
Amelioration of Experimental Autoimmune Encephalomyelitis in Alzheimer’s Disease Mouse Models: A Potential Role for Aβ

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Reagents
2.2. EAE Induction and Intraperitoneal Injection of Aβ42
2.3. Histopathological Analysis and Thioflavin-S Staining of Senile Plaques
2.4. ELISA Analysis
2.5. CD4+ T Cell Separation and In Vitro Differentiation
2.6. DC Isolation, Stimulation, IL-6, and Cell Toxicity Measurement
2.7. Flow Cytometry
2.8. Statistical Analysis
3. Results
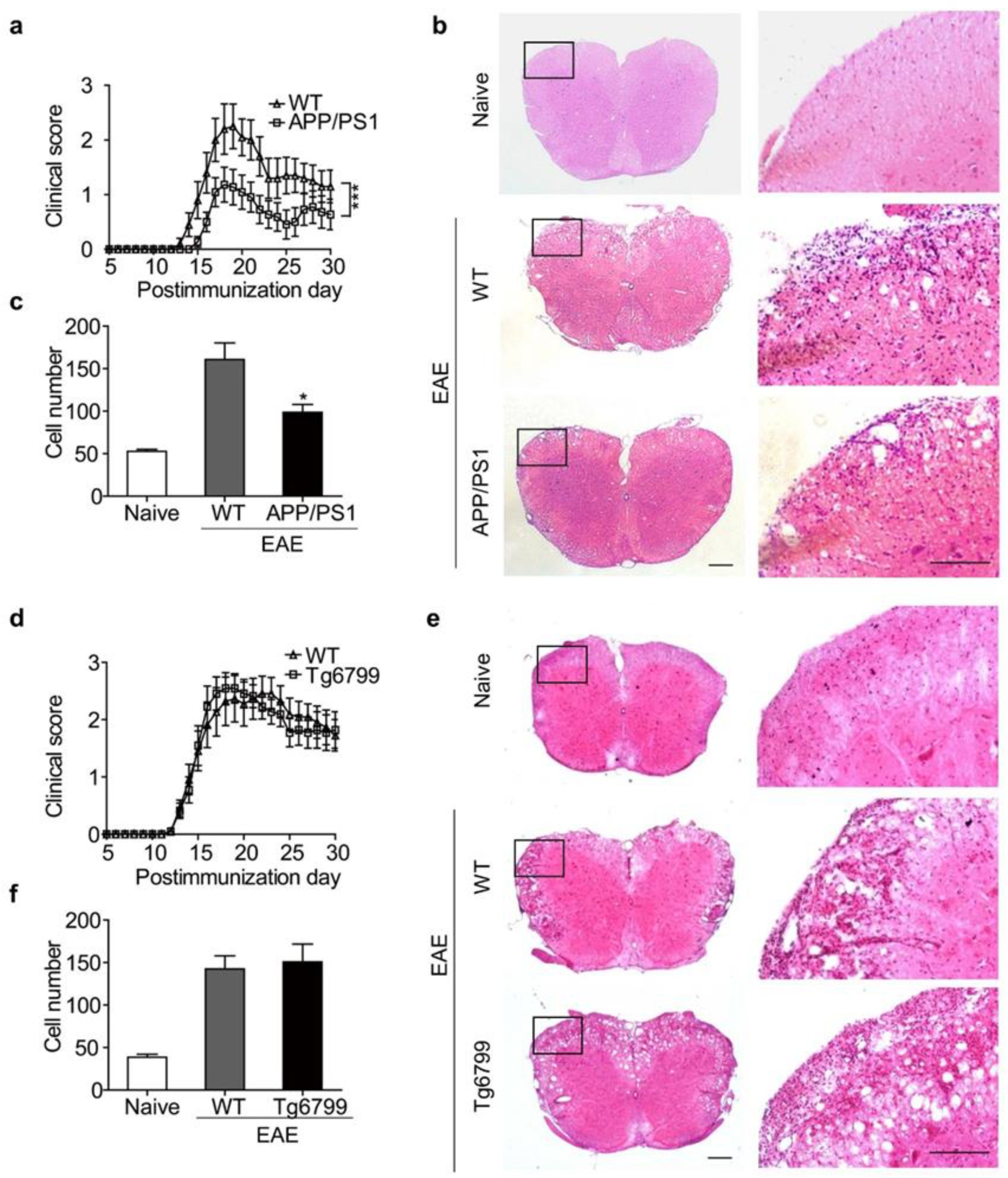
3.1. Clinical Symptoms Were Ameliorated When EAE Was Induced in APP/PS1 Mice, but Not in Tg6799 Transgenic Mice
3.2. Distinct Aβ Pathology in APP/PS1 and Tg6799 Mice
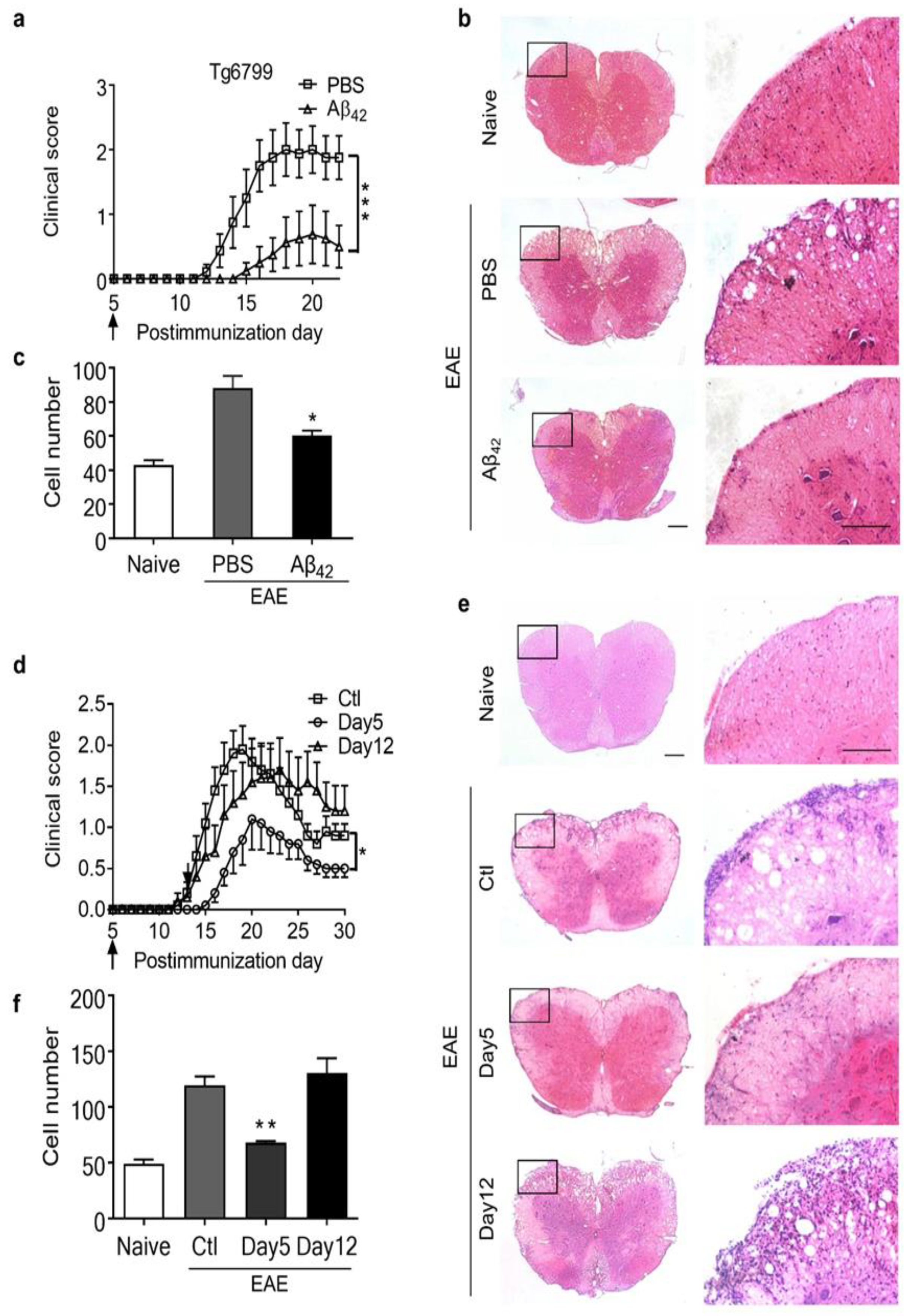
3.3. Intraperitoneal Administration of Aβ42 Peptides Ameliorated the Clinical Symptoms of EAE in Tg6799 Mice
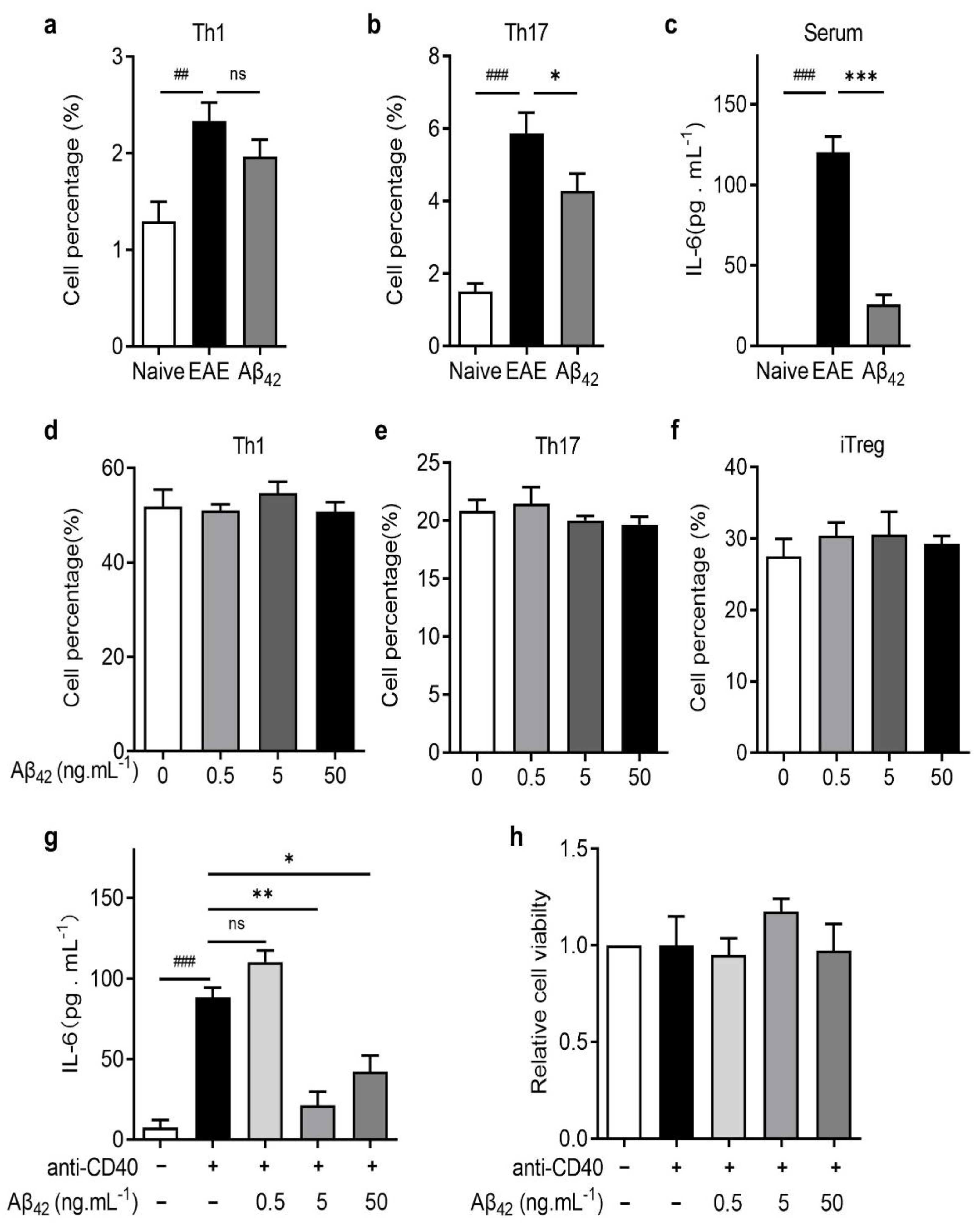
3.4. Aβ42 Regulated Th17 Development by Reducing IL-6 Production by DCs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef] [Green Version]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Hilliard, B.; Fujioka, T.; Bhopale, M.K.; Calida, D.; Rostami, A.M. Pathogenesis of neuroimmunologic diseases. Experimental models. Immunol. Res. 1998, 17, 217–227. [Google Scholar] [CrossRef]
- Steinman, L.; Zamvil, S.S. Virtues and pitfalls of EAE for the development of therapies for multiple sclerosis. Trends Immunol. 2005, 26, 565–571. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Lichtenthaler, S.F.; Wang, R.; Grimm, H.; Uljon, S.N.; Masters, C.L.; Beyreuther, K. Mechanism of the cleavage specificity of Alzheimer’s disease gamma-secretase identified by phenylalanine-scanning mutagenesis of the transmembrane domain of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 1999, 96, 3053–3058. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol. 1998, 8, 447–453. [Google Scholar] [CrossRef]
- Yang, L.B.; Lindholm, K.; Yan, R.; Citron, M.; Xia, W.; Yang, X.L.; Beach, T.; Sue, L.; Wong, P.; Price, D.; et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 2003, 9, 3–4. [Google Scholar] [CrossRef]
- Brothers, H.M.; Gosztyla, M.L.; Robinson, S.R. The Physiological Roles of Amyloid-β Peptide Hint at New Ways to Treat Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.K.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra72. [Google Scholar] [CrossRef] [Green Version]
- Grant, J.L.; Ghosn, E.E.; Axtell, R.C.; Herges, K.; Kuipers, H.F.; Woodling, N.S.; Andreasson, K.; Herzenberg, L.A.; Herzenberg, L.A.; Steinman, L. Reversal of paralysis and reduced inflammation from peripheral administration of beta-amyloid in TH1 and TH17 versions of experimental autoimmune encephalomyelitis. Sci. Transl. Med. 2012, 4, 145ra105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avitan, I.; Halperin, Y.; Saha, T.; Bloch, N.; Atrahimovich, D.; Polis, B.; Samson, A.O.; Braitbard, O. Towards a Consensus on Alzheimer’s Disease Comorbidity? J. Clin. Med. 2021, 10, 4360. [Google Scholar] [CrossRef] [PubMed]
- Luczynski, P.; Laule, C.; Hsiung, G.-Y.R.; Moore, G.R.W.; Tremlett, H. Coexistence of Multiple Sclerosis and Alzheimer’s disease: A review. Mult. Scler. Relat. Disord. 2019, 27, 232–238. [Google Scholar] [CrossRef]
- Lv, J.; Zhuang, W.; Zhang, Y.; Xie, L.; Xiang, Z.; Zhao, Q.; Jiang, X.; Shen, J.; Du, C. 9,10-Anhydrodehydroartemisinin Attenuates Experimental Autoimmune Encephalomyelitis by Inhibiting Th1 and Th17 Cell Differentiation. Inflammation 2021, 44, 1793–1802. [Google Scholar] [CrossRef]
- Wang, C.; Yang, J.; Xie, L.; Saimaier, K.; Zhuang, W.; Han, M.; Liu, G.; Lv, J.; Shi, G.; Li, N.; et al. Methyl Butyrate Alleviates Experimental Autoimmune Encephalomyelitis and Regulates the Balance of Effector T Cells and Regulatory T Cells. Inflammation 2021. [Google Scholar] [CrossRef]
- Xie, L.; Saimaier, K.; Wang, C.; Yang, J.; Han, M.; Lv, J.; Zhuang, W.; Liu, G.; Du, C. Methyl acetate arrests Th1 in peripheral immune system and alleviates CNS inflammation in EAE. Int. Immunopharmacol. 2021, 101, 108291. [Google Scholar] [CrossRef]
- Wei, W.; Du, C.; Lv, J.; Zhao, G.; Li, Z.; Wu, Z.; Hasko, G.; Xie, X. Blocking A2B adenosine receptor alleviates pathogenesis of experimental autoimmune encephalomyelitis via inhibition of IL-6 production and Th17 differentiation. J. Immunol. 2013, 190, 138–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Locci, A.; Keszycki, R.M.; Orellana, H.D.; Rodriguez, G.; Fisher, D.W. Characterization and comparison of memory and affective behavior in common transgenic mouse models of Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, e045211. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Domingues, H.S.; Mues, M.; Lassmann, H.; Wekerle, H.; Krishnamoorthy, G. Functional and pathogenic differences of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. PLoS ONE 2010, 5, e15531. [Google Scholar] [CrossRef] [Green Version]
- Burgler, S.; Ouaked, N.; Bassin, C.; Basinski, T.M.; Mantel, P.-Y.; Siegmund, K.; Meyer, N.; Akdis, C.A.; Schmidt-Weber, C.B. Differentiation and functional analysis of human TH17 cells. J. Allergy Clin. Immunol. 2009, 123, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Comabella, M.; Montalban, X.; Münz, C.; Lünemann, J.D. Targeting dendritic cells to treat multiple sclerosis. Nat. Rev. Neurol. 2010, 6, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Espay, A.J.; Sturchio, A.; Schneider, L.S.; Ezzat, K. Soluble Amyloid-β Consumption in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 82, 1403–1415. [Google Scholar] [CrossRef]
- Sturchio, A.; Dwivedi, A.K.; Young, C.B.; Malm, T.; Marsili, L.; Sharma, J.S.; Mahajan, A.; Hill, E.J.; Andaloussi, S.E.; Poston, K.L.; et al. High cerebrospinal amyloid-β 42 is associated with normal cognition in individuals with brain amyloidosis. EClinicalMedicine 2021, 38, 100988. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Liu, M.; Zhao, Z.; Yang, C.; Zhu, L.; Cai, Y.; Yang, Y.; Hu, Z. Diagnosis of Mild Cognitive Impairment and Alzheimer’s Disease by the Plasma and Serum Amyloid-beta 42 Assay through Highly Sensitive Peptoid Nanosheet Sensor. ACS Appl. Mater. Interfaces 2020, 12, 9693–9700. [Google Scholar] [CrossRef] [PubMed]
- Lopez, O.L.; Kuller, L.H.; Mehta, P.D.; Becker, J.T.; Gach, H.M.; Sweet, R.A.; Chang, Y.F.; Tracy, R.; DeKosky, S.T. Plasma amyloid levels and the risk of AD in normal subjects in the Cardiovascular Health Study. Neurology 2008, 70, 1664–1671. [Google Scholar] [CrossRef] [Green Version]
- Rissman, R.A.; Trojanowski, J.Q.; Shaw, L.M.; Aisen, P.S. Longitudinal plasma amyloid beta as a biomarker of Alzheimer’s disease. J. Neural Transm. 2012, 119, 843–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Doré, V.; Fowler, C.; Li, Q.-X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Yang, Y.-H.; Situmeang, R.F.; Ong, P.A. Can blood amyloid levels be used as a biomarker for Alzheimer’s disease? Brain Sci. Adv. 2021, 7, 17–25. [Google Scholar] [CrossRef]
- He, J.; Qiao, J.-P.; Zhu, S.; Xue, M.; Chen, W.; Wang, X.; Tempier, A.; Huang, Q.; Kong, J.; Li, X.-M. Serum β-amyloid peptide levels spike in the early stage of Alzheimer-like plaque pathology in an APP/PS1 double transgenic mouse model. Curr. Alzheimer Res. 2013, 10, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, J. Amyloid-β peptide is a substrate of the human 20S proteasome. ACS Chem. Neurosci. 2010, 1, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Flood, F.; Murphy, S.; Cowburn, R.F.; Lannfelt, L.; Walker, B.; Johnston, J.A. Proteasome-mediated effects on amyloid precursor protein processing at the gamma-secretase site. Biochem. J. 2005, 385, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Bellia, F.; Lanza, V.; García-Viñuales, S.; Ahmed, I.M.M.; Pietropaolo, A.; Iacobucci, C.; Malgieri, G.; D’Abrosca, G.; Fattorusso, R.; Nicoletti, V.G.; et al. Ubiquitin binds the amyloid β peptide and interferes with its clearance pathways. Chem. Sci. 2019, 10, 2732–2742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tundo, G.R.; Sbardella, D.; Santoro, A.M.; Coletta, A.; Oddone, F.; Grasso, G.; Milardi, D.; Lacal, P.M.; Marini, S.; Purrello, R.; et al. The proteasome as a druggable target with multiple therapeutic potentialities: Cutting and non-cutting edges. Pharmacol. Ther. 2020, 213, 107579. [Google Scholar] [CrossRef]
- Santoro, A.M.; Lanza, V.; Bellia, F.; Sbardella, D.; Tundo, G.R.; Cannizzo, A.; Grasso, G.; Arizzi, M.; Nicoletti, V.G.; Alcaro, S.; et al. Pyrazolones Activate the Proteasome by Gating Mechanisms and Protect Neuronal Cells from β-Amyloid Toxicity. ChemMedChem 2020, 15, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Zozulya, A.L.; Wiendl, H. The role of regulatory T cells in multiple sclerosis. Nat. Clin. Pract. Neurol. 2008, 4, 384–398. [Google Scholar] [CrossRef]
- Heink, S.; Yogev, N.; Garbers, C.; Herwerth, M.; Aly, L.; Gasperi, C.; Husterer, V.; Croxford, A.L.; Moller-Hackbarth, K.; Bartsch, H.S.; et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH17 cells. Nat. Immunol. 2017, 18, 74–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, Y.; Sakoda, S.; Fujimura, H.; Saeki, Y.; Kishimoto, T.; Yanagihara, T. IL-6 plays a crucial role in the induction phase of myelin oligodendrocyte glucoprotein 35–55 induced experimental autoimmune encephalomyelitis. J. Neuroimmunol. 1999, 101, 188–196. [Google Scholar] [CrossRef]
- Quintana, F.J. Old dog, new tricks: IL-6 cluster signaling promotes pathogenic TH17 cell differentiation. Nat. Immunol. 2016, 18, 8–10. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, C.; Cha, J.; Gong, J.; Wang, S.; Zeng, P.; Lian, J.; Zhang, B.; Hua, Q.; Lv, J.; Du, C.; et al. Amelioration of Experimental Autoimmune Encephalomyelitis in Alzheimer’s Disease Mouse Models: A Potential Role for Aβ. Cells 2022, 11, 1004. https://doi.org/10.3390/cells11061004
Shi C, Cha J, Gong J, Wang S, Zeng P, Lian J, Zhang B, Hua Q, Lv J, Du C, et al. Amelioration of Experimental Autoimmune Encephalomyelitis in Alzheimer’s Disease Mouse Models: A Potential Role for Aβ. Cells. 2022; 11(6):1004. https://doi.org/10.3390/cells11061004
Chicago/Turabian StyleShi, Changjie, Jiaxue Cha, Junyuan Gong, Shaodeng Wang, Peng Zeng, Junjiang Lian, Bowen Zhang, Qiuhong Hua, Jie Lv, Changsheng Du, and et al. 2022. "Amelioration of Experimental Autoimmune Encephalomyelitis in Alzheimer’s Disease Mouse Models: A Potential Role for Aβ" Cells 11, no. 6: 1004. https://doi.org/10.3390/cells11061004
APA StyleShi, C., Cha, J., Gong, J., Wang, S., Zeng, P., Lian, J., Zhang, B., Hua, Q., Lv, J., Du, C., Xie, X., & Zhang, R. (2022). Amelioration of Experimental Autoimmune Encephalomyelitis in Alzheimer’s Disease Mouse Models: A Potential Role for Aβ. Cells, 11(6), 1004. https://doi.org/10.3390/cells11061004