hpx increases total available epithelial cellular energy, as seen in an increase in cellular phoshocreatine (PCr) and ATP. Alternatively, inhibition of adenylate metabolite flux through Hpx significantly impacted barrier development, altogether identifying an important role for the purine salvage pathway in IEC function. A metabolomic screen from healthy and colitic murine colon tissue revealed a >65% decrease in Hpx during active inflammation. Furthermore, the loss of Hpx strongly correlated with disease markers (e.g., weight loss, colon length) [160].
Given the unique environment of physiological hypoxia at the intestinal epithelial–luminal interface, the role of Hpx in barrier development and energetics was explored in hypoxia. IECs were exposed to physiologically relevant hypoxia (1% O2, 40 h) in the presence and absence of Hpx. Controls suffered a significant drop in barrier strength as shown by TEER, with Hpx supplementation wholly preventing the hypoxia-induced loss of barrier function. Furthermore, hypoxia incurred a significant loss of energy pools, while Hpx supplementation salvaged this energy loss by increasing both PCr and ATP levels [160].
4.3.2. Hypoxanthine Is Also a GM Metabolite
The de novo synthesis of purines in IECs is limited due to its high energy cost, requiring 5 ATP molecules for production of one purine, in an environment of energetically-depleting hypoxia. Thus, purine metabolism typically depends on the salvage of exogenously supplied purine substrates for nucleotide biogenesis [162]. The purine salvage pathway functions to utilize nucelobases such as Hpx for the biosynthesis of ATP and equilibrating energy. A recent report sought to explore sources of purines in the murine mucosal environment. Surprisingly, water-soluble extracellular fecal metabolite analyses of conventionally raised (CR) mice revealed significant concentrations of salvageable and available purines, mostly in the form of Hpx and xanthine (Xan) (Figure 4). In contrast, fecal extracts from streptomycin-treated mice exhibited a 90% reduction in purines, identifying the microbiota as a distributor of microbiota-sourced purine (MSP) available for salvage by the intestinal epithelium [158].
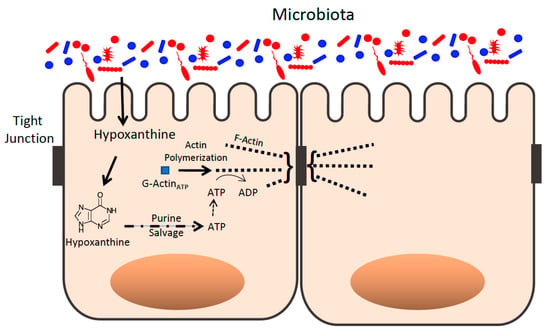
Figure 4.
Purines derived from microbial sources influence the epithelial apical tight junction actin complex. Purines such as hypoxanthine are derived, in part, from microbial sources and are salvaged as templates for ATP generation that supports actin polymerization of G-actin to F-actin in the apical epithelial TJ complex.
To assess the importance of MSP in metabolism and barrier function, control GF mice (GF-CNTL) were monocolonized with purine-producing E. coli K12 (GF-K12), and then both were subjected to DSS colitis. In contrast to GF-K12 mice, which showed minimal signs of disease, GF-CNTL mice demonstrated notable weight loss and significant mortality during the course of treatment. Through the salvage of MSP, healthy GF-K12 mice showed significant increases in purine levels versus GF-CNTL, including Hpx, Xan, Inosine (a hypoxanthine-derived nucleoside) and high-energy phosphates. Alternatively, DSS-induced colitis diminished most tissue purines in GF-K12 mice but without loss of ADP and with increases in ATP and PCr. This result suggests that colonic tissue is reliant upon MSP in order to maintain adenylate nucleotide levels and energy balance during insult. Furthermore, Hpx supplementation in streptomycin-treated mice (which show loss of MSP) subjected to DSS-induced colitis proved to be protective by reducing weight loss and colon shortening, as well as increasing tissue adenylate energy levels similar to K12-colonized mice [158]. Thus, purines (especially hypoxanthine) seem to play a vital role in intestinal barrier health in a manner tightly correlated with energy balance. Further exploration of purines in IBD patients may shine light on promising novel treatments.
4.4. Secondary Bile Acids
4.4.1. Secondary Bile Acids Metabolism
Primary bile acids (PBAs), specifically cholic acid (CA) and chenodeoxycholic acid (CDCA), are synthesized from cholesterol in the liver. PBAs are constitutively produced and, along with cholesterol and phospholipids, comprise the gallbladder-stored bile salts. Following a meal, bile salts are secreted into the small intestine. Prior to their secretion, CA and CDCA are conjugated to glycine or taurine via an amide bond [163]. This transformation allows the formation of micelles [164], which in turn emulsify lipids and fat-soluble vitamins for absorption. After digestion, approximately 95% of the PBAs are reabsorbed by the distal ileum and recycled from circulation by the liver [165]. The remainder that evade absorption reach the colon and dynamically interact with the microbiota, where they are metabolized into secondary bile acids (SBAs) produced solely by the GM.
Numerous SBAs are produced by microbial metabolism, including litocholic acid (LCA) and deoxycholic acid (DCA). A small population of intestinal species in the Firmicutes phylum and genus Clostridium, including C. scindens, C. hiranonis, C. hylemonae, and C. sordelli are capable of producing SBAs [166]. The biochemistry of this metabolism is particularly interesting, broadly encompassed by two steps: hydrolysis of the amide bond for deconjugation via bile salt hydrolases (BSH) producing free primary bile salts; followed by transformation to secondary bile acids, mainly by 7α-dehydroxylation reactions [166]. It appears that the latter reaction is restricted to free bile acids, thus the deconjugation step is a prerequisite [166]. The conversion of PBAs to SBAs by 7α-dehydroxylases is considered one of the most physiologically relevant microbial transformations in the body [166]. In addition, SBAs undergo selective microbial isomerization giving them different immunomodulatory properties, for example isoDCA (an isomer of DCA) promotes regulatory T cells (Treg) differentiation, while DCA itself cannot [167]. This suggests a possible on-demand supply of different SBAs depending on the current intestinal environment.
4.4.2. Secondary Bile Acids and Receptors
SBAs are potent nuclear receptor ligands, binding to farnesoid X receptor (FXR), vitamin D receptor (VDR), PXR, and act as endogenous agonists for the microbial G protein coupled-bile acid receptor (TGR5) [61]. These receptors play an important role in many cells, including IECs and immune cells, with response to endogenous and bacterial antigens. FXR regulate intestinal immune responses driven by the GM with altered bile acids profiles during dysbiosis [168]. Intestinal diseases such as IBD downregulate bile acids biotransformation genes mostly from the Firmicutes phylum (a.k.a main butyrate-producing bacteria), resulting in low SBAs and high PBAs levels compared to healthy individuals [169,170]. Various in vivo colonic models including FXR null mice [171], downregulation of FXR [172] and antibiotic-induced GM alterations [173] resulted in uncontrolled intestinal inflammation and premature death. Obeticholic acid (INT-747) is a semi-synthetic SBA and a potent FXR receptor agonist. In human monocytes and dendritic cells cultured in vitro, the expression of key inflammatory cytokines and chemokines are inhibited by INT-747-mediated FXR activation [174]. Likewise, the expression of pro-inflammatory genes is also repressed by FXR activation in IECs [175]. In vivo, INT-747-mediated FXR activation improves epithelial barrier integrity while decreasing inflammatory cytokines (e.g., IL-1β, IL-6) and CCL2 chemokine in DSS or TNBS-induced colitis murine models [174,175].
Furthermore, SBAs activate TGR5, unleashing a plethora of intestinal immunomodulatory and anti-inflammatory influences, mainly in macrophages and monocytes [176]. Briefly, TGR5 regulates energy metabolism and glucose homeostasis. In addition, pro-inflammatory mediators such as, IL-1, IL-6, and TNF-α are inhibited by SBA-mediated activation of TGR5 [177], while the intestinal anti-inflammatory cytokine IL-10 levels increase [178]. In vivo experiments discovered that downregulation of TGR5 results in destroyed architecture of epithelial tight junctions and abnormal distribution of zonulin-1 [179].
Lastly, VDR downregulation and vitamin D deficiency are common biomarkers in patients with IBD [180]. Using VDR as a receptor, bile acids (mainly LCA) control various physiological processes, including cell differentiation and inflammatory resolution. In various colitis models, VDR knockout mice displayed low levels of antimicrobial peptides, barrier dysregulation, and increased mortality [181,182]. Additionally in colitis models, mice with transgenic human VDR in IECs exhibit a mucosal barrier protection mechanism as elucidated by preserved TEER, reduction in IEC apoptosis, caspase-3 deactivation, and downregulation of p53-upregulated modulator of apoptosis (PUMA), an important inducer of IEC apoptosis in IBD [183].
Interestingly, bile acids play a major role in GM composition. Kakiyama et al. reported a relationship between liver health, GM populations, and fecal bile acid profiles [184]. In this study, it was observed that bacterial dysbiosis is linked to lower levels of bile acids reaching the intestine in patients with early and advanced cirrhosis as compared to control patients. This GM dysbiosis was elucidated by a significant reduction in commensal Gram-positive members (e.g., Blautia and Rumminococcaceae) and an increase in pro-inflammatory and harmful taxa Enterobacteriaceae, accompanied by decreased fecal bile acid levels [184,185]. Alternatively, a diet rich in bile acids (CA) resulted in phylum-level shifts of the GM, with Firmicutes populations vastly expanding in rats [186]. As a possible mechanism, bile acids act upon certain bacterial membranes due to their hydrophobicity and detergent properties exhibiting direct antimicrobial effects. A full report of the role of bile acids in GM composition can be found here [187].
Overall, SBAs play a pivotal role in the activation of important receptors that control inflammation and immunity. SBAs are broadly responsible for the regulation of Tregs, monocytes, and macrophages in the intestinal barrier. Furthermore, they influence GM composition in health and disease. These vital functions elicit a tremendous interest in further exploration of natural SBAs or novel synthetic derivatives as therapeutic options in inflammatory diseases.
4.5. Polyamines
Natural aliphatic biomolecules containing at least two amine groups are termed polyamines (hereafter PAs). Their carbon backbone allows them to bond to hydrophobic molecules, and their amino groups (usually carrying a positive charge under physiological pH) allow them to bind to anionic moieties. In mammalian cells, the total concentration of PAs is in the mM range. However, free intracellular PA concentration is much lower (approx. <10%), due to the fact that most of these cationic moieties are constantly binding to negatively charged molecules, including nucleic acids, proteins, and phospholipids, often modulating their function [188]. It is generally known that PAs are mostly endogenous (de novo biosynthesis, catabolism); however, the microbiome is known to be a major source of luminal polyamines. The major PAs in mammalian cells are spermidine, spermine, and their precursor putrescine [189]. PAs are essential for various cell functions from cell proliferation and viability, immune system and apoptosis [190,191,192], derived mostly from the two amino acids ornithine and methionine, and largely regulated by two enzymes ornithine decarboxylase (ODC) and S-adenosylmethionine decarboxylase (SAMDC). Studies to date support the importance of polyamines for normal gut mucosal growth and barrier function [193,194,195]. Increasing the concentration of PAs stimulates gut mucosal renewal and enhances barrier function. Alternatively, inhibiting the major PA enzymes (ODC and SAMDC) leads to compromising the gut epithelia and barrier dysbiosis [193,196]. Furthermore, evidence that PA levels are remarkably impacted in active patients with IBD [194] and other mucosa-related diseases [195] has elicited recent interest in these biomolecules as a potential novel therapeutic approach.
PAs play various important roles in intestinal health, including epithelial renewal, barrier maintenance, and immunity. Reports indicate that a sustained supply of PAs to actively dividing cells is required for normal intestinal epithelial renewal and mucosal healing [193,197]. Growth stimulation is possible through PA-mediated regulation of various genes encoding growth-promoting proteins (e.g., MYC, FOS, and JUN) [198,199,200] and control of growth-inhibiting factors such as p53, nucleophosmin, JunD, TGF-β, and Smads [201,202,203,204]. Furthermore, PAs enhance intestinal barrier function by modulating intercellular junctions. PAs (mostly spermidine) are necessary for the expression of AJ constituent E-cadherin. It has been reported that E-cadherin stabilization is partially achieved through PA-induced increases in intracellular free Ca2+, a necessary cofactor for AJ formation [205]. E-cadherin expression is also due to enhanced gene transcription by activating MYC, which directly interacts with the E-pal box located at the E-cadherin promoter [206]. PAs regulate TJ protein Zonula occludens-1 (ZO-1) expression by modulating its gene transcription via JUND [207] and other TJs including ZO-2, claudin-2, and claudin-3, though the mechanisms of this regulation are still unknown [208]. PAs are also important in regulating immune responses. As an example, spermidine restored CD8+ T cell responses in elderly mice [209]. Additionally, PA spermine inhibits LPS-mediated production of nitric oxide and pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 in macrophages and human mononuclear cells [210,211]. In the same context, biogenic amines such as spermine and histamine influence IL-18 secretion through inhibition of the NLRP6 inflammasome [212,213]. Gathering this data, it is clear yet again the important role that metabolites (including PAs) play in intestinal barrier restoration, wound healing, proliferation, and immunity.
5. Concluding Remarks
The vital role that the gut microbiota plays in intestinal health and systemic immunity is clear. The study of the complex machinery and mechanisms behind these benefits is likely in its infancy. Exploring further metabolite functions, receptors, metabolite-regulated transcriptional activity, and even novel metabolites opens a promising and vast field of research directed at ameliorating intestinal diseases. The study of microbial shifts and environment-dependent “on demand” biosynthesis of specific metabolites is also attractive.
Many questions remain; however, it is clear that metabolites interact with every cell type in the gut, from IECs to immune cells. Although a single approach (e.g., supplementation) might not be sufficient to restore barrier function, a combinatorial manipulation of receptors, metabolites, enzymes, and transcription factors might become a silver bullet—not forgetting that the simplest approach to procure a healthy gut is through a diet rich in necessary components for normal GM composition and metabolite biosynthesis.
Author Contributions
A.O., A.S.D., J.S.L. and S.P.C. contributed to writing and editing of this review. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Institutes of Health (DK05189, DK104713, DK095491, DK129410, T32AI007405-31A1), the Veterans Administration (BX002182) and the Crohn’s and Colitis Foundation (RFA #831495).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert. Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef]
- Farhadi, A.; Banan, A.; Fields, J.; Keshavarzian, A. Intestinal barrier: An interface between health and disease. J. Gastroenterol. Hepatol. 2003, 18, 479–497. [Google Scholar] [CrossRef]
- Salvo Romero, E.; Alonso Cotoner, C.; Pardo Camacho, C.; Casado Bedmar, M.; Vicario, M. The intestinal barrier function and its involvement in digestive disease. Rev. Esp. Enferm. Dig. 2015, 107, 686–696. [Google Scholar] [CrossRef]
- Nalle, S.C.; Turner, J.R. Intestinal barrier loss as a critical pathogenic link between inflammatory bowel disease and graft-versus-host disease. Mucosal. Immunol. 2015, 8, 720–730. [Google Scholar] [CrossRef]
- Caviglia, G.P.; Dughera, F.; Ribaldone, D.G.; Rosso, C.; Abate, M.L.; Pellicano, R.; Bresso, F.; Smedile, A.; Saracco, G.M.; Astegiano, M. Serum zonulin in patients with inflammatory bowel disease: A pilot study. Minerva Med. 2019, 110, 95–100. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Natividad, J.M.M.; Verdu, E.F. Modulation of intestinal barrier by intestinal microbiota: Pathological and therapeutic implications. Pharmacol. Res. 2013, 69, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, U.A.; Magnusson, M.K.; Rydström, A.; Jonstrand, C.; Hengst, J.; Johansson, M.E.; Velcich, A.; Öhman, L.; Strid, H.; Sjövall, H.; et al. Spontaneous Colitis in Muc2-Deficient Mice Reflects Clinical and Cellular Features of Active Ulcerative Colitis. PLoS ONE 2014, 9, e100217. [Google Scholar] [CrossRef] [PubMed]
- Pelaseyed, T.; Bergström, J.H.; Gustafsson, J.K.; Ermund, A.; Birchenough, G.M.H.; Schütte, A.; Van Der Post, S.; Svensson, F.; Rodríguez-Piñeiro, A.M.; Nyström, E.E.L.; et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol. Rev. 2014, 260, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Moran, A.P.; Gupta, A.; Joshi, L. Sweet-talk: Role of host glycosylation in bacterial pathogenesis of the gastrointestinal tract. Gut 2011, 60, 1412–1425. [Google Scholar] [CrossRef]
- Hooper, L.V.; Gordon, J.I. Glycans as legislators of host-microbial interactions: Spanning the spectrum from symbiosis to pathogenicity. Glycobiology 2001, 11, 1R–10R. [Google Scholar] [CrossRef]
- Barnich, N.; Carvalho, F.A.; Glasser, A.-L.; Darcha, C.; Jantscheff, P.; Allez, M.; Peeters, H.; Bommelaer, G.; Desreumaux, P.; Colombel, J.-F.; et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J. Clin. Investig. 2007, 117, 1566–1574. [Google Scholar] [CrossRef]
- Sun, W.W.; Krystofiak, E.S.; Leo-Macias, A.; Cui, R.; Sesso, A.; Weigert, R.; Ebrahim, S.; Kachar, B. Nanoarchitecture and dynamics of the mouse enteric glycocalyx examined by freeze-etching electron tomography and intravital microscopy. Commun. Biol. 2020, 3. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.V.; Larsson, J.M.H.; Hansson, G.C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 4659–4665. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.V.; Hansson, G.C. Immunological aspects of intestinal mucus and mucins. Nat. Rev. Immunol. 2016, 16, 639–649. [Google Scholar] [CrossRef]
- Schröder, J.M. Epithelial antimicrobial peptides: Innate local host response elements. Cell Mol. Life Sci. 1999, 56, 32–46. [Google Scholar] [CrossRef]
- Chu, H.; Pazgier, M.; Jung, G.; Nuccio, S.P.; Castillo, P.A.; de Jong, M.F.; Winter, M.G.; Winter, S.E.; Wehkamp, J.; Shen, B.; et al. Human α-defensin 6 promotes mucosal innate immunity through self-assembled peptide nanonets. Science 2012, 337, 477–481. [Google Scholar] [CrossRef]
- Jones, D.E.; Bevins, C.L. Paneth cells of the human small intestine express an antimicrobial peptide gene. J. Biol. Chem. 1992, 267, 23216–23225. [Google Scholar] [CrossRef]
- Wehkamp, J.; Harder, J.; Weichenthal, M.; Schwab, M.; Schäffeler, E.; Schlee, M.; Herrlinger, K.R.; Stallmach, A.; Noack, F.; Fritz, P.; et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut 2004, 53, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Wehkamp, J.; Schwind, B.; Herrlinger, K.R.; Baxmann, S.; Schmidt, K.; Duchrow, M.; Wohlschläger, C.; Feller, A.C.; Stange, E.F.; Fellermann, K. Innate immunity and colonic inflammation: Enhanced expression of epithelial α-defensins. Dig. Dis. Sci. 2002, 47, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Cobo, E.R.; Chadee, K. Antimicrobial Human β-Defensins in the Colon and Their Role in Infectious and Non-Infectious Diseases. Pathogens 2013, 2, 177–192. [Google Scholar] [CrossRef]
- Sankaran-Walters, S.; Hart, R.; Dills, C. Guardians of the Gut: Enteric Defensins. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef]
- Jakobsson, H.E.; Rodríguez-Piñeiro, A.M.; Schütte, A.; Ermund, A.; Boysen, P.; Bemark, M.; Sommer, F.; Bäckhed, F.; Hansson, G.C.; Johansson, M.E. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 2015, 16, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Salim, S.Y.; Söderholm, J.D. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm. Bowel Dis. 2011, 17, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, K.; Mall, M. Electrolyte transport in the mammalian colon: Mechanisms and implications for disease. Physiol. Rev. 2002, 82, 245–289. [Google Scholar] [CrossRef] [PubMed]
- Groschwitz, K.R.; Hogan, S.P. Intestinal barrier function: Molecular regulation and disease pathogenesis. J. Allergy Clin. Immunol. 2009, 124, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Whitley, C.S.; Haribabu, B.; Jala, V.R. Regulation of Intestinal Barrier Function by Microbial Metabolites. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 1463–1482. [Google Scholar] [CrossRef]
- Zuo, L.; Kuo, W.-T.; Turner, J.R. Tight Junctions as Targets and Effectors of Mucosal Immune Homeostasis. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 327–340. [Google Scholar] [CrossRef]
- Garcia-Hernandez, V.; Quiros, M.; Nusrat, A. Intestinal epithelial claudins: Expression and regulation in homeostasis and inflammation. Ann. N. Y. Acad. Sci. 2017, 1397, 66–79. [Google Scholar] [CrossRef]
- Johansen, F.E.; Kaetzel, C.S. Regulation of the polymeric immunoglobulin receptor and IgA transport: New advances in environmental factors that stimulate pIgR expression and its role in mucosal immunity. Mucosal Immunol. 2011, 4, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Johansen, F.-E.; Pekna, M.; Norderhaug, I.N.; Haneberg, B.; Hietala, M.A.; Krajci, P.; Betsholtz, C.; Brandtzaeg, P. Absence of Epithelial Immunoglobulin a Transport, with Increased Mucosal Leakiness, in Polymeric Immunoglobulin Receptor/Secretory Component–Deficient Mice. J. Exp. Med. 1999, 190, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Shulzhenko, N.; Morgun, A.; Hsiao, W.; Battle, M.; Yao, M.; Gavrilova, O.; Orandle, M.; Mayer, L.; Macpherson, A.J.; McCoy, K.D.; et al. Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat. Med. 2011, 17, 1585–1593. [Google Scholar] [CrossRef]
- Bunker, J.J.; Flynn, T.M.; Koval, J.C.; Shaw, D.G.; Meisel, M.; McDonald, B.D.; Ishizuka, I.; Dent, A.L.; Wilson, P.C.; Jabri, B.; et al. Innate and Adaptive Humoral Responses Coat Distinct Commensal Bacteria with Immunoglobulin A. Immunity 2015, 43, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Palm, N.W.; Zoete, M.R.d.; Cullen, T.W.; Barry, N.A.; Stefanowski, J.; Hao, L.; Degnan, P.H.; Hu, J.; Peter, I.; Zhang, W.; et al. Immunoglobulin A Coating Identifies Colitogenic Bacteria in Inflammatory Bowel Disease. Cell 2014, 158, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.L.; Greenstein, A.J.; Geller, S.A.; Gordon, R.E.; Aufses, A.H., Jr. A freeze fracture study of Crohn’s disease of the terminal ileum: Changes in epithelial tight junction organization. Am. J. Gastroenterol. 1983, 78, 537–547. [Google Scholar] [PubMed]
- Gassler, N.; Rohr, C.; Schneider, A.; Kartenbeck, J.; Bach, A.; Obermüller, N.; Otto, H.F.; Autschbach, F. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G216–G228. [Google Scholar] [CrossRef]
- Wyatt, J.; Vogelsang, H.; Hübl, W.; Waldhöer, T.; Lochs, H. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet 1993, 341, 1437–1439. [Google Scholar] [CrossRef]
- D’Incà, R.; Di Leo, V.; Corrao, G.; Martines, D.; D’Odorico, A.; Mestriner, C.; Venturi, C.; Longo, G.; Sturniolo, G.C. Intestinal permeability test as a predictor of clinical course in Crohn’s disease. Am. J. Gastroenterol. 1999, 94, 2956–2960. [Google Scholar] [CrossRef]
- Mehandru, S.; Colombel, J.-F. The intestinal barrier, an arbitrator turned provocateur in IBD. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.A.; Rodrigues, B.L.; Ayrizono, M.L.; Leal, R.F. The Immunological Basis of Inflammatory Bowel Disease. Gastroenterol. Res. Pract. 2016, 2016, 2097274. [Google Scholar] [CrossRef] [PubMed]
- Glover, L.E.; Lee, J.S.; Colgan, S.P. Oxygen metabolism and barrier regulation in the intestinal mucosa. J. Clin. Investig. 2016, 126, 3680–3688. [Google Scholar] [CrossRef]
- Colgan, S.P.; Taylor, C.T. Hypoxia: An alarm signal during intestinal inflammation. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Karhausen, J.; Furuta, G.T.; Tomaszewski, J.E.; Johnson, R.S.; Colgan, S.P.; Haase, V.H. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J. Clin. Investig. 2004, 114, 1098–1106. [Google Scholar] [CrossRef]
- Shepherd, A.P. Metabolic control of intestinal oxygenation and blood flow. Fed. Proc. 1982, 41, 2084–2089. [Google Scholar] [PubMed]
- Albenberg, L.; Esipova, T.V.; Judge, C.P.; Bittinger, K.; Chen, J.; Laughlin, A.; Grunberg, S.; Baldassano, R.N.; Lewis, J.D.; Li, H.; et al. Correlation Between Intraluminal Oxygen Gradient and Radial Partitioning of Intestinal Microbiota. Gastroenterology 2014, 18, 1055–1063. [Google Scholar] [CrossRef]
- Colgan, S.P.; Campbell, E.L.; Kominsky, D.J. Hypoxia and Mucosal Inflammation. Ann. Rev. Pathol. 2016, 11, 77–100. [Google Scholar] [CrossRef]
- Konjar, Š.; Pavšič, M.; Veldhoen, M. Regulation of Oxygen Homeostasis at the Intestinal Epithelial Barrier Site. Int. J. Mol. Sci. 2021, 22, 9170. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.M.; Hahn, S.; Pook, D.R.; Jenkins, W.T.; Chalian, A.A.; Zhang, P.; Stevens, C.; Weber, R.; Weinstein, G.; Benjamin, I.; et al. Detection of hypoxia in human squamous cell carcinoma by EF5 binding. Cancer Res. 2000, 60, 2018–2024. [Google Scholar]
- Lord, E.M.; Harwell, L.; Koch, C.J. Detection of hypoxic cells by monoclonal antibody recognizing 2-nitroimidazole adducts. Cancer Res. 1993, 53, 5721–5726. [Google Scholar] [PubMed]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Furuta, G.T.; Turner, J.R.; Taylor, C.T.; Hershberg, R.M.; Comerford, K.; Narravula, S.; Podolsky, D.K.; Colgan, S.P. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J. Exp. Med. 2001, 193, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002, 62, 3387–3394. [Google Scholar] [PubMed]
- Synnestvedt, K.; Furuta, G.T.; Comerford, K.M.; Louis, N.; Karhausen, J.; Eltzschig, H.K.; Hansen, K.R.; Thompson, L.F.; Colgan, S.P. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J. Clin. Investig. 2002, 110, 993–1002. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eltzschig, H.K.; Ibla, J.C.; Furuta, G.T.; Leonard, M.O.; Jacobson, K.A.; Enjyoji, K.; Robson, S.C.; Colgan, S.P. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: Role of ectonucleotidases and adenosine A2B receptors. J. Exp. Med. 2003, 198, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Regulation of Metabolism by Hypoxia-Inducible Factor 1. Cold Spring Harb. Symp. Quant. Biol. 2011, 2011, 22. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1, O(2), and the 3 PHDs: How animal cells signal hypoxia to the nucleus. Cell 2001, 107, 1–3. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Tanimoto, K.; Makino, Y.; Pereira, T.; Poellinger, L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. Embo J. 2000, 19, 4298–4309. [Google Scholar] [CrossRef] [PubMed]
- Gasaly, N.; de Vos, P.; Hermoso, M.A. Impact of Bacterial Metabolites on Gut Barrier Function and Host Immunity: A Focus on Bacterial Metabolism and Its Relevance for Intestinal Inflammation. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.; Gasbarrini, A.; Mele, M. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.J.; Lynch, D.B.; Murphy, K.; Ulaszewska, M.; Jeffery, I.B.; O’Shea, C.A.; Watkins, C.; Dempsey, E.; Mattivi, F.; Tuohy, K.; et al. Evolution of gut microbiota composition from birth to 24 weeks in the INFANTMET Cohort. Microbiome 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Jandhyala, S.M. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787. [Google Scholar] [CrossRef]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Quan, G.; Jiang, X.; Yang, Y.; Ding, X.; Zhang, D.; Wang, X.; Hardwidge, P.R.; Ren, W.; Zhu, G. Effects of Metabolites Derived From Gut Microbiota and Hosts on Pathogens. Front. Cell. Infect. Microbiol. 2018, 8. [Google Scholar] [CrossRef]
- Donia, M.S.; Fischbach, M.A. Small molecules from the human microbiota. Science 2015, 349, 1254766. [Google Scholar] [CrossRef]
- Anders, H.-J.; Andersen, K.; Stecher, B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013, 83, 1010–1016. [Google Scholar] [CrossRef]
- Blumberg, R.; Powrie, F. Microbiota, Disease, and Back to Health: A Metastable Journey. Sci. Transl. Med. 2012, 4, 137rv7. [Google Scholar] [CrossRef]
- Thorburn, A.N.; Macia, L.; Mackay, C.R. Diet, Metabolites, and “Western-Lifestyle” Inflammatory Diseases. Immunity 2014, 40, 833–842. [Google Scholar] [CrossRef]
- Hamard, A.; Sève, B.; Le Floc’H, N. Intestinal development and growth performance of early-weaned piglets fed a low-threonine diet. Animal 2007, 1, 1134–1142. [Google Scholar] [CrossRef]
- Wang, M.; Wichienchot, S.; He, X.; Fu, X.; Huang, Q.; Zhang, B. In vitro colonic fermentation of dietary fibers: Fermentation rate, short-chain fatty acid production and changes in microbiota. Trends Food Sci. Technol. 2019, 88, 1–9. [Google Scholar] [CrossRef]
- Fardet, A. New hypotheses for the health-protective mechanisms of whole-grain cereals: What is beyond fibre? Nutr. Res. Rev. 2010, 23, 65–134. [Google Scholar] [CrossRef] [PubMed]
- Aune, D.; Chan, D.S.M.; Lau, R.; Vieira, R.; Greenwood, D.C.; Kampman, E.; Norat, T. Dietary fibre, whole grains, and risk of colorectal cancer: Systematic review and dose-response meta-analysis of prospective studies. BMJ 2011, 343, d6617. [Google Scholar] [CrossRef]
- Reynolds, A.; Mann, J.; Cummings, J.; Winter, N.; Mete, E.; Te Morenga, L. Carbohydrate quality and human health: A series of systematic reviews and meta-analyses. Lancet 2019, 393, 434–445. [Google Scholar] [CrossRef]
- Mu, Q.; Kirby, J.; Reilly, C.M.; Luo, X.M. Leaky Gut As a Danger Signal for Autoimmune Diseases. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- McGuckin, M.A.; Eri, R.; Simms, L.A.; Florin, T.H.J.; Radford-Smith, G. Intestinal barrier dysfunction in inflammatory bowel diseases. Inflamm. Bowel Dis. 2009, 15, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, E.D.; Sonnenburg, J.R. Starving our Microbial Self: The Deleterious Consequences of a Diet Deficient in Microbiota-Accessible Carbohydrates. Cell Metab. 2014, 20, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Beukema, M.; Faas, M.M.; De Vos, P. The effects of different dietary fiber pectin structures on the gastrointestinal immune barrier: Impact via gut microbiota and direct effects on immune cells. Exp. Mol. Med. 2020, 52, 1364–1376. [Google Scholar] [CrossRef] [PubMed]
- Topping, D.L.; Clifton, P.M. Short-chain fatty acids and human colonic function: Roles of resistant starch and nonstarch polysaccharides. Physiol. Rev. 2001, 81, 1031–1064. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef]
- Sivaprakasam, S.; Bhutia, Y.D.; Yang, S.; Ganapathy, V. Short-Chain Fatty Acid Transporters: Role in Colonic Homeostasis. Compr. Physiol. 2017, 8, 299–314. [Google Scholar] [CrossRef]
- Dankert, J.; Zijlstra, J.B.; Wolthers, B.G. Volatile fatty acids in human peripheral and portal blood: Quantitative determination vacuum distillation and gas chromatography. Clin. Chim. Acta 1981, 110, 301–307. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Meredith, D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflug. Arch. 2004, 447, 619–628. [Google Scholar] [CrossRef]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Boets, E.; Gomand, S.V.; Deroover, L.; Preston, T.; Vermeulen, K.; De Preter, V.; Hamer, H.M.; Van den Mooter, G.; De Vuyst, L.; Courtin, C.M.; et al. Systemic availability and metabolism of colonic-derived short-chain fatty acids in healthy subjects: A stable isotope study. J. Physiol. 2017, 595, 541–555. [Google Scholar] [CrossRef]
- Karaki, S.-I.; Mitsui, R.; Hayashi, H.; Kato, I.; Sugiya, H.; Iwanaga, T.; Furness, J.B.; Kuwahara, A. Short-chain fatty acid receptor, GPR43, is expressed by enteroendocrine cells and mucosal mast cells in rat intestine. Cell Tissue Res. 2006, 324, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Thangaraju, M.; Cresci, G.A.; Liu, K.; Ananth, S.; Gnanaprakasam, J.P.; Browning, D.D.; Mellinger, J.D.; Smith, S.B.; Digby, G.J.; Lambert, N.A.; et al. GPR109A Is a G-protein–Coupled Receptor for the Bacterial Fermentation Product Butyrate and Functions as a Tumor Suppressor in Colon. Cancer Res. 2009, 69, 2826–2832. [Google Scholar] [CrossRef] [PubMed]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian Mckenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshini, M.; Kotlo, K.U.; Dudeja, P.K.; Layden, B.T. Role of Short Chain Fatty Acid Receptors in Intestinal Physiology and Pathophysiology. Compr. Physiol. 2018, 1091–1115. [Google Scholar] [CrossRef]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, Receptor for Niacin and the Commensal Metabolite Butyrate, Suppresses Colonic Inflammation and Carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Kaisar, M.M.M.; Pelgrom, L.R.; van der Ham, A.J.; Yazdanbakhsh, M.; Everts, B. Butyrate Conditions Human Dendritic Cells to Prime Type 1 Regulatory T Cells via both Histone Deacetylase Inhibition and G Protein-Coupled Receptor 109A Signaling. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Nastasi, C.; Fredholm, S.; Willerslev-Olsen, A.; Hansen, M.; Bonefeld, C.M.; Geisler, C.; Andersen, M.H.; Ødum, N.; Woetmann, A. Butyrate and propionate inhibit antigen-specific CD8+ T cell activation by suppressing IL-12 production by antigen-presenting cells. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Davie, J.R. Inhibition of Histone Deacetylase Activity by Butyrate. J. Nutr. 2003, 133, 2485S–2493S. [Google Scholar] [CrossRef] [PubMed]
- Shakespear, M.R.; Halili, M.A.; Irvine, K.M.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011, 32, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Roediger, W.E. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 1980, 21, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The Microbiome and Butyrate Regulate Energy Metabolism and Autophagy in the Mammalian Colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 2011, 474, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Kelly, C.J.; Battista, K.D.; Schaefer, R.; Lanis, J.M.; Alexeev, E.E.; Wang, R.X.; Onyiah, J.C.; Kominsky, D.J.; Colgan, S.P. Microbial-Derived Butyrate Promotes Epithelial Barrier Function through IL-10 Receptor-Dependent Repression of Claudin-2. J. Immunol. 2017, 199, 2976–2984. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, M.; Singh, A.B.; Smith, J.J.; Sharma, A.; Chen, X.; Eschrich, S.; Yeatman, T.J.; Beauchamp, R.D.; Dhawan, P. HDAC inhibitors regulate claudin-1 expression in colon cancer cells through modulation of mRNA stability. Oncogene 2010, 29, 305–312. [Google Scholar] [CrossRef]
- Wang, R.X.; Lee, J.S.; Campbell, E.L.; Colgan, S.P. Microbiota-derived butyrate dynamically regulates intestinal homeostasis through regulation of actin-associated protein synaptopodin. Proc. Natl. Acad. Sci. USA 2020, 117, 11648–11657. [Google Scholar] [CrossRef]
- Kelly, C.J.; Colgan, S.P. Breathless in the Gut: Implications of Luminal O 2 for Microbial Pathogenicity. Cell Host Microbe 2016, 19, 427–428. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.X.; Henen, M.A.; Lee, J.S.; Vogeli, B.; Colgan, S.P. Microbiota-derived butyrate is an endogenous HIF prolyl hydroxylase inhibitor. Gut Microbes 2021, 13, 1938380. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, B.J.; Kao, D.J.; Kitzenberg, D.A.; Dobrinskikh, E.; Schwisow, K.D.; Masterson, J.C.; Kendrick, A.A.; Kelly, C.J.; Bayless, A.J.; Kominsky, D.J.; et al. HIF-dependent regulation of claudin-1 is central to intestinal epithelial tight junction integrity. Mol. Biol. Cell 2015, 26, 2252–2262. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.; Pandey, R.; Chauhan, N.S. Hypoxia Inducible Factor-1α: The Curator of Gut Homeostasis. Front. Cell. Infect. Microbiol. 2020, 10, 227. [Google Scholar] [CrossRef] [PubMed]
- Finnie, I.A.; Dwarakanath, A.D.; Taylor, B.A.; Rhodes, J.M. Colonic mucin synthesis is increased by sodium butyrate. Gut 1995, 36, 93–99. [Google Scholar] [CrossRef]
- Glover, L.E.; Bowers, B.E.; Saeedi, B.; Ehrentraut, S.F.; Campbell, E.L.; Bayless, A.J.; Dobrinskikh, E.; Kendrick, A.A.; Kelly, C.J.; Burgess, A. Control of creatine metabolism by HIF is an endogenous mechanism of barrier regulation in colitis. Proc. Natl. Acad. Sci. USA 2013, 110, 19820–19825. [Google Scholar] [CrossRef]
- Hall, C.H.T.; Lee, J.S.; Murphy, E.M.; Gerich, M.E.; Dran, R.; Glover, L.E.; Abdulla, Z.I.; Skelton, M.R.; Colgan, S.P. Creatine Transporter, Reduced in Colon Tissues From Patients With Inflammatory Bowel Diseases, Regulates Energy Balance in Intestinal Epithelial Cells, Epithelial Integrity, and Barrier Function. Gastroenterology 2020, 159, 984–998. [Google Scholar] [CrossRef] [PubMed]
- Singhal, R.; Shah, Y.M. Oxygen battle in the gut: Hypoxia and hypoxia-inducible factors in metabolic and inflammatory responses in the intestine. J. Biol. Chem. 2020, 295, 10493–10505. [Google Scholar] [CrossRef]
- Kumari, R. Fluctuations in butyrate-producing bacteria in ulcerative colitis patients of North India. World J. Gastroenterol. 2013, 19, 3404. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, L.; Zhou, R.; Wang, X.; Song, L.; Huang, S.; Wang, G. Increased Proportions of Bifidobacterium and the Lactobacillus Group and Loss of Butyrate-Producing Bacteria in Inflammatory Bowel Disease. J. Clin. Microbiol. 2014, 52, 398–406. [Google Scholar] [CrossRef]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2013. [Google Scholar] [CrossRef]
- Eeckhaut, V.; Machiels, K.; Perrier, C.; Romero, C.; Maes, S.; Flahou, B.; Steppe, M.; Haesebrouck, F.; Sas, B.; Ducatelle, R.; et al. Butyricicoccus pullicaecorum in inflammatory bowel disease. Gut 2013, 62, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Seksik, P.; Furet, J.P.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Dore, J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel Dis. 2009, 15, 1183–1189. [Google Scholar] [CrossRef]
- Hallert, C.; Bjorck, I.; Nyman, M.; Pousette, A.; Granno, C.; Svensson, H. Increasing fecal butyrate in ulcerative colitis patients by diet: Controlled pilot study. Inflamm. Bowel Dis. 2003, 9, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Dong, Y.; Huang, W.; Zhu, D.; Mao, H.; Su, P. Fecal Microbiota Transplantation for Ulcerative Colitis: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0157259. [Google Scholar]
- De Preter, V.; Arijs, I.; Windey, K.; Vanhove, W.; Vermeire, S.; Schuit, F.; Rutgeerts, P.; Verbeke, K. Impaired butyrate oxidation in ulcerative colitis is due to decreased butyrate uptake and a defect in the oxidation pathway. Inflamm. Bowel Dis. 2012, 18, 1127–1136. [Google Scholar] [CrossRef]
- Planell, N.; Lozano, J.J.; Mora-Buch, R.; Masamunt, M.C.; Jimeno, M.; Ordás, I.; Esteller, M.; Ricart, E.; Piqué, J.M.; Panés, J.; et al. Transcriptional analysis of the intestinal mucosa of patients with ulcerative colitis in remission reveals lasting epithelial cell alterations. Gut 2013, 62, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, O.; Creanza, T.M.; Bossa, F.; Palumbo, O.; Maglietta, R.; Ancona, N.; Corritore, G.; Latiano, T.; Martino, G.; Biscaglia, G.; et al. Genome-wide Pathway Analysis Using Gene Expression Data of Colonic Mucosa in Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 1, 1260–1268. [Google Scholar] [CrossRef]
- Borthakur, A.; Anbazhagan, A.N.; Kumar, A.; Raheja, G.; Sing, V.; Ramaswamy, K.; Dudeja, P.K. The probiotic Lactobacillus plantarum counteracts TNF-α-induced downregulation of SMCT1 expression and function. Am. J. Physiol. Gastrointest. 2010, 299, 928–934. [Google Scholar] [CrossRef]
- Nancey, S.; Moussata, D.; Graber, I.; Claudel, S.; Saurin, J.-C.; Flourié, B. Tumor Necrosis Factor α Reduces Butyrate Oxidation In Vitro in Human Colonic Mucosa: A Link from Inflammatory Process to Mucosal Damage? Inflamm. Bowel Dis. 2005, 11, 559–566. [Google Scholar] [CrossRef]
- Thibault, R.; De Coppet, P.; Daly, K.; Bourreille, A.; Cuff, M.; Bonnet, C.; Mosnier, J.F.; Galmiche, J.P.; Shirazi–Beechey, S.; Segain, J.P. Down-Regulation of the Monocarboxylate Transporter 1 Is Involved in Butyrate Deficiency During Intestinal Inflammation. Gastroenterology 2007, 133, 1916–1927. [Google Scholar] [CrossRef] [PubMed]
- Caviglia, G.P.; De Blasio, F.; Vernero, M.; Armandi, A.; Rosso, C.; Saracco, G.M.; Bugianesi, E.; Astegiano, M.; Ribaldone, D.G. Efficacy of a Preparation Based on Calcium Butyrate, Bifidobacterium bifidum, Bifidobacterium lactis, and Fructooligosaccharides in the Prevention of Relapse in Ulcerative Colitis: A Prospective Observational Study. J. Clin. Med. 2021, 10, 4961. [Google Scholar] [CrossRef] [PubMed]
- Vernero, M.; De Blasio, F.; Ribaldone, D.G.; Bugianesi, E.; Pellicano, R.; Saracco, G.M.; Astegiano, M.; Caviglia, G.P. The Usefulness of Microencapsulated Sodium Butyrate Add-On Therapy in Maintaining Remission in Patients with Ulcerative Colitis: A Prospective Observational Study. J. Clin. Med. 2020, 9, 3941. [Google Scholar] [CrossRef] [PubMed]
- Newton, W.A.; Snell, E.E. Formation and Interrelationships of Tryptophanase and Tryptophan Synthetases in Escherichia coli. J. Bacteriol. 1965, 89, 355–364. [Google Scholar] [CrossRef]
- Newton, W.A.; Morino, Y.; Snell, E.E. PROPERTIES OF CRYSTALLINE TRYPTOPHANASE. J. Biol. Chem. 1965, 240, 1211–1218. [Google Scholar] [CrossRef]
- Lee, J.-H.; Lee, J. Indole as an intercellular signal in microbial communities. FEMS Microbiol. Rev. 2010, 34, 426–444. [Google Scholar] [CrossRef] [PubMed]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Duca, D.R.; Glick, B.R. Indole-3-acetic acid biosynthesis and its regulation in plant-associated bacteria. Appl. Microbiol. Biotechnol. 2020, 104, 8607–8619. [Google Scholar] [CrossRef] [PubMed]
- Darkoh, C.; Chappell, C.; Gonzales, C.; Okhuysen, P. A Rapid and Specific Method for the Detection of Indole in Complex Biological Samples. Appl. Environ. Microbiol. 2015, 81, 8093–8097. [Google Scholar] [CrossRef] [PubMed]
- Chappell, C.L.; Darkoh, C.; Shimmin, L.; Farhana, N.; Kim, D.-K.; Okhuysen, P.C.; Hixson, J. Fecal Indole as a Biomarker of Susceptibility to Cryptosporidium Infection. Infect. Immun. 2016, 84, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sperandio, V. Indole Signaling at the Host-Microbiota-Pathogen Interface. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-G.; Lee, J.-H.; Cho, M.; Lee, J. Indole and 3-indolylacetonitrile inhibit spore maturation in Paenibacillus alvei. BMC Microbiol. 2011, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Chant, E.L.; Summers, D.K. Indole signalling contributes to the stable maintenance of Escherichia coli multicopy plasmids. Mol. Microbiol. 2007, 63, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Chimerel, C.; Field, C.M.; Piñero-Fernandez, S.; Keyser, U.F.; Summers, D.K. Indole prevents Escherichia coli cell division by modulating membrane potential. Biochim. Biophys. Acta 2012, 1818, 1590–1594. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jayaraman, A.; Wood, T.K. Indole is an inter-species biofilm signal mediated by SdiA. BMC Microbiol. 2007, 7, 42. [Google Scholar] [CrossRef]
- Lee, J.H.; Wood, T.K.; Lee, J. Roles of indole as an interspecies and interkingdom signaling molecule. Trends Microbiol. 2015, 23, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Bansal, T.; Alaniz, R.C.; Wood, T.K.; Jayaraman, A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 228–233. [Google Scholar] [CrossRef]
- Valenzano, M.C.; Diguilio, K.; Mercado, J.; Teter, M.; To, J.; Ferraro, B.; Mixson, B.; Manley, I.; Baker, V.; Moore, B.A.; et al. Remodeling of Tight Junctions and Enhancement of Barrier Integrity of the CACO-2 Intestinal Epithelial Cell Layer by Micronutrients. PLoS ONE 2015, 10, e0133926. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.A.; Fu, J.; Chang, P.V. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 19376–19387. [Google Scholar] [CrossRef] [PubMed]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J.; et al. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am. J. Pathol. 2018, 188, 1183–1194. [Google Scholar] [CrossRef]
- Stockinger, B.; Shah, K.; Wincent, E. AHR in the intestinal microenvironment: Safeguarding barrier function. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 559–570. [Google Scholar] [CrossRef]
- Lanis, J.M.; Alexeev, E.E.; Curtis, V.F.; Kitzenberg, D.A.; Kao, D.J.; Battista, K.D.; Gerich, M.E.; Glover, L.E.; Kominsky, D.J.; Colgan, S.P. Tryptophan metabolite activation of the aryl hydrocarbon receptor regulates IL-10 receptor expression on intestinal epithelia. Mucosal Immunol. 2017, 10, 1133–1144. [Google Scholar] [CrossRef]
- Turner, J.R.; Rill, B.K.; Carlson, S.L.; Carnes, D.; Kerner, R.; Mrsny, R.J.; Madara, J.L. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am. J. Physiol. 1997, 273, C1378–C1385. [Google Scholar] [CrossRef]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Shah, Y.M.; Ma, X.; Morimura, K.; Kim, I.; Gonzalez, F.J. Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1114–G1122. [Google Scholar] [CrossRef] [PubMed]
- Alexeev, E.E.; Dowdell, A.S.; Henen, M.A.; Lanis, J.M.; Lee, J.S.; Cartwright, I.M.; Schaefer, R.E.M.; Ornelas, A.; Onyiah, J.C.; Vögeli, B.; et al. Microbial-derived indoles inhibit neutrophil myeloperoxidase to diminish bystander tissue damage. Faseb J. 2021, 35, e21552. [Google Scholar] [CrossRef] [PubMed]
- Ximenes, V.F.; Paino, I.M.; Faria-Oliveira, O.M.; Fonseca, L.M.; Brunetti, I.L. Indole ring oxidation by activated leukocytes prevents the production of hypochlorous acid. Braz. J. Med. Biol Res. 2005, 38, 1575–1583. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Davies, M.J. Role of myeloperoxidase and oxidant formation in the extracellular environment in inflammation-induced tissue damage. Free Radic. Biol. Med. 2021, 172, 633–651. [Google Scholar] [CrossRef] [PubMed]
- Whitfield-Cargile, C.M.; Cohen, N.D.; Chapkin, R.S.; Weeks, B.R.; Davidson, L.A.; Goldsby, J.S.; Hunt, C.L.; Steinmeyer, S.H.; Menon, R.; Suchodolski, J.S.; et al. The microbiota-derived metabolite indole decreases mucosal inflammation and injury in a murine model of NSAID enteropathy. Gut Microbes 2016, 7, 246–261. [Google Scholar] [CrossRef] [PubMed]
- Busbee, P.B.; Menzel, L.; Alrafas, H.; Dopkins, N.; Becker, W.; Miranda, K.; Tang, C.; Chatterjee, S.; Singh, U.; Nagarkatti, M.; et al. Indole-3-carbinol prevents colitis and associated microbial dysbiosis in an IL-22–dependent manner. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Darkoh, C.; Plants-Paris, K.; Bishoff, D.; Dupont, H.L. Clostridium difficile Modulates the Gut Microbiota by Inducing the Production of Indole, an Interkingdom Signaling and Antimicrobial Molecule. mSystems 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Hibbard, J.; Jiang, Z.D.; DuPont, H.L. Fecal calprotectin and fecal indole predict outcome of fecal microbiota transplantation in subjects with recurrent Clostridium difficile infection. Anaerobe 2019, 56, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Wang, R.X.; Goldberg, M.S.; Clifford, G.P.; Kao, D.J.; Colgan, S.P. Microbiota-Sourced Purines Support Wound Healing and Mucous Barrier Function. iScience 2020, 23, 101226. [Google Scholar] [CrossRef] [PubMed]
- Dignass, A.U.; Becker, A.; Spiegler, S.; Goebell, H. Adenine nucleotides modulate epithelial wound healing in vitro. Eur J. Clin. Investig. 1998, 28, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Wang, R.X.; Alexeev, E.E.; Lanis, J.M.; Battista, K.D.; Glover, L.E.; Colgan, S.P. Hypoxanthine is a checkpoint stress metabolite in colonic epithelial energy modulation and barrier function. J. Biol. Chem. 2018, 293, 6039–6051. [Google Scholar] [CrossRef]
- Olivier, S.; Leclerc, J.; Grenier, A.; Foretz, M.; Tamburini, J.; Viollet, B. AMPK Activation Promotes Tight Junction Assembly in Intestinal Epithelial Caco-2 Cells. Int. J. Mol. Sci. 2019, 20, 5171. [Google Scholar] [CrossRef]
- Biaggioni, I.; Freeman, R.; Mathias, C.J.; Low, P.; Hewitt, L.A.; Kaufmann, H. Randomized Withdrawal Study of Patients With Symptomatic Neurogenic Orthostatic Hypotension Responsive to Droxidopa. Hypertension 2015, 65, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Hagey, L.R.; Krasowski, M.D. Bile salts of vertebrates: Structural variation and possible evolutionary significance. J. Lipid Res. 2010, 51, 226–246. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A. The function of bile salts in fat absorption. The solvent properties of dilute micellar solutions of conjugated bile salts. Biochem. J. 1963, 89, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L. Bile Acid Metabolism and Signaling. Compr. Physiol. 2013. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; McKenney, P.T.; Konstantinovsky, D.; Isaeva, O.I.; Schizas, M.; Verter, J.; Mai, C.; Jin, W.-B.; Guo, C.-J.; Violante, S.; et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 2020, 581, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 1679–1694. [Google Scholar] [CrossRef]
- Das, P.; Marcišauskas, S.; Ji, B.; Nielsen, J. Metagenomic analysis of bile salt biotransformation in the human gut microbiome. BMC Genom. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Heinken, A.; Ravcheev, D.A.; Baldini, F.; Heirendt, L.; Fleming, R.M.T.; Thiele, I. Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome 2019, 7. [Google Scholar] [CrossRef]
- Carr, R.M.; Reid, A.E. FXR agonists as therapeutic agents for non-alcoholic fatty liver disease. Curr. Atheroscler. Rep. 2015, 17, 500. [Google Scholar] [CrossRef] [PubMed]
- Modica, S.; Murzilli, S.; Salvatore, L.; Schmidt, D.R.; Moschetta, A. Nuclear bile acid receptor FXR protects against intestinal tumorigenesis. Cancer Res. 2008, 68, 9589–9594. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.M.; Bowman, A.A.; Young, V.B. Antibiotic-Induced Alterations of the Gut Microbiota Alter Secondary Bile Acid Production and Allow for Clostridium difficile Spore Germination and Outgrowth in the Large Intestine. mSphere 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.; Renooij, W.; Murzilli, S.; Klomp, L.W.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The Bile Acid Receptor FXR Is a Modulator of Intestinal Innate Immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef]
- Jia, E.-T.; Liu, Z.-Y.; Pan, M.; Lu, J.-F.; Ge, Q.-Y. Regulation of bile acid metabolism-related signaling pathways by gut microbiota in diseases. J. Zhejiang Univ.-Sci. B 2019, 20, 781–792. [Google Scholar] [CrossRef]
- Biagioli, M.; Carino, A.; Cipriani, S.; Francisci, D.; Marchianò, S.; Scarpelli, P.; Sorcini, D.; Zampella, A.; Fiorucci, S. The Bile Acid Receptor GPBAR1 Regulates the M1/M2 Phenotype of Intestinal Macrophages and Activation of GPBAR1 Rescues Mice from Murine Colitis. J. Immunol. 2017, 199, 718–733. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, S.; Mencarelli, A.; Chini, M.G.; Distrutti, E.; Renga, B.; Bifulco, G.; Baldelli, F.; Donini, A.; Fiorucci, S. The Bile Acid Receptor GPBAR-1 (TGR5) Modulates Integrity of Intestinal Barrier and Immune Response to Experimental Colitis. PLoS ONE 2011, 6, e25637. [Google Scholar] [CrossRef] [PubMed]
- Simmons, J.D.; Mullighan, C.; Welsh, K.I.; Jewell, D.P. Vitamin D receptor gene polymorphism: Association with Crohn’s disease susceptibility. Gut 2000, 47, 211–214. [Google Scholar] [CrossRef]
- Kim, J.-H.; Yamaori, S.; Tanabe, T.; Johnson, C.H.; Krausz, K.W.; Kato, S.; Gonzalez, F.J. Implication of intestinal VDR deficiency in inflammatory bowel disease. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 2118–2128. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Zhang, Z.; Musch, M.W.; Ning, G.; Sun, J.; Hart, J.; Bissonnette, M.; Li, Y.C. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G208–G216. [Google Scholar] [CrossRef]
- Li, Y.C.; Chen, Y.; Du, J. Critical roles of intestinal epithelial vitamin D receptor signaling in controlling gut mucosal inflammation. J. Steroid Biochem. Mol. Biol. 2015, 148, 179–183. [Google Scholar] [CrossRef]
- Kakiyama, G.; Pandak, W.M.; Gillevet, P.M.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; Takei, H.; Muto, A.; Nittono, H.; Ridlon, J.M.; et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J. Hepatol. 2013, 58, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Heuman, D.M.; Hylemon, P.B.; Sanyal, A.J.; White, M.B.; Monteith, P.; Noble, N.A.; Unser, A.B.; Daita, K.; Fisher, A.R.; et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 2014, 60, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Islam, K.B.; Fukiya, S.; Hagio, M.; Fujii, N.; Ishizuka, S.; Ooka, T.; Ogura, Y.; Hayashi, T.; Yokota, A. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 2011, 141, 1773–1781. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef]
- Igarashi, K.; Kashiwagi, K. Polyamines: Mysterious modulators of cellular functions. Biochem. Biophys. Res. Commun. 2000, 271, 559–564. [Google Scholar] [CrossRef]
- Tabor, C.W.; Tabor, H. Polyamines. Annu. Rev. Biochem. 1984, 53, 749–790. [Google Scholar] [CrossRef]
- McCormack, S.A.; Viar, M.J.; Johnson, L.R. Polyamines are necessary for cell migration by a small intestinal crypt cell line. Am. J. Physiol.-Gastrointest. Liver Physiol. 1993, 264, G367–G374. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Jiménez, F.; Medina, M.Á.; Villalobos-Rueda, L.; Urdiales, J.L. Polyamines in mammalian pathophysiology. Cell. Mol. Life Sci. 2019, 76, 3987–4008. [Google Scholar] [CrossRef]
- Seiler, N.; Raul, F. Polyamines and apoptosis. J. Cell. Mol. Med. 2005, 9, 623–642. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; McCormack, S.A.; Viar, M.J.; Johnson, L.R. Stimulation of proximal small intestinal mucosal growth by luminal polyamines. Am. J. Physiol.-Gastrointest. Liver Physiol. 1991, 261, G504–G511. [Google Scholar] [CrossRef] [PubMed]
- Lan, A.; Blachier, F.; Benamouzig, R.; Beaumont, M.; Barrat, C.; Coelho, D.; Lancha, A.; Kong, X.; Yin, Y.; Marie, J.-C.; et al. Mucosal Healing in Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2015, 21, 198–207. [Google Scholar] [CrossRef]
- Ray, R.M.; Johnson, L.R. Regulation of intestinal mucosal growth by amino acids. Amino Acids 2014, 46, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Timmons, J.; Chang, E.T.; Wang, J.Y.; Rao, J.N. Polyamines and Gut Mucosal Homeostasis. J. Gastrointest. Dig. Syst. 2012, 2, 1. [Google Scholar] [CrossRef]
- Wang, J.-y.; Johnson, L.R. Polyamines and ornithine decarboxylase during repair of duodenal mucosa after stress in rats. Gastroenterology 1991, 100, 333–343. [Google Scholar] [CrossRef]
- Patel, A.R.; Wang, J.Y. Polyamines modulate transcription but not posttranscription of c-myc and c-jun in IEC-6 cells. Am. J. Physiol.-Cell Physiol. 1997, 273, C1020–C1029. [Google Scholar] [CrossRef]
- Wang, J.Y.; McCormack, S.A.; Viar, M.J.; Wang, H.; Tzen, C.Y.; Scott, R.E.; Johnson, L.R. Decreased expression of protooncogenes c-fos, c-myc, and c-jun following polyamine depletion in IEC-6 cells. Am. J. Physiol.-Gastrointest. Liver Physiol. 1993, 265, G331–G338. [Google Scholar] [CrossRef]
- Wang, J.Y.; Johnson, L.R. Expression of protooncogenes c-fos and c-myc in healing of gastric mucosal stress ulcers. Am. J. Physiol.-Gastrointest. Liver Physiol. 1994, 266, G878–G886. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Santora, R.; Rao, J.N.; Guo, X.; Zou, T.; Zhang, H.M.; Turner, D.J.; Wang, J.-Y. Activation of TGF-β-Smad signaling pathway following polyamine depletion in intestinal epithelial cells. Am. J. Physiol.-Gastrointest. Liver Physiol. 2003, 285, G1056–G1067. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.N.; Li, L.; Bass, B.L.; Wang, J.-Y. Expression of the TGF-β receptor gene and sensitivity to growth inhibition following polyamine depletion. Am. J. Physiol.-Cell Physiol. 2000, 279, C1034–C1044. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y. Polyamines and mRNA stability in regulation of intestinal mucosal growth. Amino Acids 2007, 33, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Rao, J.N.; Liu, L.; Xiao, L.; Yu, T.-X.; Jiang, P.; Gorospe, M.; Wang, J.-Y. Polyamines Regulate the Stability of JunD mRNA by Modulating the Competitive Binding of Its 3′ Untranslated Region to HuR and AUF1. Mol. Cell. Biol. 2010, 30, 5021–5032. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Rao, J.N.; Liu, L.; Zou, T.-T.; Turner, D.J.; Bass, B.L.; Wang, J.-Y. Regulation of adherens junctions and epithelial paracellular permeability: A novel function for polyamines. Am. J. Physiol.-Cell Physiol. 2003, 285, C1174–C1187. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Guo, X.; Rao, J.N.; Zou, T.; Xiao, L.; Yu, T.; Timmons, J.A.; Turner, D.J.; Wang, J.-Y. Polyamines regulate E-cadherin transcription through c-Myc modulating intestinal epithelial barrier function. Am. J. Physiol.-Cell Physiol. 2009, 296, C801–C810. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xiao, L.; Rao, J.N.; Zou, T.; Liu, L.; Bellavance, E.; Gorospe, M.; Wang, J.-Y. JunD Represses Transcription and Translation of the Tight Junction Protein Zona Occludens-1 Modulating Intestinal Epithelial Barrier Function. Mol. Biol. Cell 2008, 19, 3701–3712. [Google Scholar] [CrossRef]
- Rao, J.N.; Xiao, L.; Wang, J.-Y. Polyamines in Gut Epithelial Renewal and Barrier Function. Physiology 2020, 35, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Puleston, D.J.; Zhang, H.; Powell, T.J.; Lipina, E.; Sims, S.; Panse, I.; Watson, A.S.; Cerundolo, V.; Townsend, A.R.; Klenerman, P.; et al. Autophagy is a critical regulator of memory CD8+ T cell formation. eLife 2014, 3. [Google Scholar] [CrossRef]
- Szabó, C.; Southan, G.J.; Wood, E.; Thiemermann, C.; Vane, J.R. Inhibition by spermine of the induction of nitric oxide synthase in J774.2 macrophages: Requirement of a serum factor. Br. J. Pharm. 1994, 112, 355–356. [Google Scholar] [CrossRef]
- Zhang, M.; Caragine, T.; Wang, H.; Cohen, P.S.; Botchkina, G.; Soda, K.; Bianchi, M.; Ulrich, P.; Cerami, A.; Sherry, B.; et al. Spermine Inhibits Proinflammatory Cytokine Synthesis in Human Mononuclear Cells: A Counterregulatory Mechanism that Restrains the Immune Response. J. Exp. Med. 1997, 185, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalová, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Thaiss, C.A.; Elinav, E. Metabolites: Messengers between the microbiota and the immune system. Genes Dev. 2016, 30, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).