
Loss of Human Beta Cell Identity in a Reconstructed Omental Stromal Cell Environment

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture of EndoC-βH1 Cells
2.2. Isolation, Culture of Adipose-Derived Stromal Cells and Production of Conditioned Media (CM)
2.3. SiRNA Transfection
2.4. RNA Isolation, Reverse Transcription, and qPCR
2.5. Transcriptome Analysis
2.6. Immunoblotting
2.7. Glucose-Stimulated Insulin Secretion (GSIS)
2.8. Statistics
3. Results
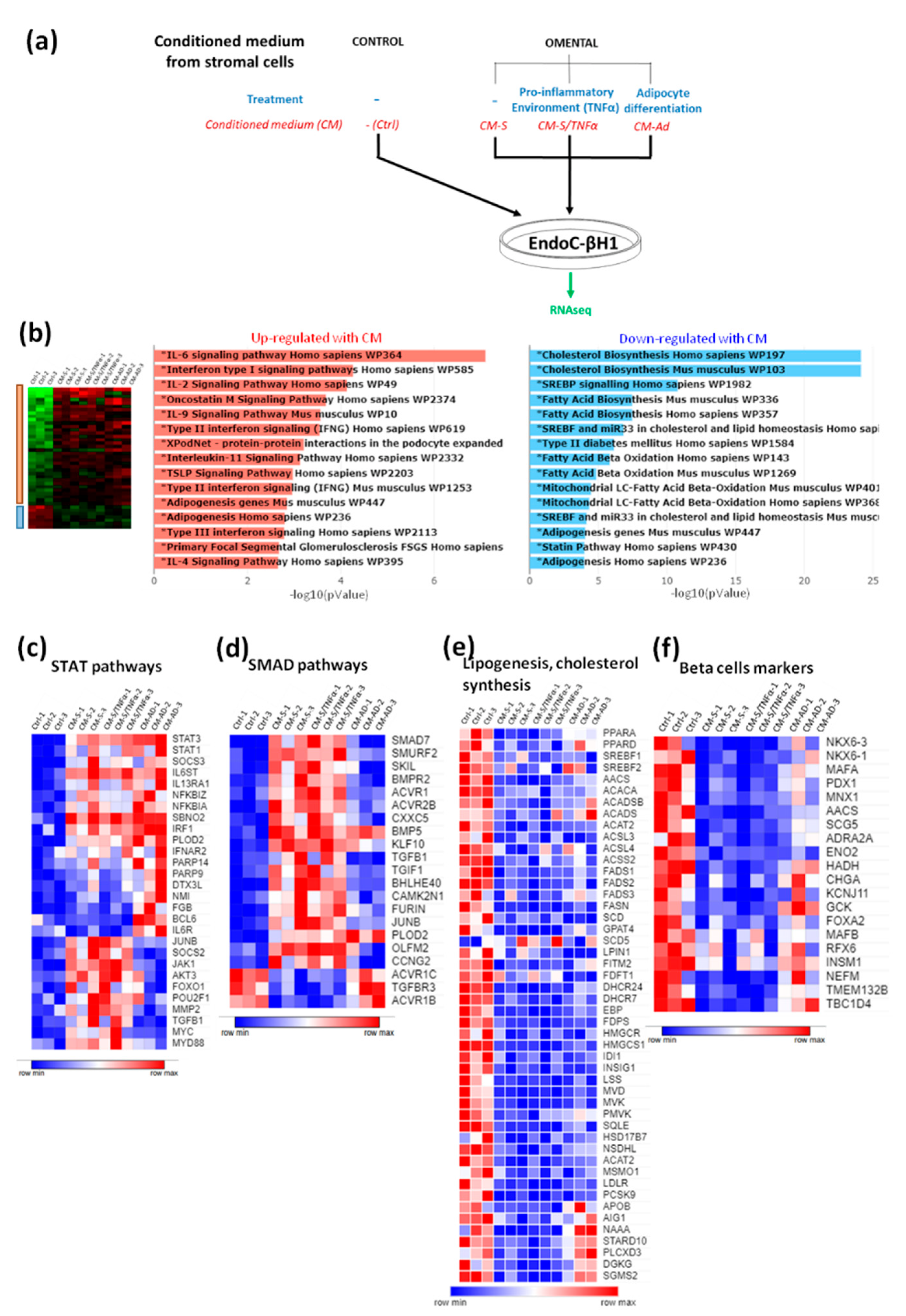
3.1. EndoC-βH1 Cells Are Sensitive to Conditioned Medium from Human Omental Stromal Cells
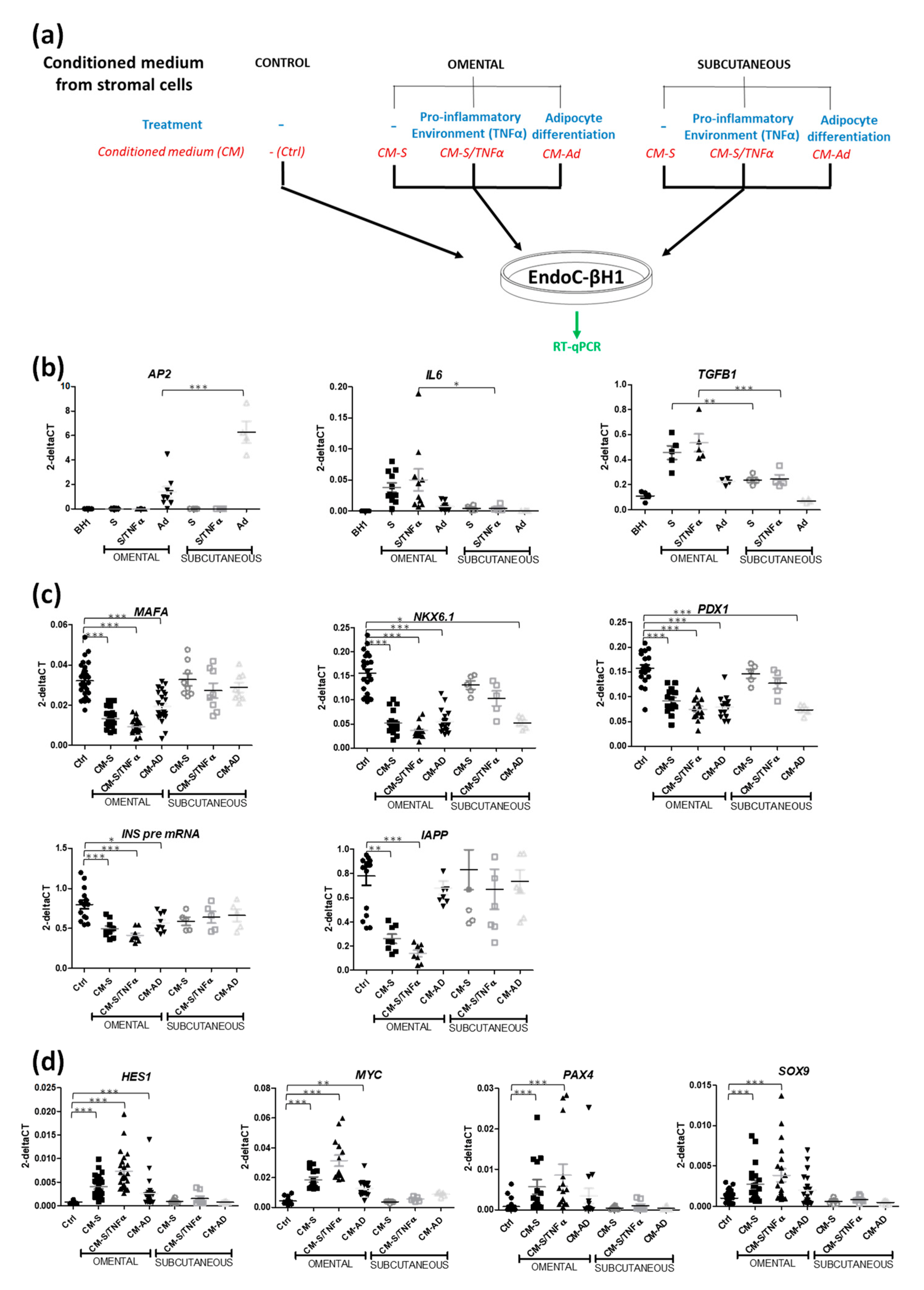
3.2. CM from Subcutaneous Stromal Cells Does Not Recapitulate the Effects of CM from Omental Stromal Cells
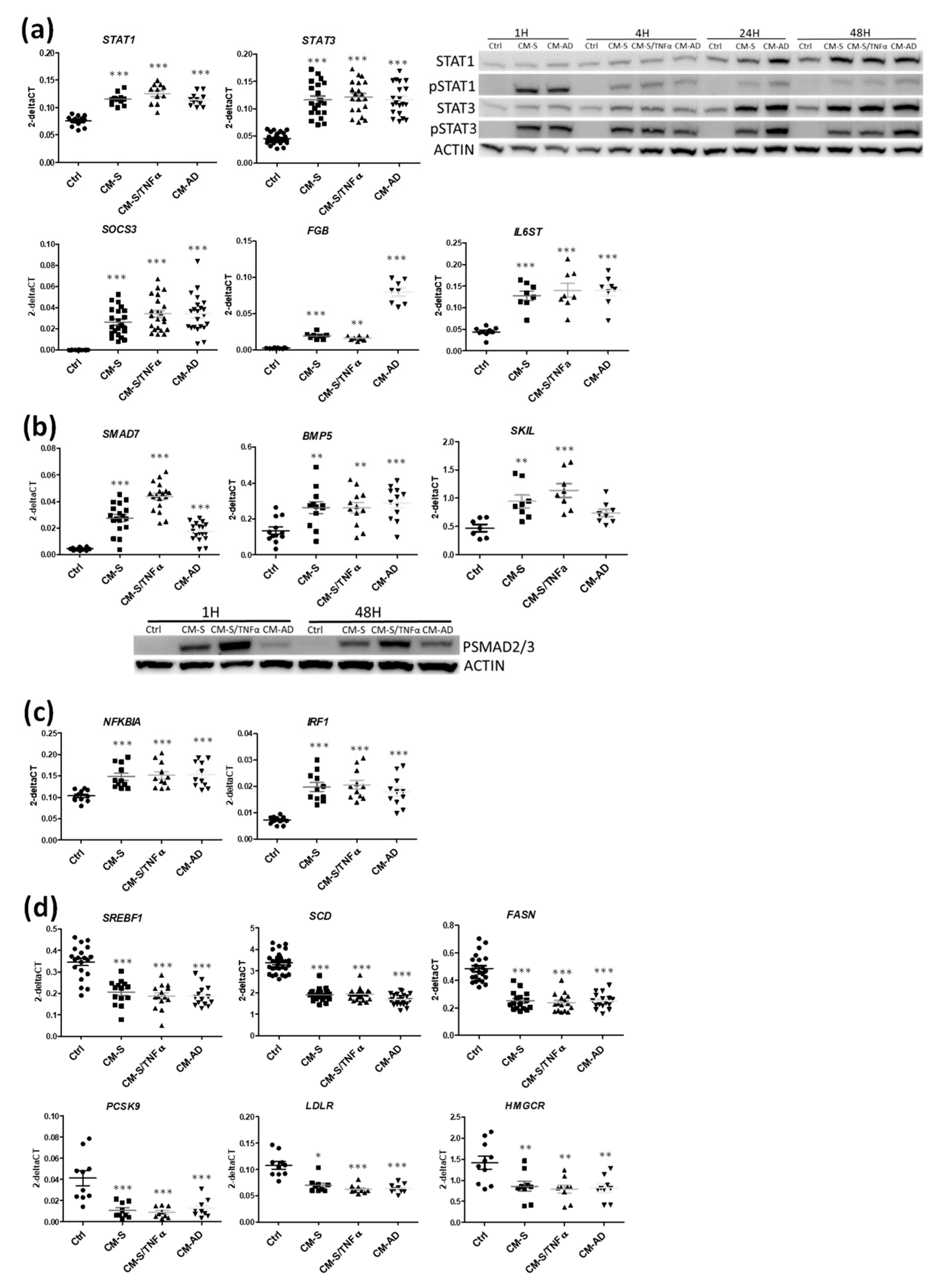
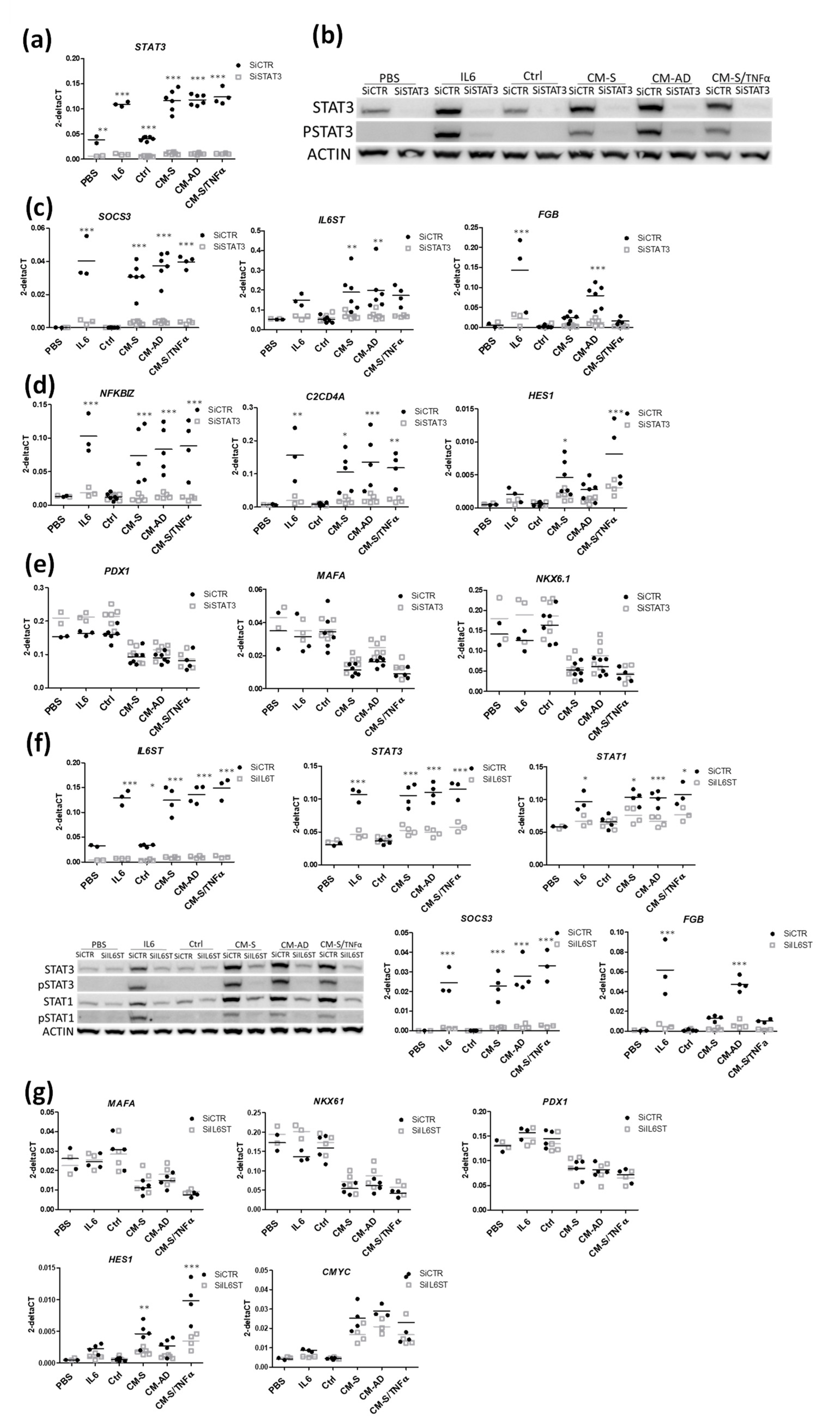
3.3. Implication of STAT1 and STAT3 Pathways in the Effects of CM from Omental Stromal Cells on Beta Cell Identity
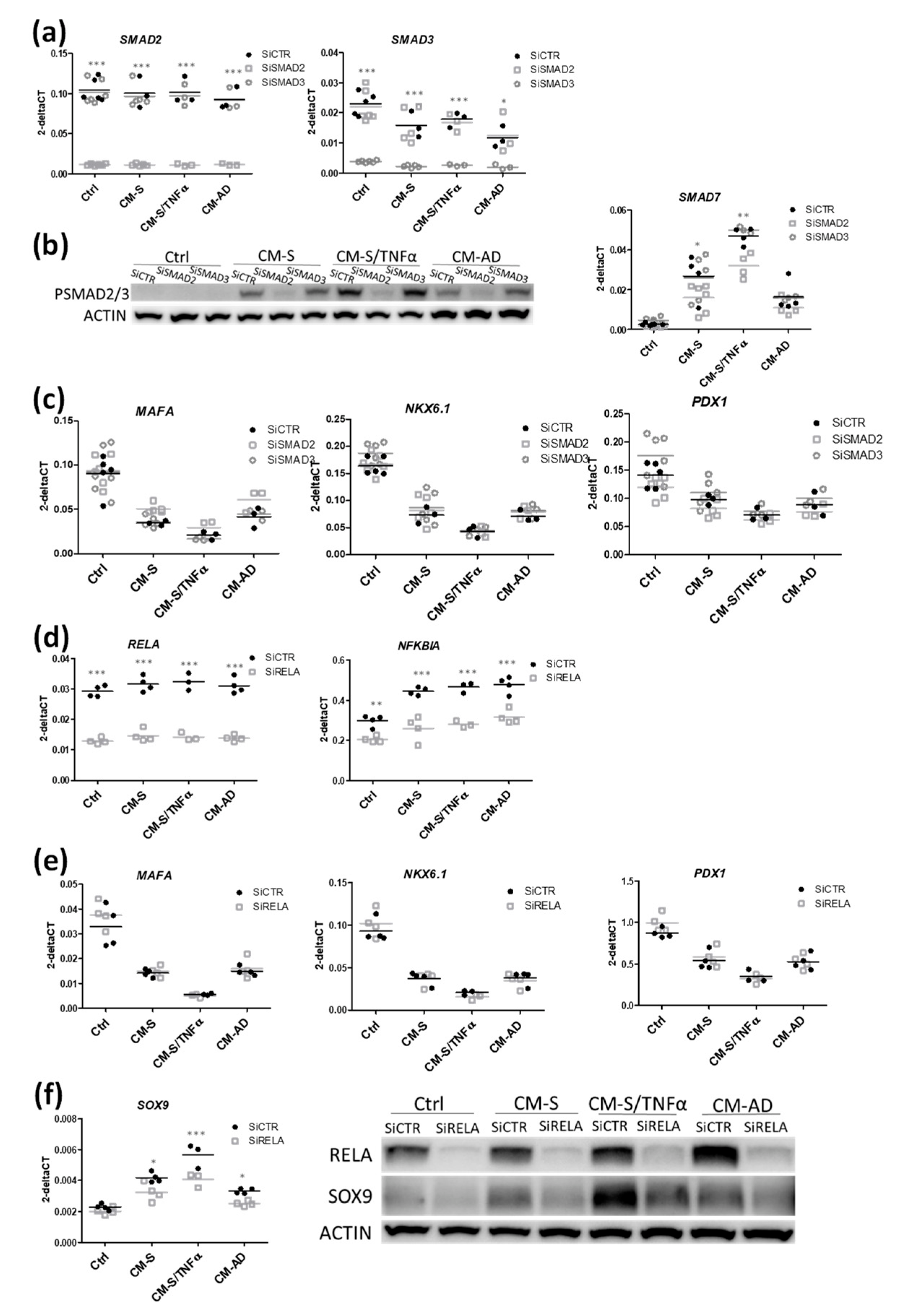
3.4. Implication of SMAD and RELA Pathways in the Effects of CM from Omental Stromal Cells on Beta Cell Identity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Mallone, R.; Eizirik, D.L. Presumption of innocence for beta cells: Why are they vulnerable autoimmune targets in type 1 diabetes? Diabetologia 2020, 63, 1999–2006. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, G.C.; Gaglia, J.; Bonner-Weir, S. Inadequate beta-cell mass is essential for the pathogenesis of type 2 diabetes. Lancet Diabetes Endocrinol. 2020, 8, 249–256. [Google Scholar] [CrossRef]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.C. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 2008, 10 (Suppl. S4), 32–42. [Google Scholar] [CrossRef]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Yoon, K.H.; Ko, S.H.; Cho, J.H.; Lee, J.M.; Ahn, Y.B.; Song, K.H.; Yoo, S.J.; Kang, M.I.; Cha, B.Y.; Lee, K.W.; et al. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J. Clin. Endocrinol. Metab. 2003, 88, 2300–2308. [Google Scholar] [CrossRef]
- Ferrannini, E. The stunned beta cell: A brief history. Cell Metab. 2010, 11, 349–352. [Google Scholar] [CrossRef]
- Wajchenberg, B.L. beta-cell failure in diabetes and preservation by clinical treatment. Endocr. Rev. 2007, 28, 187–218. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Cardozo, A.K.; Cnop, M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 2008, 29, 42–61. [Google Scholar] [CrossRef] [Green Version]
- Jonas, J.C.; Sharma, A.; Hasenkamp, W.; Ilkova, H.; Patane, G.; Laybutt, R.; Bonner-Weir, S.; Weir, G.C. Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J. Biol. Chem. 1999, 274, 14112–14121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efrat, S. Beta-Cell Dedifferentiation in Type 2 Diabetes: Concise Review. Stem Cells 2019, 37, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Bensellam, M.; Jonas, J.C.; Laybutt, D.R. Mechanisms of beta-cell dedifferentiation in diabetes: Recent findings and future research directions. J. Endocrinol. 2018, 236, R109–R143. [Google Scholar] [CrossRef] [Green Version]
- Hunter, C.S.; Stein, R.W. Evidence for Loss in Identity, De-Differentiation, and Trans-Differentiation of Islet beta-Cells in Type 2 Diabetes. Front. Genet. 2017, 8, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moin, A.S.M.; Butler, A.E. Alterations in Beta Cell Identity in Type 1 and Type 2 Diabetes. Curr. Diab. Rep. 2019, 19, 83. [Google Scholar] [CrossRef] [Green Version]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of beta-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef] [Green Version]
- Ravassard, P.; Hazhouz, Y.; Pechberty, S.; Bricout-Neveu, E.; Armanet, M.; Czernichow, P.; Scharfmann, R. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J. Clin. Investig. 2011, 121, 3589–3597. [Google Scholar] [CrossRef]
- Scharfmann, R.; Staels, W.; Albagli, O. The supply chain of human pancreatic beta cell lines. J. Clin. Investig. 2019, 129, 3511–3520. [Google Scholar] [CrossRef] [Green Version]
- Diedisheim, M.; Oshima, M.; Albagli, O.; Huldt, C.W.; Ahlstedt, I.; Clausen, M.; Menon, S.; Aivazidis, A.; Andreasson, A.C.; Haynes, W.G.; et al. Modeling human pancreatic beta cell dedifferentiation. Mol. Metab. 2018, 10, 74–86. [Google Scholar] [CrossRef]
- Oshima, M.; Knoch, K.P.; Diedisheim, M.; Petzold, A.; Cattan, P.; Bugliani, M.; Marchetti, P.; Choudhary, P.; Huang, G.C.; Bornstein, S.R.; et al. Virus-like infection induces human beta cell dedifferentiation. JCI Insight 2018, 3, e97732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kita, S.; Maeda, N.; Shimomura, I. Interorgan communication by exosomes, adipose tissue, and adiponectin in metabolic syndrome. J. Clin. Investig. 2019, 129, 4041–4049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romacho, T.; Elsen, M.; Rohrborn, D.; Eckel, J. Adipose tissue and its role in organ crosstalk. Acta Physiol. 2014, 210, 733–753. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Marcelin, G.; Silveira, A.L.M.; Martins, L.B.; Ferreira, A.V.; Clement, K. Deciphering the cellular interplays underlying obesity-induced adipose tissue fibrosis. J. Clin. Investig. 2019, 129, 4032–4040. [Google Scholar] [CrossRef]
- Drareni, K.; Ballaire, R.; Alzaid, F.; Goncalves, A.; Chollet, C.; Barilla, S.; Nguewa, J.L.; Dias, K.; Lemoine, S.; Riveline, J.P.; et al. Adipocyte Reprogramming by the Transcriptional Coregulator GPS2 Impacts Beta Cell Insulin Secretion. Cell Rep. 2020, 32, 108141. [Google Scholar] [CrossRef]
- Wu, C.L.; Diekman, B.O.; Jain, D.; Guilak, F. Diet-induced obesity alters the differentiation potential of stem cells isolated from bone marrow, adipose tissue and infrapatellar fat pad: The effects of free fatty acids. Int. J. Obes. 2013, 37, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Andersen, E.; Ingerslev, L.R.; Fabre, O.; Donkin, I.; Altintas, A.; Versteyhe, S.; Bisgaard, T.; Kristiansen, V.B.; Simar, D.; Barres, R. Preadipocytes from obese humans with type 2 diabetes are epigenetically reprogrammed at genes controlling adipose tissue function. Int. J. Obes. 2019, 43, 306–318. [Google Scholar] [CrossRef]
- Heni, M.; Machann, J.; Staiger, H.; Schwenzer, N.F.; Peter, A.; Schick, F.; Claussen, C.D.; Stefan, N.; Haring, H.U.; Fritsche, A. Pancreatic fat is negatively associated with insulin secretion in individuals with impaired fasting glucose and/or impaired glucose tolerance: A nuclear magnetic resonance study. Diabetes Metab. Res. Rev. 2010, 26, 200–205. [Google Scholar] [CrossRef]
- Begovatz, P.; Koliaki, C.; Weber, K.; Strassburger, K.; Nowotny, B.; Nowotny, P.; Mussig, K.; Bunke, J.; Pacini, G.; Szendrodi, J.; et al. Pancreatic adipose tissue infiltration, parenchymal steatosis and beta cell function in humans. Diabetologia 2015, 58, 1646–1655. [Google Scholar] [CrossRef]
- Guglielmi, V.; Sbraccia, P. Type 2 diabetes: Does pancreatic fat really matter? Diabetes Metab. Res. Rev. 2018, 34, e2955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdez, I.A.; Dirice, E.; Gupta, M.K.; Shirakawa, J.; Teo, A.K.K.; Kulkarni, R.N. Proinflammatory Cytokines Induce Endocrine Differentiation in Pancreatic Ductal Cells via STAT3-Dependent NGN3 Activation. Cell Rep. 2016, 15, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Groef, S.; Renmans, D.; Cai, Y.; Leuckx, G.; Roels, S.; Staels, W.; Gradwohl, G.; Baeyens, L.; Heremans, Y.; Martens, G.A.; et al. STAT3 modulates beta-cell cycling in injured mouse pancreas and protects against DNA damage. Cell Death Dis. 2016, 7, e2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, F.; Naamane, N.; Colli, M.L.; Bouckenooghe, T.; Ortis, F.; Gurzov, E.N.; Igoillo-Esteve, M.; Mathieu, C.; Bontempi, G.; Thykjaer, T.; et al. STAT1 is a master regulator of pancreatic {beta}-cell apoptosis and islet inflammation. J. Biol. Chem. 2011, 286, 929–941. [Google Scholar] [CrossRef] [Green Version]
- Saleh, M.; Mohamed, N.A.; Sehrawat, A.; Zhang, T.; Thomas, M.; Wang, Y.; Kalsi, R.; Molitoris, J.; Prasadan, K.; Gittes, G.K. β-cell Smad2 null mice have improved β-cell function and are protected from diet-induced hyperglycemia. J. Biol. Chem. 2021, 297, 101235. [Google Scholar] [CrossRef]
- Arda, H.E.; Benitez, C.M.; Kim, S.K. Gene regulatory networks governing pancreas development. Dev. Cell 2013, 25, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Seymour, P.A. Sox9: A master regulator of the pancreatic program. Rev. Diabet. Stud. 2014, 11, 51–83. [Google Scholar] [CrossRef] [Green Version]
- Rachdi, L.; Maugein, A.; Pechberty, S.; Armanet, M.; Hamroune, J.; Ravassard, P.; Marullo, S.; Albagli, O.; Scharfmann, R. Regulated expression and function of the GABAB receptor in human pancreatic beta cell line and islets. Sci. Rep. 2020, 10, 13469. [Google Scholar] [CrossRef]
- Perrin, S.; Firmo, C.; Lemoine, S.; Le Crom, S.; Jourdren, L. Aozan: An automated post-sequencing data-processing pipeline. Bioinformatics 2017, 33, 2212–2213. [Google Scholar] [CrossRef]
- Torre, D.; Lachmann, A.; Ma’ayan, A. BioJupies: Automated Generation of Interactive Notebooks for RNA-Seq Data Analysis in the Cloud. Cell Syst 2018, 7, 556–561.e3. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvillie, B.; Attali, M.; Aiello, V.; Quemeneur, E.; Scharfmann, R. Label-retaining cells in the rat pancreas: Location and differentiation potential in vitro. Diabetes 2003, 52, 2035–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraro, M.J.; Dharmadhikari, G.; Grun, D.; Groen, N.; Dielen, T.; Jansen, E.; van Gurp, L.; Engelse, M.A.; Carlotti, F.; de Koning, E.J.; et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016, 3, 385–394.e383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, G.A.; Jiang, L.; Hellemans, K.H.; Stange, G.; Heimberg, H.; Nielsen, F.C.; Sand, O.; Van Helden, J.; Van Lommel, L.; Schuit, F.; et al. Clusters of conserved beta cell marker genes for assessment of beta cell phenotype. PLoS ONE 2011, 6, e24134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nica, A.C.; Ongen, H.; Irminger, J.C.; Bosco, D.; Berney, T.; Antonarakis, S.E.; Halban, P.A.; Dermitzakis, E.T. Cell-type, allelic, and genetic signatures in the human pancreatic beta cell transcriptome. Genome Res. 2013, 23, 1554–1562. [Google Scholar] [CrossRef] [Green Version]
- Segerstolpe, A.; Palasantza, A.; Eliasson, P.; Andersson, E.M.; Andreasson, A.C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Klochendler, A.; Caspi, I.; Corem, N.; Moran, M.; Friedlich, O.; Elgavish, S.; Nevo, Y.; Helman, A.; Glaser, B.; Eden, A.; et al. The Genetic Program of Pancreatic beta-Cell Replication In Vivo. Diabetes 2016, 65, 2081–2093. [Google Scholar] [CrossRef] [Green Version]
- Camunas-Soler, J.; Dai, X.Q.; Hang, Y.; Bautista, A.; Lyon, J.; Suzuki, K.; Kim, S.K.; Quake, S.R.; MacDonald, P.E. Patch-Seq Links Single-Cell Transcriptomes to Human Islet Dysfunction in Diabetes. Cell Metab. 2020, 31, 1017–1031.e1014. [Google Scholar] [CrossRef]
- Lorenzo, P.I.; Juarez-Vicente, F.; Cobo-Vuilleumier, N.; Garcia-Dominguez, M.; Gauthier, B.R. The Diabetes-Linked Transcription Factor PAX4: From Gene to Functional Consequences. Genes 2017, 8, 101. [Google Scholar] [CrossRef] [Green Version]
- Kuo, T.; Kraakman, M.J.; Damle, M.; Gill, R.; Lazar, M.A.; Accili, D. Identification of C2CD4A as a human diabetes susceptibility gene with a role in beta cell insulin secretion. Proc. Natl. Acad. Sci. USA 2019, 116, 20033–20042. [Google Scholar] [CrossRef] [Green Version]
- Strawbridge, R.J.; Dupuis, J.; Prokopenko, I.; Barker, A.; Ahlqvist, E.; Rybin, D.; Petrie, J.R.; Travers, M.E.; Bouatia-Naji, N.; Dimas, A.S.; et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes 2011, 60, 2624–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, R.; Brown, D.; Churchwell, L.; Lee, T.J.; Kodeboyina, S.K.; Bloom, J.; Sharma, A.; Sharma, S. RNA-Seq analysis reveals gene expression changes induced by IL-6 trans-signaling activation in retinal endothelial cells. Cytokine 2021, 139, 155375. [Google Scholar] [CrossRef]
- Zhu, F.; Wang, K.B.; Rui, L. STAT3 Activation and Oncogenesis in Lymphoma. Cancers 2019, 12, 19. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Muller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naka, T.; Nishimoto, N.; Kishimoto, T. The paradigm of IL-6: From basic science to medicine. Arthritis Res. 2002, 4 (Suppl. S3), S233–S242. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, O.P.; Mandrup-Poulsen, T. Interleukin-6 and diabetes: The good, the bad, or the indifferent? Diabetes 2005, 54 (Suppl. S2), S114–S124. [Google Scholar] [CrossRef] [Green Version]
- Guzman-Ayala, M.; Lee, K.L.; Mavrakis, K.J.; Goggolidou, P.; Norris, D.P.; Episkopou, V. Graded Smad2/3 activation is converted directly into levels of target gene expression in embryonic stem cells. PLoS ONE 2009, 4, e4268. [Google Scholar] [CrossRef]
- Nicklas, D.; Saiz, L. Computational modelling of Smad-mediated negative feedback and crosstalk in the TGF-beta superfamily network. J. R. Soc. Interface 2013, 10, 20130363. [Google Scholar] [CrossRef] [Green Version]
- Tsonkova, V.G.; Sand, F.W.; Wolf, X.A.; Grunnet, L.G.; Kirstine Ringgaard, A.; Ingvorsen, C.; Winkel, L.; Kalisz, M.; Dalgaard, K.; Bruun, C.; et al. The EndoC-betaH1 cell line is a valid model of human beta cells and applicable for screenings to identify novel drug target candidates. Mol. Metab. 2018, 8, 144–157. [Google Scholar] [CrossRef]
- Oshima, M.; Pechberty, S.; Bellini, L.; Gopel, S.O.; Campana, M.; Rouch, C.; Dairou, J.; Cosentino, C.; Fantuzzi, F.; Toivonen, S.; et al. Stearoyl CoA desaturase is a gatekeeper that protects human beta cells against lipotoxicity and maintains their identity. Diabetologia 2020, 63, 395–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marroqui, L.; Dos Santos, R.S.; Op de Beeck, A.; Coomans de Brachene, A.; Marselli, L.; Marchetti, P.; Eizirik, D.L. Interferon-alpha mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia 2017, 60, 656–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, M.A.; Cooper, A.C.; Dhayal, S.; Morgan, N.G. Differential effects of interleukin-13 and interleukin-6 on Jak/STAT signaling and cell viability in pancreatic beta-cells. Islets 2013, 5, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cnop, M.; Hughes, S.J.; Igoillo-Esteve, M.; Hoppa, M.B.; Sayyed, F.; van de Laar, L.; Gunter, J.H.; de Koning, E.J.; Walls, G.V.; Gray, D.W.; et al. The long lifespan and low turnover of human islet beta cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia 2010, 53, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Rorsman, P.; Ashcroft, F.M. Pancreatic beta-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol. Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef]
- Rui, J.; Deng, S.; Arazi, A.; Perdigoto, A.L.; Liu, Z.; Herold, K.C. beta Cells that Resist Immunological Attack Develop during Progression of Autoimmune Diabetes in NOD Mice. Cell Metab. 2017, 25, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Tchkonia, T.; Giorgadze, N.; Pirtskhalava, T.; Thomou, T.; DePonte, M.; Koo, A.; Forse, R.A.; Chinnappan, D.; Martin-Ruiz, C.; von Zglinicki, T.; et al. Fat depot-specific characteristics are retained in strains derived from single human preadipocytes. Diabetes 2006, 55, 2571–2578. [Google Scholar] [CrossRef] [Green Version]
- Silva, K.R.; Baptista, L.S. Adipose-derived stromal/stem cells from different adipose depots in obesity development. World J. Stem Cells 2019, 11, 147–166. [Google Scholar] [CrossRef]
- Blum, B.; Roose, A.N.; Barrandon, O.; Maehr, R.; Arvanites, A.C.; Davidow, L.S.; Davis, J.C.; Peterson, Q.P.; Rubin, L.L.; Melton, D.A. Reversal of beta cell de-differentiation by a small molecule inhibitor of the TGFbeta pathway. eLife 2014, 3, e02809. [Google Scholar] [CrossRef]
- Casteels, T.; Zhang, Y.; Frogne, T.; Sturtzel, C.; Lardeau, C.H.; Sen, I.; Liu, X.; Hong, S.; Pauler, F.M.; Penz, T.; et al. An inhibitor-mediated beta-cell dedifferentiation model reveals distinct roles for FoxO1 in glucagon repression and insulin maturation. Mol. Metab. 2021, 54, 101329. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Secco, B.; Saitoski, K.; Drareni, K.; Soprani, A.; Pechberty, S.; Rachdi, L.; Venteclef, N.; Scharfmann, R. Loss of Human Beta Cell Identity in a Reconstructed Omental Stromal Cell Environment. Cells 2022, 11, 924. https://doi.org/10.3390/cells11060924
Secco B, Saitoski K, Drareni K, Soprani A, Pechberty S, Rachdi L, Venteclef N, Scharfmann R. Loss of Human Beta Cell Identity in a Reconstructed Omental Stromal Cell Environment. Cells. 2022; 11(6):924. https://doi.org/10.3390/cells11060924
Chicago/Turabian StyleSecco, Blandine, Kevin Saitoski, Karima Drareni, Antoine Soprani, Severine Pechberty, Latif Rachdi, Nicolas Venteclef, and Raphaël Scharfmann. 2022. "Loss of Human Beta Cell Identity in a Reconstructed Omental Stromal Cell Environment" Cells 11, no. 6: 924. https://doi.org/10.3390/cells11060924
APA StyleSecco, B., Saitoski, K., Drareni, K., Soprani, A., Pechberty, S., Rachdi, L., Venteclef, N., & Scharfmann, R. (2022). Loss of Human Beta Cell Identity in a Reconstructed Omental Stromal Cell Environment. Cells, 11(6), 924. https://doi.org/10.3390/cells11060924