
Cardiovascular Inflammaging: Mechanisms and Translational Aspects

 ,
,  , ,
, , 
Abstract
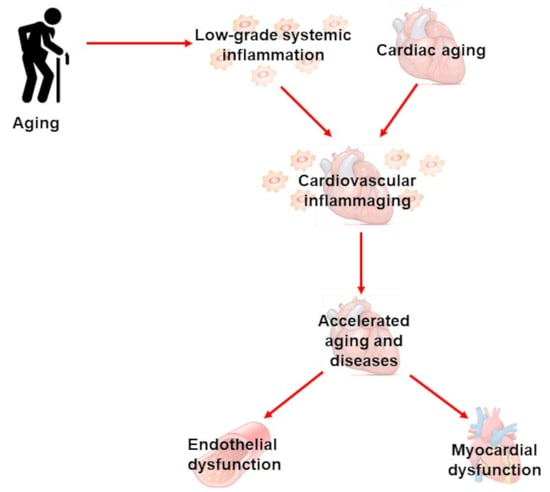
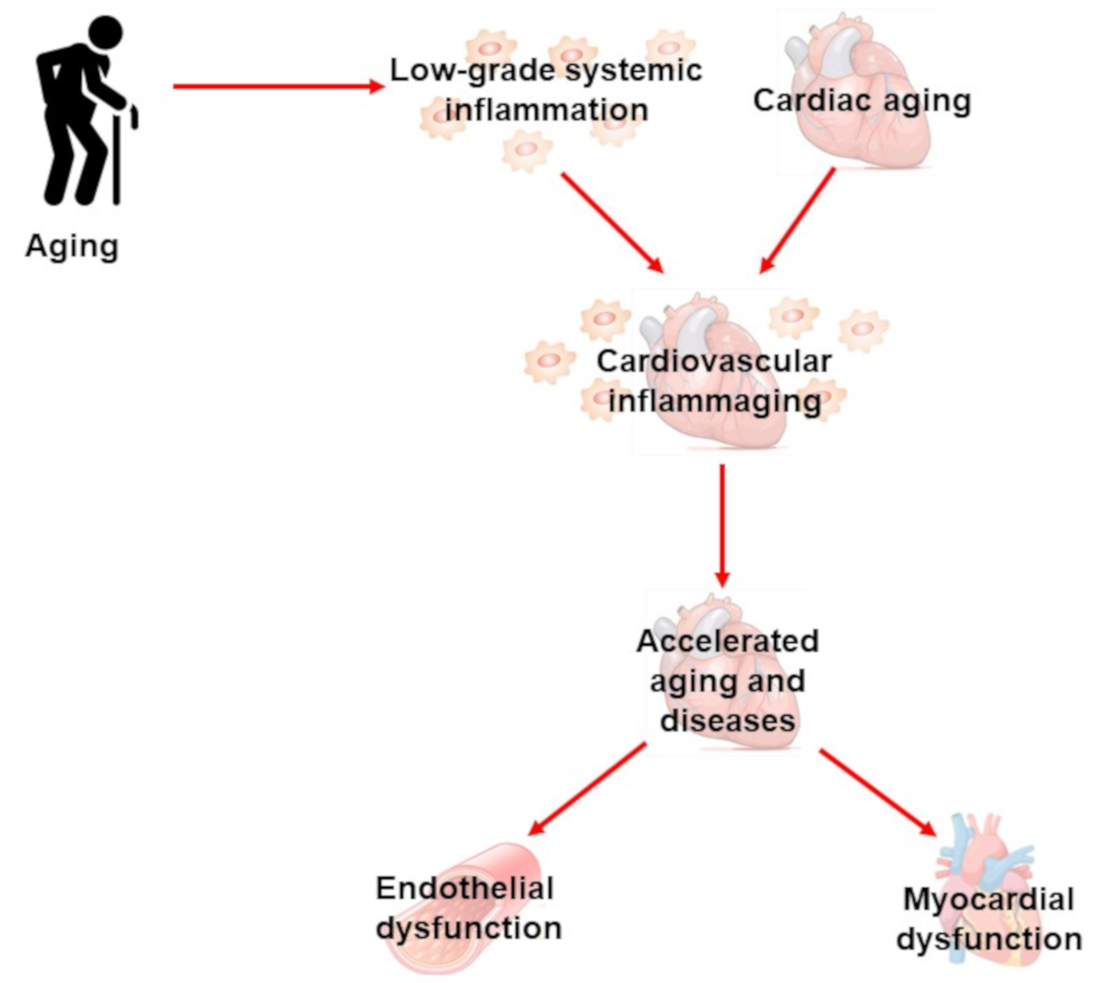{kind=link}
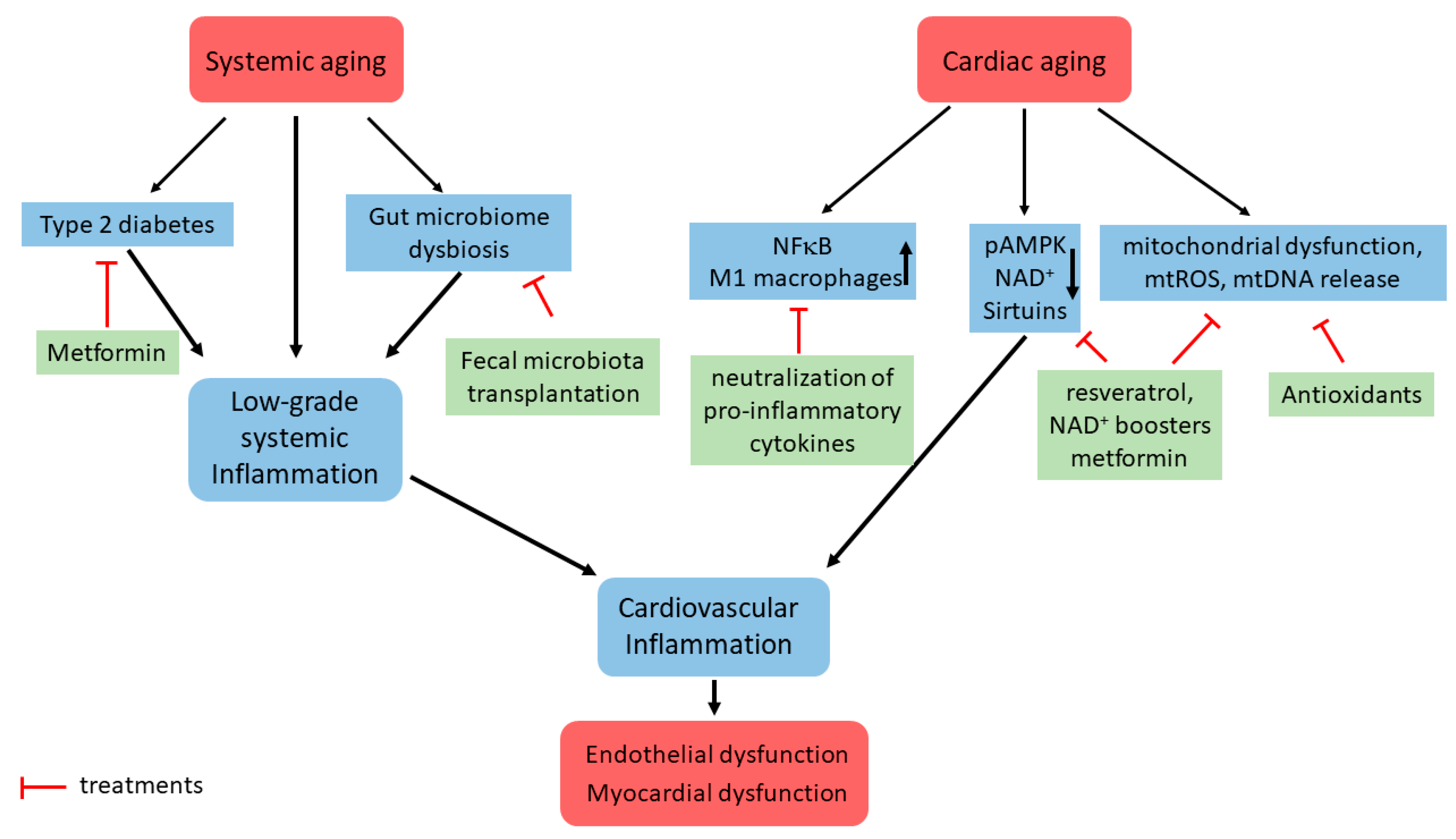{kind=link}
1. Inflammaging
2. Cardiac Inflammaging
2.1. Low-Grade Systemic Inflammation in Aging Heart, Sex-Related Differences
2.2. Role of AMPK and Sirtuins in Aging Heart
2.3. Mitochondria and Cardiac Inflammaging
3. Vascular Inflammaging
4. Inflammaging and the Microbiome
5. Translational Aspects of Cardiovascular Inflammaging
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Toba, H.; de Castro Bras, L.E.; Baicu, C.F.; Zile, M.R.; Lindsey, M.L.; Bradshaw, A.D. Secreted protein acidic and rich in cysteine facilitates age-related cardiac inflammation and macrophage M1 polarization. Am. J. Physiol. Cell Physiol. 2015, 308, C972–C982. [Google Scholar] [CrossRef] [PubMed]
- Kinn, P.M.; Holdren, G.O.; Westermeyer, B.A.; Abuissa, M.; Fischer, C.L.; Fairley, J.A.; Brogden, K.A.; Brogden, N.K. Age-dependent variation in cytokines, chemokines, and biologic analytes rinsed from the surface of healthy human skin. Sci. Rep. 2015, 5, 10472. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, E.; An, Y.; Zoli, M.; Simonsick, E.M.; Guralnik, J.M.; Bandinelli, S.; Boyd, C.M. Aging and the burden of multimorbidity: Associations with inflammatory and anabolic hormonal biomarkers. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Ballou, S.P.; Lozanski, F.B.; Hodder, S.; Rzewnicki, D.L.; Mion, L.C.; Sipe, J.D.; Ford, A.B.; Kushner, I. Quantitative and qualitative alterations of acute-phase proteins in healthy elderly persons. Age Ageing 1996, 25, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Xu, H.; Davies, J.L.; Hemmings, G.P. Increase of plasma IL-6 concentration with age in healthy subjects. Life Sci. 1992, 51, 1953–1956. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Howcroft, T.K.; Campisi, J.; Louis, G.B.; Smith, M.T.; Wise, B.; Wyss-Coray, T.; Augustine, A.D.; McElhaney, J.E.; Kohanski, R.; Sierra, F. The role of inflammation in age-related disease. Aging 2013, 5, 84–93. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Suuronen, T.; Kaarniranta, K. SIRT1 longevity factor suppresses NF-kappaB -driven immune responses: Regulation of aging via NF-kappaB acetylation? Bioessays 2008, 30, 939–942. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Ruparelia, N.; Chai, J.T.; Fisher, E.A.; Choudhury, R.P. Inflammatory processes in cardiovascular disease: A route to targeted therapies. Nat. Rev. Cardiol. 2017, 14, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and ‘Garb-aging’. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef] [PubMed]
- Jezek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Tavenier, J.; Rasmussen, L.J.H.; Houlind, M.B.; Andersen, A.L.; Panum, I.; Andersen, O.; Petersen, J.; Langkilde, A.; Nehlin, J.O. Alterations of monocyte NF-kappaB p65/RelA signaling in a cohort of older medical patients, age-matched controls, and healthy young adults. Immun. Ageing 2020, 17, 25. [Google Scholar] [CrossRef]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef]
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 2, 4172. [Google Scholar] [CrossRef]
- Bernal, G.M.; Wahlstrom, J.S.; Crawley, C.D.; Cahill, K.E.; Pytel, P.; Liang, H.; Kang, S.; Weichselbaum, R.R.; Yamini, B. Loss of Nfkb1 leads to early onset aging. Aging 2014, 6, 931–943. [Google Scholar] [CrossRef][Green Version]
- Garcia-Garcia, V.A.; Alameda, J.P.; Page, A.; Casanova, M.L. Role of NF-kappaB in Ageing and Age-Related Diseases: Lessons from Genetically Modified Mouse Models. Cells 2021, 10, 1906. [Google Scholar] [CrossRef]
- Youm, Y.H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef]
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.M.; Longo, D.L. The origins of age-related proinflammatory state. Blood 2005, 105, 2294–2299. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Barcena de Arellano, M.L.; Pozdniakova, S.; Kuhl, A.A.; Baczko, I.; Ladilov, Y.; Regitz-Zagrosek, V. Sex differences in the aging human heart: Decreased sirtuins, pro-inflammatory shift and reduced anti-oxidative defense. Aging 2019, 11, 1918–1933. [Google Scholar] [CrossRef]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med. 1997, 336, 973–979. [Google Scholar] [CrossRef]
- Ikonomidis, I.; Andreotti, F.; Economou, E.; Stefanadis, C.; Toutouzas, P.; Nihoyannopoulos, P. Increased proinflammatory cytokines in patients with chronic stable angina and their reduction by aspirin. Circulation 1999, 100, 793–798. [Google Scholar] [CrossRef]
- Liuzzo, G.; Biasucci, L.M.; Gallimore, J.R.; Caligiuri, G.; Buffon, A.; Rebuzzi, A.G.; Pepys, M.B.; Maseri, A. Enhanced inflammatory response in patients with preinfarction unstable angina. J. Am. Coll. Cardiol. 1999, 34, 1696–1703. [Google Scholar] [CrossRef]
- Muller-Werdan, U.; Nuding, S.; Ost, M. Assessing inflammageing. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 346–348. [Google Scholar] [CrossRef] [PubMed]
- Stice, J.P.; Chen, L.; Kim, S.C.; Jung, J.S.; Tran, A.L.; Liu, T.T.; Knowlton, A.A. 17β-Estradiol, aging, inflammation, and the stress response in the female heart. Endocrinology 2011, 152, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Duckles, S.P.; Weiss, J.H.; Li, X.; Krause, D.N. 17β-Estradiol prevents cell death and mitochondrial dysfunction by an estrogen receptor-dependent mechanism in astrocytes after oxygen-glucose deprivation/reperfusion. Free Radic. Biol. Med. 2012, 52, 2151–2160. [Google Scholar] [CrossRef] [PubMed]
- Klinge, C.M. Estrogenic control of mitochondrial function and biogenesis. J. Cell Biochem. 2008, 105, 1342–1351. [Google Scholar] [CrossRef]
- Barcena, M.L.; Pozdniakova, S.; Haritonow, N.; Breiter, P.; Kuhl, A.A.; Milting, H.; Baczko, I.; Ladilov, Y.; Regitz-Zagrosek, V. Dilated cardiomyopathy impairs mitochondrial biogenesis and promotes inflammation in an age- and sex-dependent manner. Aging 2020, 12, 24117–24133. [Google Scholar] [CrossRef]
- Ghisays, F.; Brace, C.S.; Yackly, S.M.; Kwon, H.J.; Mills, K.F.; Kashentseva, E.; Dmitriev, I.P.; Curiel, D.T.; Imai, S.I.; Ellenberger, T. The N-Terminal Domain of SIRT1 Is a Positive Regulator of Endogenous SIRT1-Dependent Deacetylation and Transcriptional Outputs. Cell Rep. 2015, 10, 1665–1673. [Google Scholar] [CrossRef]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
- Epelman, S.; Liu, P.P.; Mann, D.L. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat. Rev. Immunol. 2015, 15, 117–129. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated protein kinase: Maintaining energy homeostasis at the cellular and whole-body levels. Annu. Rev. Nutr. 2014, 34, 31–55. [Google Scholar] [CrossRef]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Age-related changes in AMPK activation: Role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling pathways. Ageing Res. Rev. 2016, 28, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Pang, J.; Han, Y.; Dai, Y.; Dai, D.; Cai, J.; Zhang, T.M. Age-dependent tissue expression patterns of Sirt1 in senescence-accelerated mice. Mol. Med. Rep. 2014, 10, 3296–3302. [Google Scholar] [CrossRef]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE 2011, 6, e19194. [Google Scholar] [CrossRef]
- Xu, H.; Gan, C.; Gao, Z.; Huang, Y.; Wu, S.; Zhang, D.; Wang, X.; Sheng, J. Caffeine Targets SIRT3 to Enhance SOD2 Activity in Mitochondria. Front. Cell Dev. Biol. 2020, 8, 822. [Google Scholar] [CrossRef]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef]
- Parihar, P.; Solanki, I.; Mansuri, M.L.; Parihar, M.S. Mitochondrial sirtuins: Emerging roles in metabolic regulations, energy homeostasis and diseases. Exp. Gerontol. 2015, 61, 130–141. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarrago, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef]
- Jayarajan, V.; Appukuttan, A.; Aslam, M.; Reusch, P.; Regitz-Zagrosek, V.; Ladilov, Y. Regulation of AMPK activity by type 10 adenylyl cyclase: Contribution to the mitochondrial biology, cellular redox and energy homeostasis. Cell Mol. Life Sci. 2019, 76, 4945–4959. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Brenmoehl, J.; Hoeflich, A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion 2013, 13, 755–761. [Google Scholar] [CrossRef] [PubMed]
- El Assar, M.; Angulo, J.; Rodriguez-Manas, L. Oxidative stress and vascular inflammation in aging. Free Radic. Biol. Med. 2013, 65, 380–401. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef]
- Bess, E.; Fisslthaler, B.; Fromel, T.; Fleming, I. Nitric oxide-induced activation of the AMP-activated protein kinase alpha2 subunit attenuates IkappaB kinase activity and inflammatory responses in endothelial cells. PLoS ONE 2011, 6, e20848. [Google Scholar] [CrossRef]
- Salminen, A.; Hyttinen, J.M.; Kaarniranta, K. AMP-activated protein kinase inhibits NF-kappaB signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med. 2011, 89, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Peairs, A.; Radjavi, A.; Davis, S.; Li, L.; Ahmed, A.; Giri, S.; Reilly, C.M. Activation of AMPK inhibits inflammation in MRL/lpr mouse mesangial cells. Clin. Exp. Immunol. 2009, 156, 542–551. [Google Scholar] [CrossRef]
- Hoogendijk, A.J.; Pinhancos, S.S.; van der Poll, T.; Wieland, C.W. AMP-activated protein kinase activation by 5-aminoimidazole-4-carbox-amide-1-beta-D-ribofuranoside (AICAR) reduces lipoteichoic acid-induced lung inflammation. J. Biol. Chem. 2013, 288, 7047–7052. [Google Scholar] [CrossRef]
- Gurd, B.J. Deacetylation of PGC-1alpha by SIRT1: Importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl Physiol. Nutr. Metab. 2011, 36, 589–597. [Google Scholar] [CrossRef]
- Soto-Heredero, G.; Gomez de Las Heras, M.M.; Gabande-Rodriguez, E.; Oller, J.; Mittelbrunn, M. Glycolysis—A key player in the inflammatory response. FEBS J. 2020, 287, 3350–3369. [Google Scholar] [CrossRef] [PubMed]
- Palsson-McDermott, E.M.; O’Neill, L.A.J. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 2020, 30, 300–314. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, S.K.; Naik, P.P.; Panigrahi, D.P.; Bhol, C.S.; Mahapatra, K.K. Mitophagy, Diseases, and Aging. In Models, Molecules and Mechanisms in Biogerontology; Rath, P.C., Ed.; Springer Nature: Singapore, 2019; pp. 177–191. [Google Scholar]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, F.; Preston, C.C.; Emelyanova, L.; Yousufuddin, M.; Viqar, M.; Dakwar, O.; Ross, G.R.; Faustino, R.S.; Holmuhamedov, E.L.; Jahangir, A. Effects of Aging on Cardiac Oxidative Stress and Transcriptional Changes in Pathways of Reactive Oxygen Species Generation and Clearance. J. Am. Heart Assoc. 2021, 10, e019948. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1552–1562. [Google Scholar] [CrossRef]
- Dela Cruz, C.S.; Kang, M.J. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 2018, 41, 37–44. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches. EMBO Rep. 2020, 21, e49612. [Google Scholar] [CrossRef] [PubMed]
- Elorza, A.A.; Soffia, J.P. mtDNA Heteroplasmy at the Core of Aging-Associated Heart Failure. An Integrative View of OXPHOS and Mitochondrial Life Cycle in Cardiac Mitochondrial Physiology. Front. Cell Dev. Biol. 2021, 9, 625020. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; Whitehall, J.C.; Bradshaw, C.; Gay, D.; Robertson, F.; Blain, A.P.; Hudson, G.; Pyle, A.; Houghton, D.; Hunt, M.; et al. Age-associated mitochondrial DNA mutations cause metabolic remodelling that contributes to accelerated intestinal tumorigenesis. Nat. Cancer 2020, 1, 976–989. [Google Scholar] [CrossRef] [PubMed]
- Tabara, L.C.; Morris, J.L.; Prudent, J. The Complex Dance of Organelles during Mitochondrial Division. Trends Cell Biol. 2021, 31, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Duicu, O.M.; Mirica, S.N.; Gheorgheosu, D.E.; Privistirescu, A.I.; Fira-Mladinescu, O.; Muntean, D.M. Ageing-induced decrease in cardiac mitochondrial function in healthy rats. Can. J. Physiol. Pharmacol. 2013, 91, 593–600. [Google Scholar] [CrossRef]
- Assar, M.E.; Angulo, J.; Rodriguez-Manas, L. Diabetes and ageing-induced vascular inflammation. J. Physiol. 2016, 594, 2125–2146. [Google Scholar] [CrossRef]
- Scuteri, A.; Cunha, P.G.; Agabiti Rosei, E.; Badariere, J.; Bekaert, S.; Cockcroft, J.R.; Cotter, J.; Cucca, F.; De Buyzere, M.L.; De Meyer, T.; et al. Arterial stiffness and influences of the metabolic syndrome: A cross-countries study. Atherosclerosis 2014, 233, 654–660. [Google Scholar] [CrossRef]
- Guzik, T.J.; Cosentino, F. Epigenetics and Immunometabolism in Diabetes and Aging. Antioxid. Redox Signal. 2018, 29, 257–274. [Google Scholar] [CrossRef]
- Lakatta, E.G. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part III: Cellular and molecular clues to heart and arterial aging. Circulation 2003, 107, 490–497. [Google Scholar] [CrossRef]
- Laina, A.; Stellos, K.; Stamatelopoulos, K. Vascular ageing: Underlying mechanisms and clinical implications. Exp. Gerontol. 2018, 109, 16–30. [Google Scholar] [CrossRef]
- Long, D.A.; Newaz, M.A.; Prabhakar, S.S.; Price, K.L.; Truong, L.D.; Feng, L.; Mu, W.; Oyekan, A.O.; Johnson, R.J. Loss of nitric oxide and endothelial-derived hyperpolarizing factor-mediated responses in aging. Kidney Int. 2005, 68, 2154–2163. [Google Scholar] [CrossRef] [PubMed]
- Luscher, T.F.; Corti, R. Flow: The signal of life. Circ. Res. 2004, 95, 749–751. [Google Scholar] [CrossRef] [PubMed]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arter. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef] [PubMed]
- Trinity, J.D.; Groot, H.J.; Layec, G.; Rossman, M.J.; Ives, S.J.; Morgan, D.E.; Gmelch, B.S.; Bledsoe, A.; Richardson, R.S. Passive leg movement and nitric oxide-mediated vascular function: The impact of age. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H672–H679. [Google Scholar] [CrossRef] [PubMed]
- Lerman, A.; Burnett, J.C., Jr. Intact and altered endothelium in regulation of vasomotion. Circulation 1992, 86 (Suppl. 6), III12–III19. [Google Scholar] [PubMed]
- Kitta, Y.; Obata, J.E.; Nakamura, T.; Hirano, M.; Kodama, Y.; Fujioka, D.; Saito, Y.; Kawabata, K.; Sano, K.; Kobayashi, T.; et al. Persistent impairment of endothelial vasomotor function has a negative impact on outcome in patients with coronary artery disease. J. Am. Coll. Cardiol. 2009, 53, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.J. Assessment and treatment of endothelial dysfunction in humans. J. Am. Coll. Cardiol. 1999, 34, 631–638. [Google Scholar] [CrossRef]
- Bonetti, P.O.; Pumper, G.M.; Higano, S.T.; Holmes, D.R.; Kuvin, J.T., Jr.; Lerman, A. Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia. J. Am. Coll. Cardiol. 2004, 44, 2137–2141. [Google Scholar] [CrossRef]
- Sverdlov, A.L.; Ngo, D.T.; Chan, W.P.; Chirkov, Y.Y.; Horowitz, J.D. Aging of the nitric oxide system: Are we as old as our NO? J. Am. Heart Assoc. 2014, 3, e000973. [Google Scholar] [CrossRef]
- Celermajer, D.S.; Sorensen, K.E.; Spiegelhalter, D.J.; Georgakopoulos, D.; Robinson, J.; Deanfield, J.E. Aging is associated with endothelial dysfunction in healthy men years before the age-related decline in women. J. Am. Coll. Cardiol. 1994, 24, 471–476. [Google Scholar] [CrossRef]
- Eskurza, I.; Monahan, K.D.; Robinson, J.A.; Seals, D.R. Effect of acute and chronic ascorbic acid on flow-mediated dilatation with sedentary and physically active human ageing. J. Physiol. 2004, 556, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Andrew, P.J.; Mayer, B. Enzymatic function of nitric oxide synthases. Cardiovasc. Res. 1999, 43, 521–531. [Google Scholar] [CrossRef]
- Cernadas, M.R.; Sanchez de Miguel, L.; Garcia-Duran, M.; Gonzalez-Fernandez, F.; Millas, I.; Monton, M.; Rodrigo, J.; Rico, L.; Fernandez, P.; de Frutos, T.; et al. Expression of constitutive and inducible nitric oxide synthases in the vascular wall of young and aging rats. Circ. Res. 1998, 83, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.R.; Visioli, F.; Hagen, T.M. Plasma membrane-associated endothelial nitric oxide synthase and activity in aging rat aortic vascular endothelia markedly decline with age. Arch. Biochem. Biophys. 2006, 454, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Dumont, O.; Pinaud, F.; Guihot, A.L.; Baufreton, C.; Loufrani, L.; Henrion, D. Alteration in flow (shear stress)-induced remodelling in rat resistance arteries with aging: Improvement by a treatment with hydralazine. Cardiovasc. Res. 2008, 77, 600–608. [Google Scholar] [CrossRef]
- Berkowitz, D.E.; White, R.; Li, D.; Minhas, K.M.; Cernetich, A.; Kim, S.; Burke, S.; Shoukas, A.A.; Nyhan, D.; Champion, H.C.; et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation 2003, 108, 2000–2006. [Google Scholar] [CrossRef]
- Sakai, Y.; Masuda, H.; Kihara, K.; Kurosaki, E.; Yamauchi, Y.; Azuma, H. Involvement of increased arginase activity in impaired cavernous relaxation with aging in the rabbit. J. Urol. 2004, 172, 369–373. [Google Scholar] [CrossRef]
- Garate-Carrillo, A.; Navarrete-Yanez, V.; Ortiz-Vilchis, P.; Guevara, G.; Castillo, C.; Mendoza-Lorenzo, P.; Ceballos, G.; Ortiz-Flores, M.; Najera, N.; Bustamante-Pozo, M.M.; et al. Arginase inhibition by (-)-Epicatechin reverses endothelial cell aging. Eur. J. Pharmacol. 2020, 885, 173442. [Google Scholar] [CrossRef]
- van der Loo, B.; Labugger, R.; Skepper, J.N.; Bachschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callender, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T.; et al. Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 2000, 192, 1731–1744. [Google Scholar] [CrossRef]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Salvetti, G.; Bernini, G.; Magagna, A.; Salvetti, A. Age-related reduction of NO availability and oxidative stress in humans. Hypertension 2001, 38, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, C.A.; Brosnan, M.J.; McIntyre, M.; Graham, D.; Dominiczak, A.F. Superoxide excess in hypertension and aging: A common cause of endothelial dysfunction. Hypertension 2001, 37, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Fleenor, B.S.; Seals, D.R.; Zigler, M.L.; Sindler, A.L. Superoxide-lowering therapy with TEMPOL reverses arterial dysfunction with aging in mice. Aging Cell 2012, 11, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Alp, N.J.; Channon, K.M. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Delp, M.D.; Behnke, B.J.; Spier, S.A.; Wu, G.; Muller-Delp, J.M. Ageing diminishes endothelium-dependent vasodilatation and tetrahydrobiopterin content in rat skeletal muscle arterioles. J. Physiol. 2008, 586, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Eskurza, I.; Myerburgh, L.A.; Kahn, Z.D.; Seals, D.R. Tetrahydrobiopterin augments endothelium-dependent dilatation in sedentary but not in habitually exercising older adults. J. Physiol. 2005, 568 Pt 3, 1057–1065. [Google Scholar] [CrossRef]
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Wong, C.; Song, Y.; Shen, H.; Mori, D.; Rotllan, N.; Price, N.; Dobrian, A.D.; Meng, H.; Kleinstein, S.H.; et al. Age-associated vascular inflammation promotes monocytosis during atherogenesis. Aging Cell 2016, 15, 766–777. [Google Scholar] [CrossRef]
- Liu, Y.; Jacobowitz, D.M.; Barone, F.; McCarron, R.; Spatz, M.; Feuerstein, G.; Hallenbeck, J.M.; Siren, A.L. Quantitation of perivascular monocytes and macrophages around cerebral blood vessels of hypertensive and aged rats. J. Cereb. Blood Flow Metab. 1994, 14, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Nosalski, R.; Mikolajczyk, T.; Siedlinski, M.; Saju, B.; Koziol, J.; Maffia, P.; Guzik, T.J. Nox1/4 inhibition exacerbates age dependent perivascular inflammation and fibrosis in a model of spontaneous hypertension. Pharmacol. Res. 2020, 161, 105235. [Google Scholar] [CrossRef]
- Desdin-Mico, G.; Soto-Heredero, G.; Aranda, J.F.; Oller, J.; Carrasco, E.; Gabande-Rodriguez, E.; Blanco, E.M.; Alfranca, A.; Cusso, L.; Desco, M.; et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 2020, 368, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Ramos, G.C.; van den Berg, A.; Nunes-Silva, V.; Weirather, J.; Peters, L.; Burkard, M.; Friedrich, M.; Pinnecker, J.; Abesser, M.; Heinze, K.G.; et al. Myocardial aging as a T-cell-mediated phenomenon. Proc. Natl. Acad. Sci. USA 2017, 114, E2420–E2429. [Google Scholar] [CrossRef] [PubMed]
- Delgobo, M.; Heinrichs, M.; Hapke, N.; Ashour, D.; Appel, M.; Srivastava, M.; Heckel, T.; Spyridopoulos, I.; Hofmann, U.; Frantz, S.; et al. Terminally Differentiated CD4+ T Cells Promote Myocardial Inflammaging. Front. Immunol. 2021, 12, 584538. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Verges, M.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Zhao, J.; Wu, L.; Carru, C.; Biagi, E.; Franceschi, C. Microbiomes other than the gut: Inflammaging and age-related diseases. Semin. Immunopathol. 2020, 42, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Zeng, D.N.; Chi, L.; Tan, Y.; Galzote, C.; Cardona, C.; Lax, S.; Gilbert, J.; Quan, Z.X. The Influence of Age and Gender on Skin-Associated Microbial Communities in Urban and Rural Human Populations. PLoS ONE 2015, 10, e0141842. [Google Scholar]
- Kim, H.J.; Kim, J.J.; Myeong, N.R.; Kim, T.; Kim, D.; An, S.; Kim, H.; Park, T.; Jang, S.I.; Yeon, J.H.; et al. Segregation of age-related skin microbiome characteristics by functionality. Sci. Rep. 2019, 9, 16748. [Google Scholar] [CrossRef] [PubMed]
- Remond, D.; Shahar, D.R.; Gille, D.; Pinto, P.; Kachal, J.; Peyron, M.A.; Dos Santos, C.N.; Walther, B.; Bordoni, A.; Dupont, D.; et al. Understanding the gastrointestinal tract of the elderly to develop dietary solutions that prevent malnutrition. Oncotarget 2015, 6, 13858–13898. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, R.; Yadav, H. Bacterial Translocation from the Gut to the Distant Organs: An Overview. Ann. Nutr. Metab. 2017, 71 (Suppl. 1), 11–16. [Google Scholar] [CrossRef]
- Berg, R.D. Bacterial Translocation from the Gastrointestinal Tract. In Mechanisms in the Pathogenesis of Enteric Diseases 2. Advances in Experimental Medicine and Biology; Prem, S.P., David, H.F., Eds.; Springer: Boston, MA, USA, 1999; Volume 473, pp. 11–30. [Google Scholar]
- Biagi, E.; Candela, M.; Franceschi, C.; Brigidi, P. The aging gut microbiota: New perspectives. Ageing Res. Rev. 2011, 10, 428–429. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, R.; Mainali, R.; Ahmadi, S.; Wang, S.; Singh, R.; Kavanagh, K.; Kitzman, D.W.; Kushugulova, A.; Marotta, F.; Yadav, H. Gut microbiome and aging: Physiological and mechanistic insights. Nutr. Healthy Aging 2018, 4, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Vinolo, M.A.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of inflammation by short chain fatty acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef] [PubMed]
- Inan, M.S.; Rasoulpour, R.J.; Yin, L.; Hubbard, A.K.; Rosenberg, D.W.; Giardina, C. The luminal short-chain fatty acid butyrate modulates NF-kappaB activity in a human colonic epithelial cell line. Gastroenterology 2000, 118, 724–734. [Google Scholar] [CrossRef]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; Gonzalez, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Chen, S.; Deng, B.; Tan, C.; Deng, J.; Zhu, G.; Yin, Y.; Ren, W. Implication of G Protein-Coupled Receptor 43 in Intestinal Inflammation: A Mini-Review. Front. Immunol. 2018, 9, 1434. [Google Scholar] [CrossRef] [PubMed]
- Ragonnaud, E.; Biragyn, A. Gut microbiota as the key controllers of “healthy” aging of elderly people. Immun. Ageing 2021, 18, 2. [Google Scholar] [CrossRef]
- Biagi, E.; Nylund, L.; Candela, M.; Ostan, R.; Bucci, L.; Pini, E.; Nikkila, J.; Monti, D.; Satokari, R.; Franceschi, C.; et al. Through ageing, and beyond: Gut microbiota and inflammatory status in seniors and centenarians. PLoS ONE 2010, 5, e10667. [Google Scholar] [CrossRef]
- Shintouo, C.M.; Mets, T.; Beckwee, D.; Bautmans, I.; Ghogomu, S.M.; Souopgui, J.; Leemans, L.; Meriki, H.D.; Njemini, R. Is inflammageing influenced by the microbiota in the aged gut? A systematic review. Exp. Gerontol. 2020, 141, 111079. [Google Scholar] [CrossRef]
- Fransen, F.; van Beek, A.A.; Borghuis, T.; Aidy, S.E.; Hugenholtz, F.; van der Gaast-de Jongh, C.; Savelkoul, H.F.J.; De Jonge, M.I.; Boekschoten, M.V.; Smidt, H.; et al. Aged Gut Microbiota Contributes to Systemical Inflammaging after Transfer to Germ-Free Mice. Front. Immunol. 2017, 8, 1385. [Google Scholar] [CrossRef]
- Ahmad, A.F.; Dwivedi, G.; O’Gara, F.; Caparros-Martin, J.; Ward, N.C. The gut microbiome and cardiovascular disease: Current knowledge and clinical potential. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H923–H938. [Google Scholar] [CrossRef]
- Yoshida, N.; Yamashita, T.; Hirata, K.I. Gut Microbiome and Cardiovascular Diseases. Diseases 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, G.; MacDonald, K.; Reid, G.; Burton, J.P. The influence of the human microbiome and probiotics on cardiovascular health. Gut Microbes 2014, 5, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Ahmadmehrabi, S.; Tang, W.H.W. Gut microbiome and its role in cardiovascular diseases. Curr. Opin. Cardiol. 2017, 32, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Troseid, M.; Andersen, G.O.; Broch, K.; Hov, J.R. The gut microbiome in coronary artery disease and heart failure: Current knowledge and future directions. EBioMedicine 2020, 52, 102649. [Google Scholar] [CrossRef] [PubMed]
- Kazemian, N.; Mahmoudi, M.; Halperin, F.; Wu, J.C.; Pakpour, S. Gut microbiota and cardiovascular disease: Opportunities and challenges. Microbiome 2020, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, X.; Yang, F.; Zhao, R.; Pan, X.; Liang, J.; Tian, L.; Li, X.; Liu, L.; Xing, Y.; et al. Gut Microbiota-Dependent Marker TMAO in Promoting Cardiovascular Disease: Inflammation Mechanism, Clinical Prognostic, and Potential as a Therapeutic Target. Front. Pharmacol. 2019, 10, 1360. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Luo, Y.; Liu, J.P.; Sun, N.; Guo, D.; Cui, L.L.; Zheng, P.P.; Yao, S.M.; Yang, J.F.; Wang, H. Trimethylamine N-Oxide, a Gut Microbiota-Dependent Metabolite, is Associated with Frailty in Older Adults with Cardiovascular Disease. Clin. Interv. Aging 2020, 15, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Roncal, C.; Martinez-Aguilar, E.; Orbe, J.; Ravassa, S.; Fernandez-Montero, A.; Saenz-Pipaon, G.; Ugarte, A.; Estrella-Hermoso de Mendoza, A.; Rodriguez, J.A.; Fernandez-Alonso, S.; et al. Trimethylamine-N-Oxide (TMAO) Predicts Cardiovascular Mortality in Peripheral Artery Disease. Sci. Rep. 2019, 9, 15580. [Google Scholar] [CrossRef] [PubMed]
- Leshem, A.; Horesh, N.; Elinav, E. Fecal Microbial Transplantation and Its Potential Application in Cardiometabolic Syndrome. Front. Immunol. 2019, 10, 1341. [Google Scholar] [CrossRef]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef]
- Hu, X.F.; Zhang, W.Y.; Wen, Q.; Chen, W.J.; Wang, Z.M.; Chen, J.; Zhu, F.; Liu, K.; Cheng, L.X.; Yang, J.; et al. Fecal microbiota transplantation alleviates myocardial damage in myocarditis by restoring the microbiota composition. Pharmacol. Res. 2019, 139, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, T.S.; Rampelli, S.; Jeffery, I.B.; Santoro, A.; Neto, M.; Capri, M.; Giampieri, E.; Jennings, A.; Candela, M.; Turroni, S.; et al. Mediterranean diet intervention alters the gut microbiome in older people reducing frailty and improving health status: The NU-AGE 1-year dietary intervention across five European countries. Gut 2020, 69, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, A.; Santoro, A.; Pini, E.; Cevenini, E.; Ostan, R.; Pietruszka, B.; Rolf, K.; Cano, N.; Caille, A.; Lyon-Belgy, N.; et al. Reprint of: A parallel randomized trial on the effect of a healthful diet on inflammageing and its consequences in European elderly people: Design of the NU-AGE dietary intervention study. Mech. Ageing Dev. 2014, 136–137, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Martin-Ruiz, C.M.; Takayama, M.; Abe, Y.; Takebayashi, T.; Koyasu, S.; Suematsu, M.; Hirose, N.; von Zglinicki, T. Inflammation, But Not Telomere Length, Predicts Successful Ageing at Extreme Old Age: A Longitudinal Study of Semi-supercentenarians. EBioMedicine 2015, 2, 1549–1558. [Google Scholar] [CrossRef]
- Goldeck, D.; Pawelec, G.; Norman, K.; Steinhagen-Thiessen, E.; Oettinger, L.; Haehnel, K.; Demuth, I. No strong correlations between serum cytokine levels, CMV serostatus and hand-grip strength in older subjects in the Berlin BASE-II cohort. Biogerontology 2016, 17, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Waaijer, M.E.; Westendorp, R.G.; Goldeck, D.; Gunn, D.A.; Pawelec, G.; Stijntjes, M.; Slagboom, P.E.; Maier, A.B. Assessment of health status by molecular measures in adults ranging from middle-aged to old: Ready for clinical use? Exp. Gerontol. 2017, 87, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Ladilov, Y. Emerging Role of cAMP/AMPK Signaling. Cells 2022, 11, 308. [Google Scholar] [CrossRef]
- Gupta, S.C.; Kunnumakkara, A.B.; Aggarwal, S.; Aggarwal, B.B. Inflammation, a Double-Edge Sword for Cancer and Other Age-Related Diseases. Front. Immunol. 2018, 9, 2160. [Google Scholar] [CrossRef]
- Park, S.J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Yi, L.; Jin, X.; Liang, X.Y.; Zhou, Y.; Zhang, T.; Xie, Q.; Zhou, X.; Chan, H.; Fu, Y.J.; et al. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy 2013, 9, 2033–2045. [Google Scholar] [CrossRef]
- Diaz-Casado, M.E.; Quiles, J.L.; Barriocanal-Casado, E.; Gonzalez-Garcia, P.; Battino, M.; Lopez, L.C.; Varela-Lopez, A. The Paradox of Coenzyme Q10 in Aging. Nutrients 2019, 11, 2221. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, J.J.; Quiles, J.L.; Huertas, J.R.; Mataix, J. Coenzyme Q10 protects from aging-related oxidative stress and improves mitochondrial function in heart of rats fed a polyunsaturated fatty acid (PUFA)-rich diet. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, H.; Ikeda, K.; Shinozaki, S.; Yasuhara, S.; Yu, Y.M.; Martyn, J.A.J.; Tompkins, R.G.; Yorozu, T.; Inoue, S.; Kaneki, M. Coenzyme Q10 protects against burn-induced mitochondrial dysfunction and impaired insulin signaling in mouse skeletal muscle. FEBS Open Biol. 2019, 9, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, S.A.; Rosenfeldt, F.; Kumar, A.; Dolliner, P.; Filipiak, K.J.; Pella, D.; Alehagen, U.; Steuer, G.; Littarru, G.P.; Symbio, Q. Study Investigators.The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: Results from Q-SYMBIO: A randomized double-blind trial. JACC Heart Fail. 2014, 2, 641–649. [Google Scholar] [CrossRef]
- Forini, F.; Canale, P.; Nicolini, G.; Iervasi, G. Mitochondria-Targeted Drug Delivery in Cardiovascular Disease: A Long Road to Nano-Cardio Medicine. Pharmaceutics 2020, 12, 1122. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barcena, M.L.; Aslam, M.; Pozdniakova, S.; Norman, K.; Ladilov, Y. Cardiovascular Inflammaging: Mechanisms and Translational Aspects. Cells 2022, 11, 1010. https://doi.org/10.3390/cells11061010
Barcena ML, Aslam M, Pozdniakova S, Norman K, Ladilov Y. Cardiovascular Inflammaging: Mechanisms and Translational Aspects. Cells. 2022; 11(6):1010. https://doi.org/10.3390/cells11061010
Chicago/Turabian StyleBarcena, Maria Luisa, Muhammad Aslam, Sofya Pozdniakova, Kristina Norman, and Yury Ladilov. 2022. "Cardiovascular Inflammaging: Mechanisms and Translational Aspects" Cells 11, no. 6: 1010. https://doi.org/10.3390/cells11061010
APA StyleBarcena, M. L., Aslam, M., Pozdniakova, S., Norman, K., & Ladilov, Y. (2022). Cardiovascular Inflammaging: Mechanisms and Translational Aspects. Cells, 11(6), 1010. https://doi.org/10.3390/cells11061010