
The Spermidine Synthase Gene SPD1: A Novel Auxotrophic Marker for Chlamydomonas reinhardtii Designed by Enhanced CRISPR/Cas9 Gene Editing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. C. reinhardtii Cultivation and Nuclear Transformation
2.2. CRISPR/Cas9-Based Gene Editing
2.3. Vector Design and Cloning
2.4. Fluorescence Screening and Imaging
2.5. Starch Assay and Calculation of Editing Frequency
2.6. Polyamine Extraction and Quantification
3. Results and Discussion
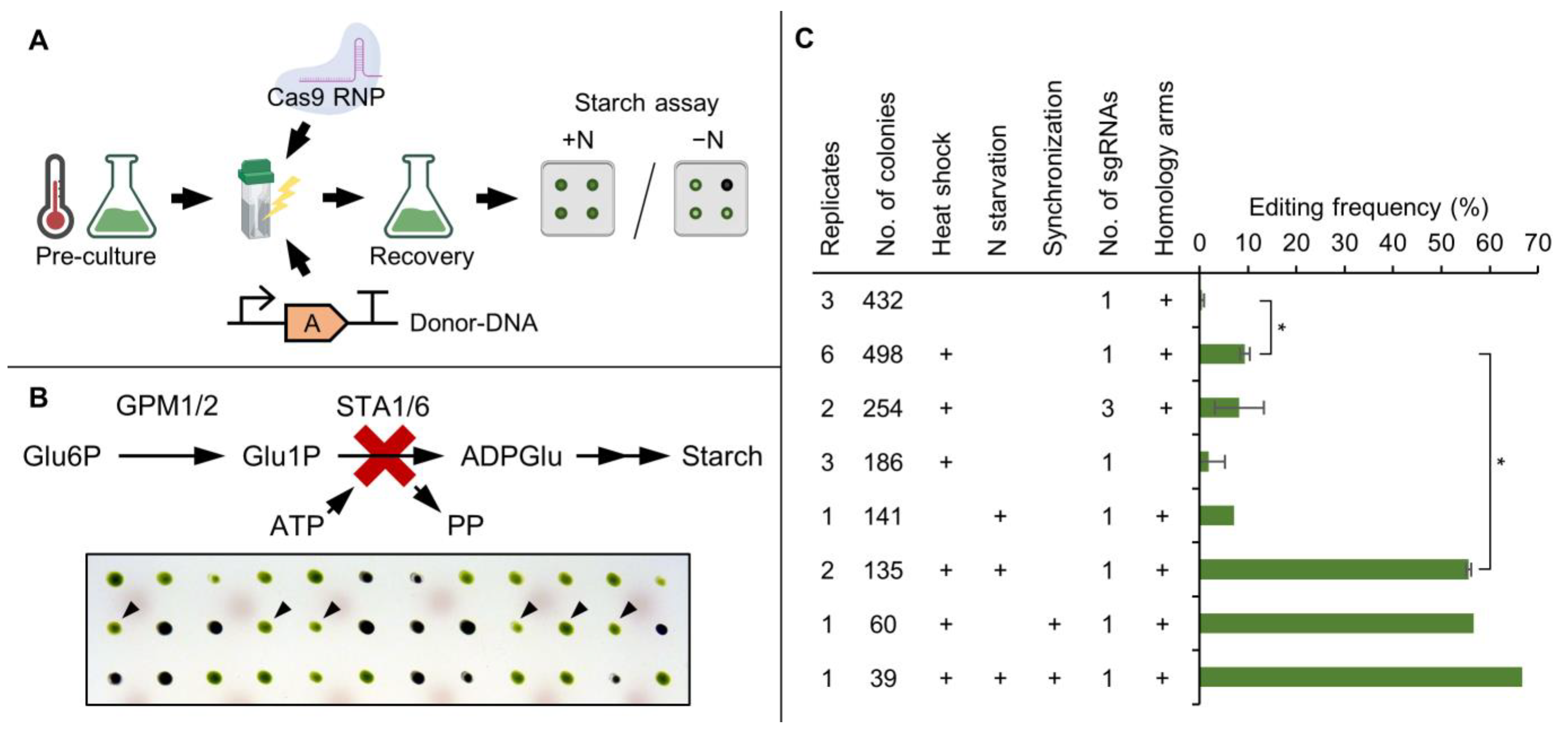
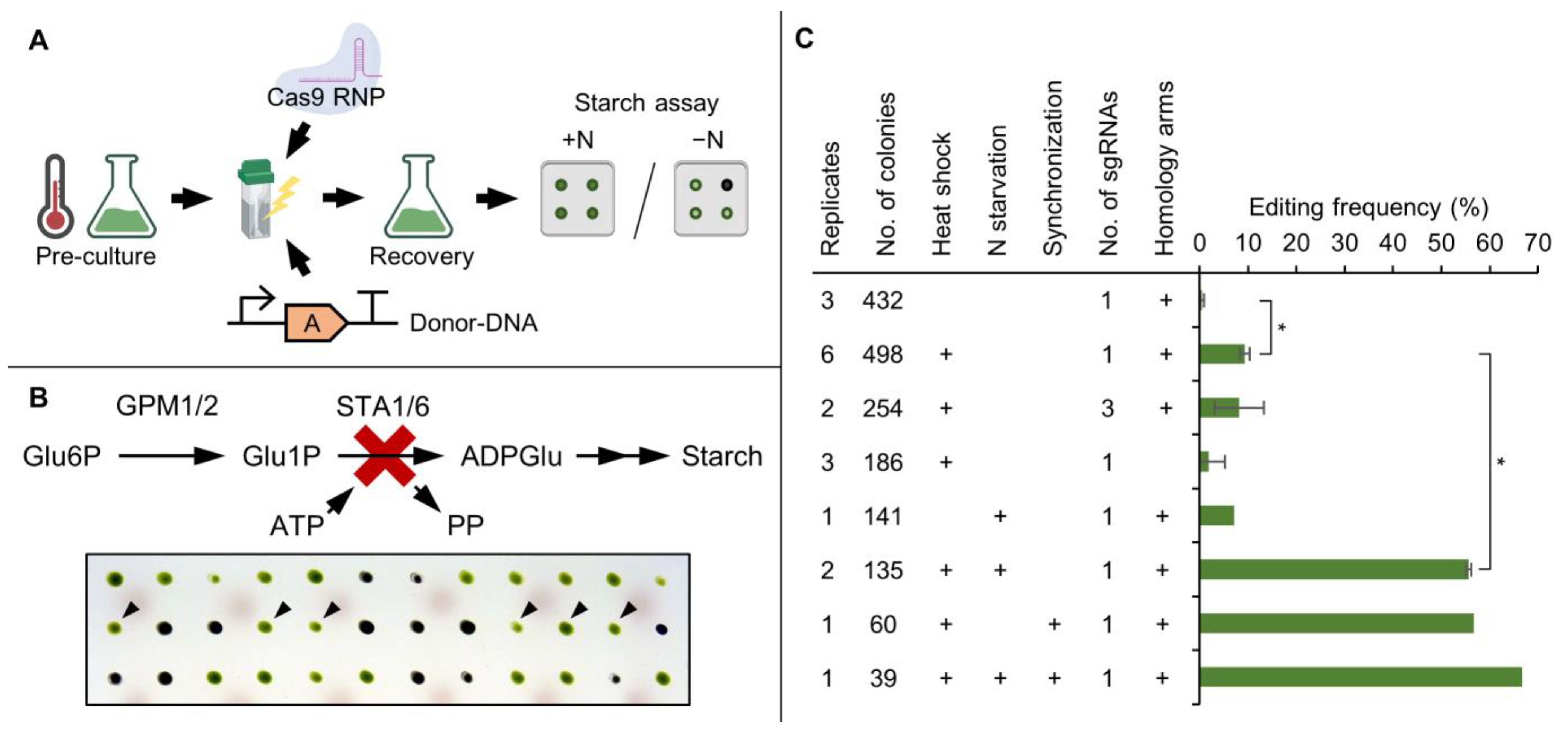
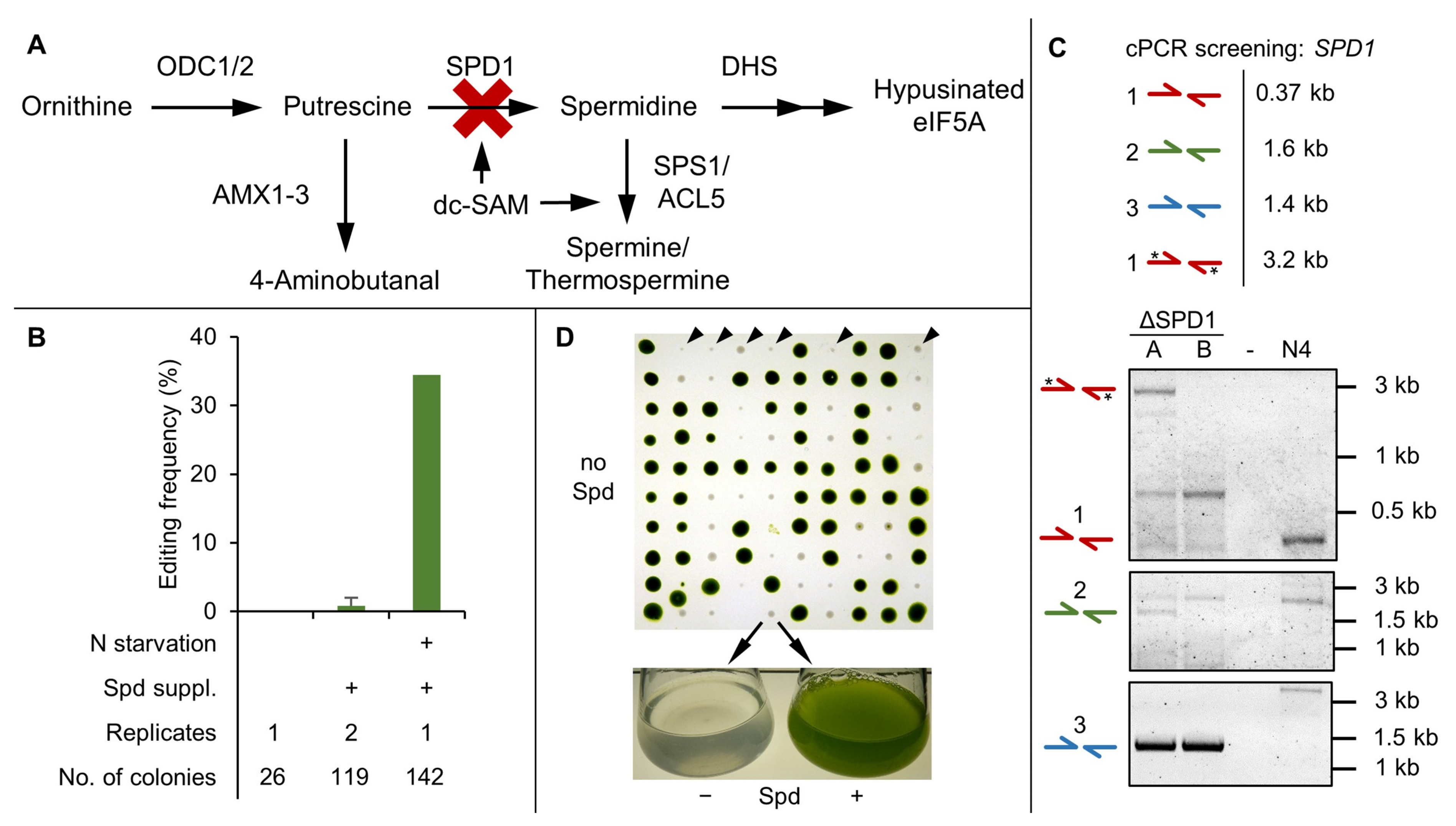
3.1. Nitrogen Starvation of the Pre-Culture Substantially Increases Editing Frequency of CRISPR/Cas9-Based Gene Editing
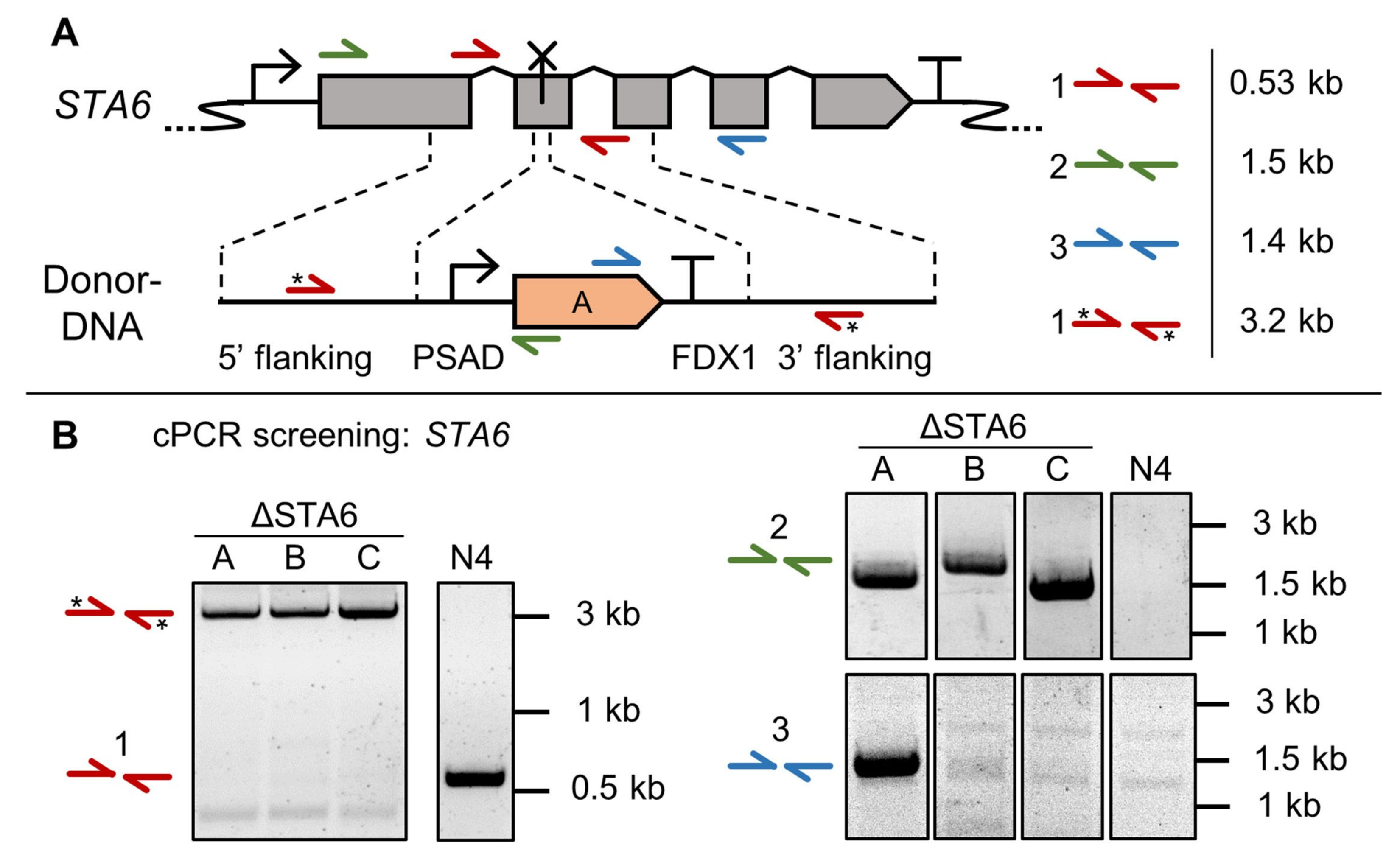
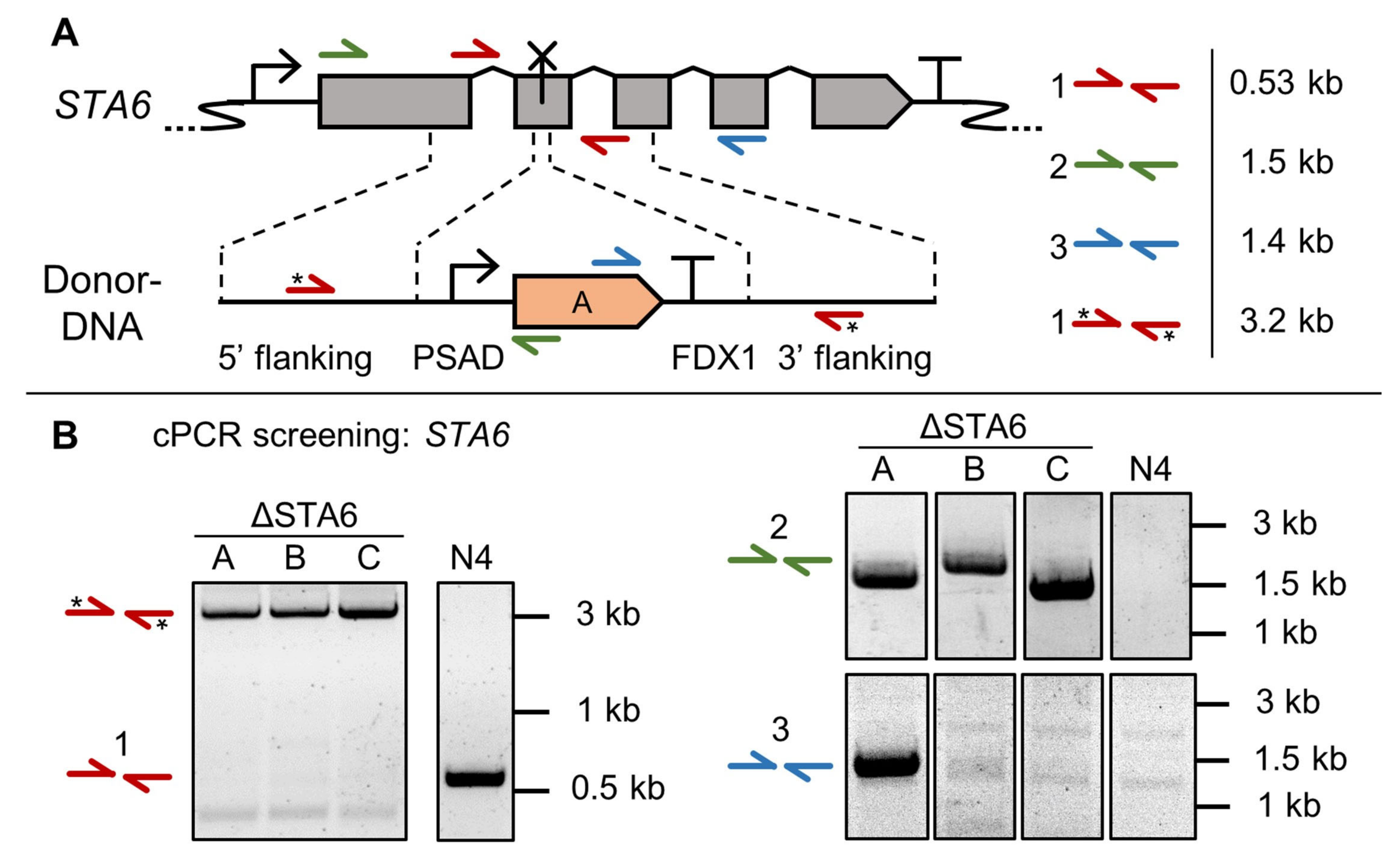
3.2. High Throughput Confirmation of Successful Gene Edits by Colony PCR
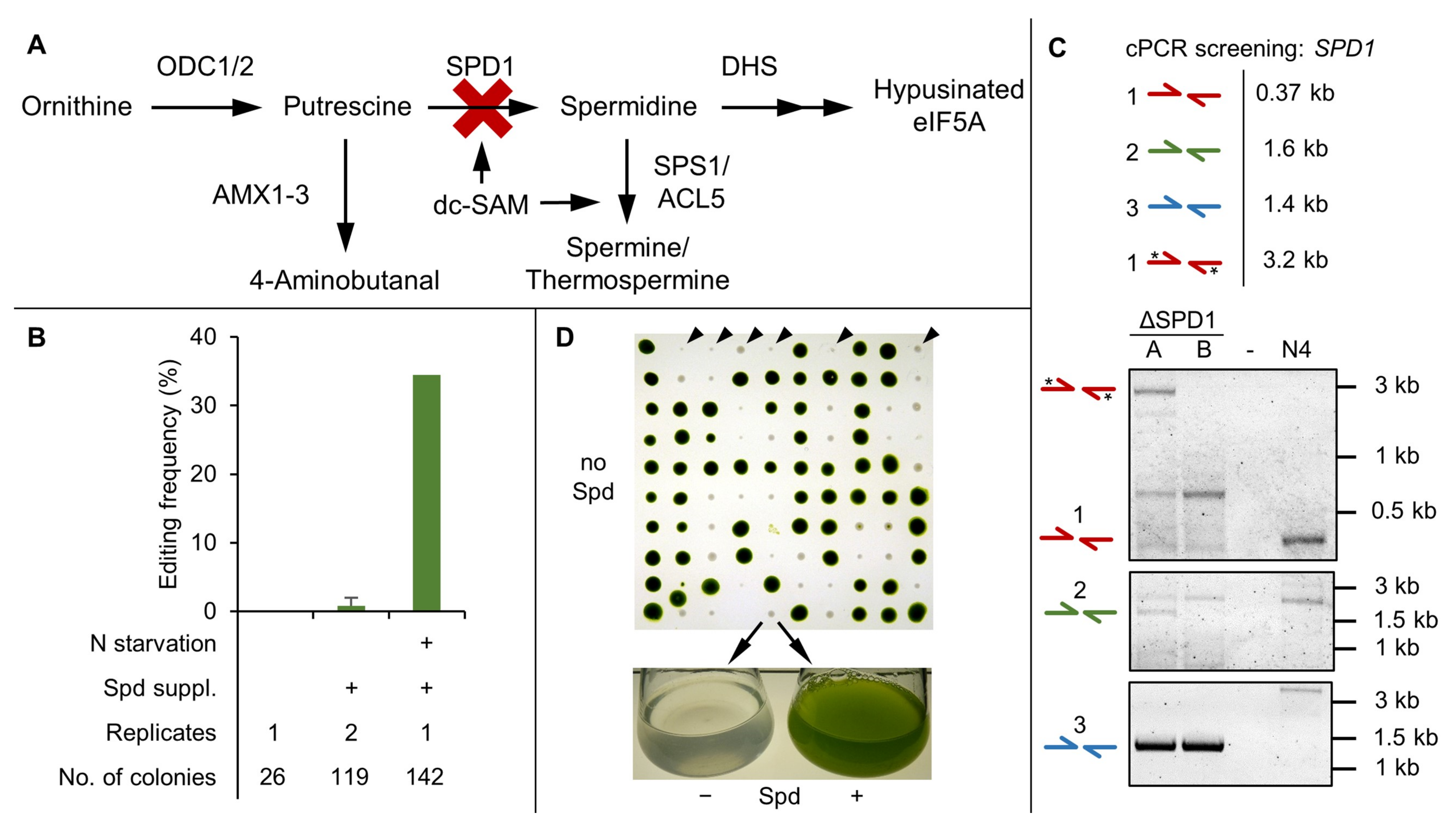
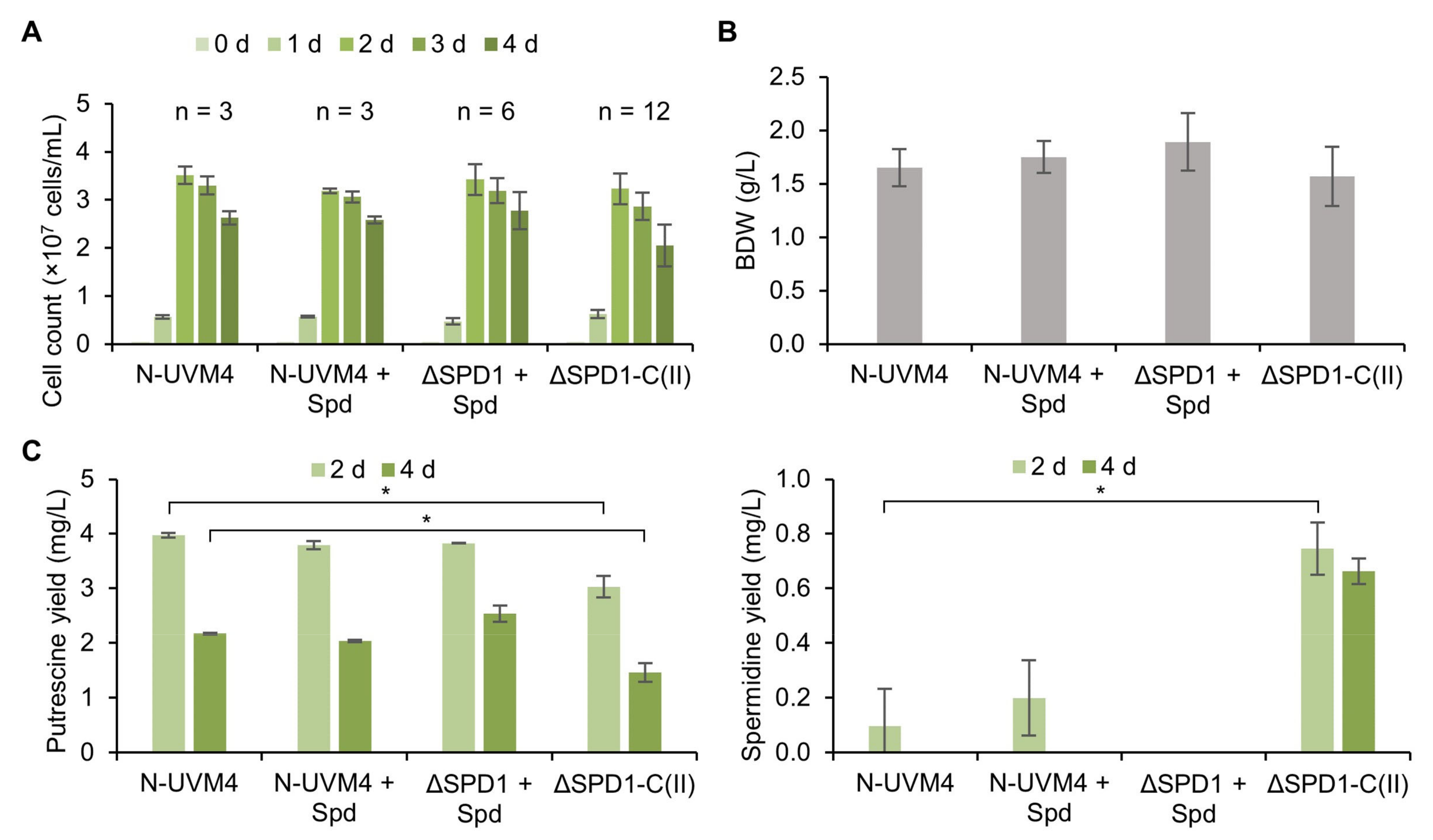
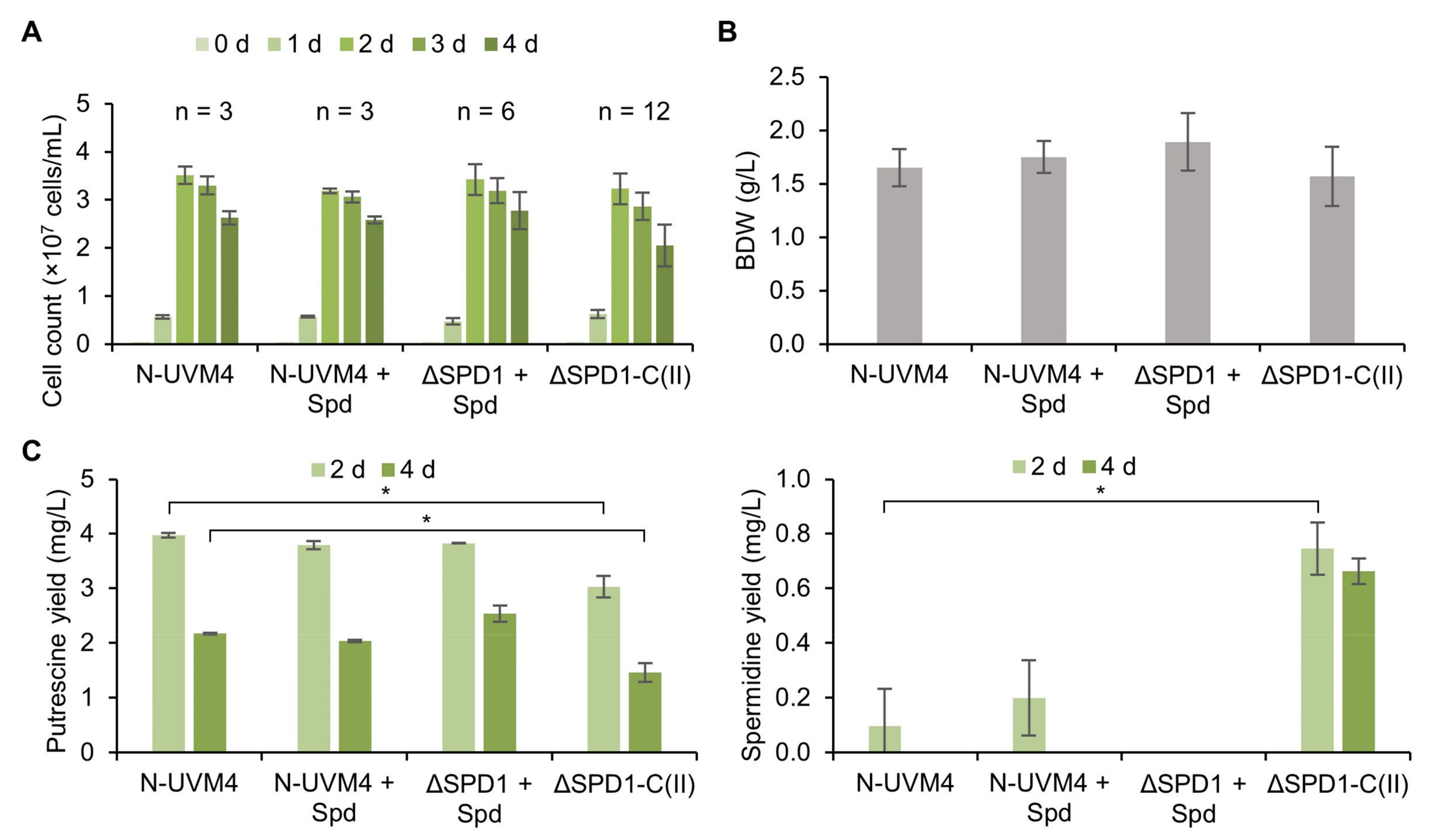
3.3. SPD1 Is Essential for C. reinhardtii and Its Knockout Creates a Novel Spermidine Auxotrophy
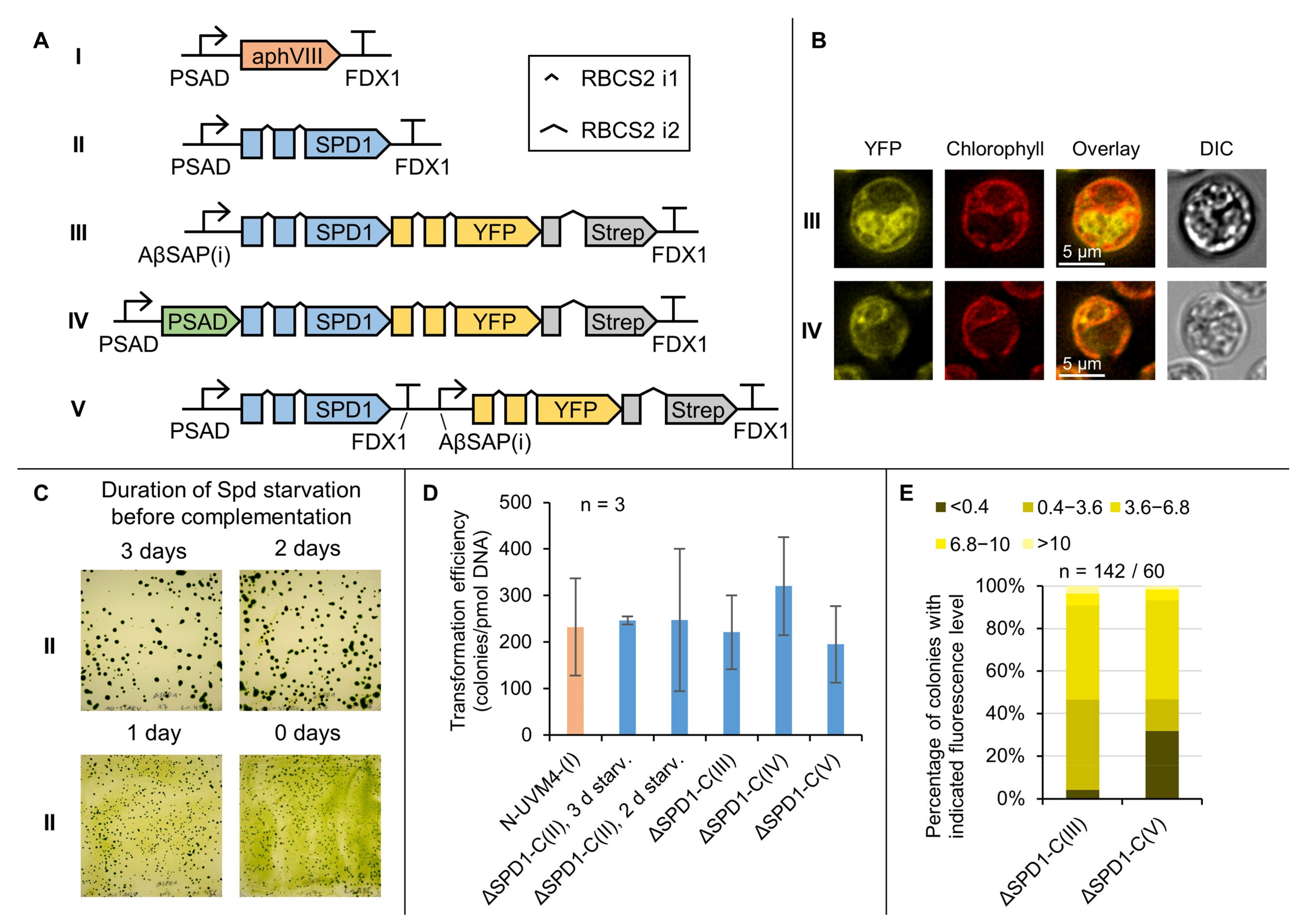
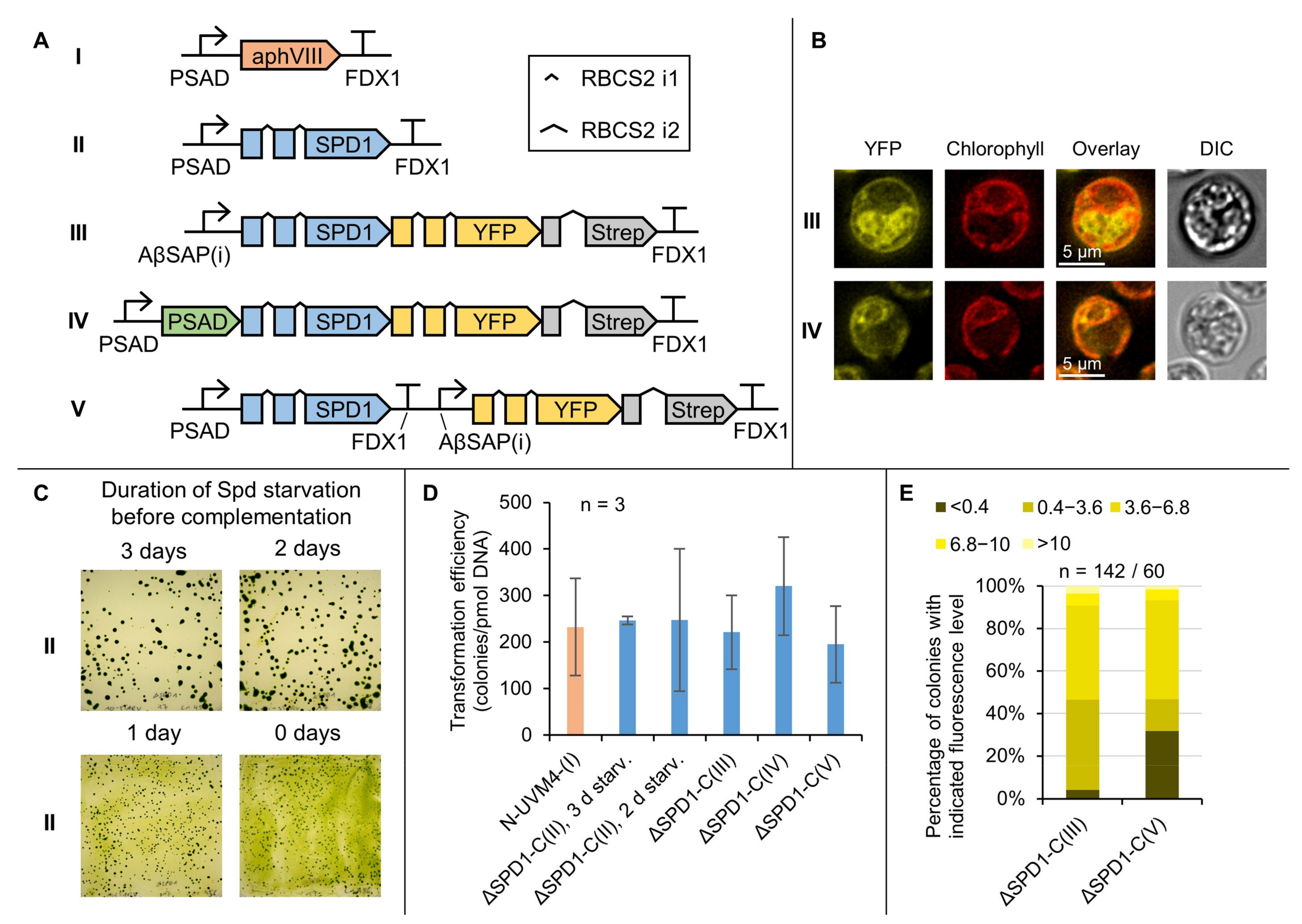
3.4. Complementation of the ΔSPD1 Mutant Has Versatile Biotechnological Utility
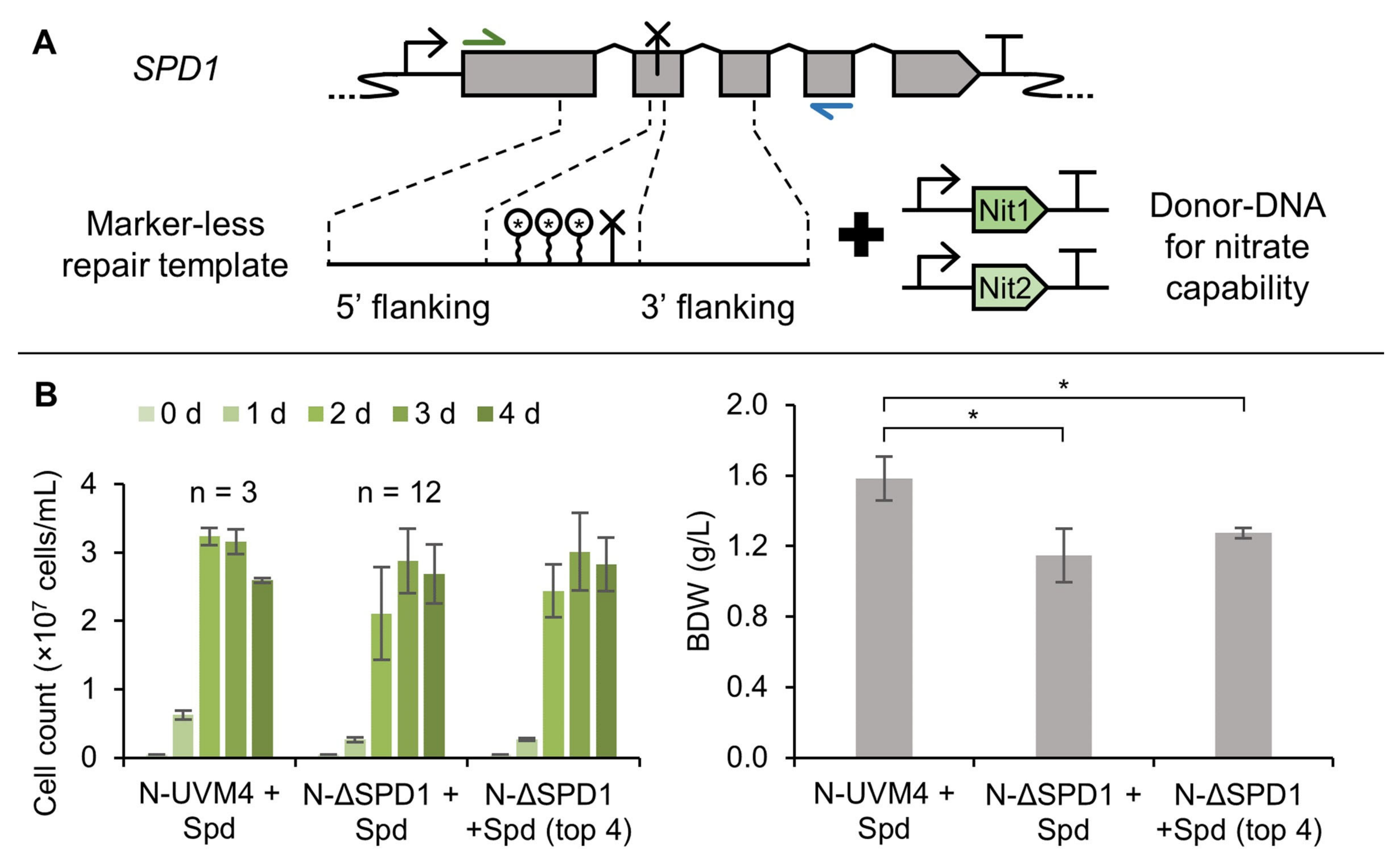
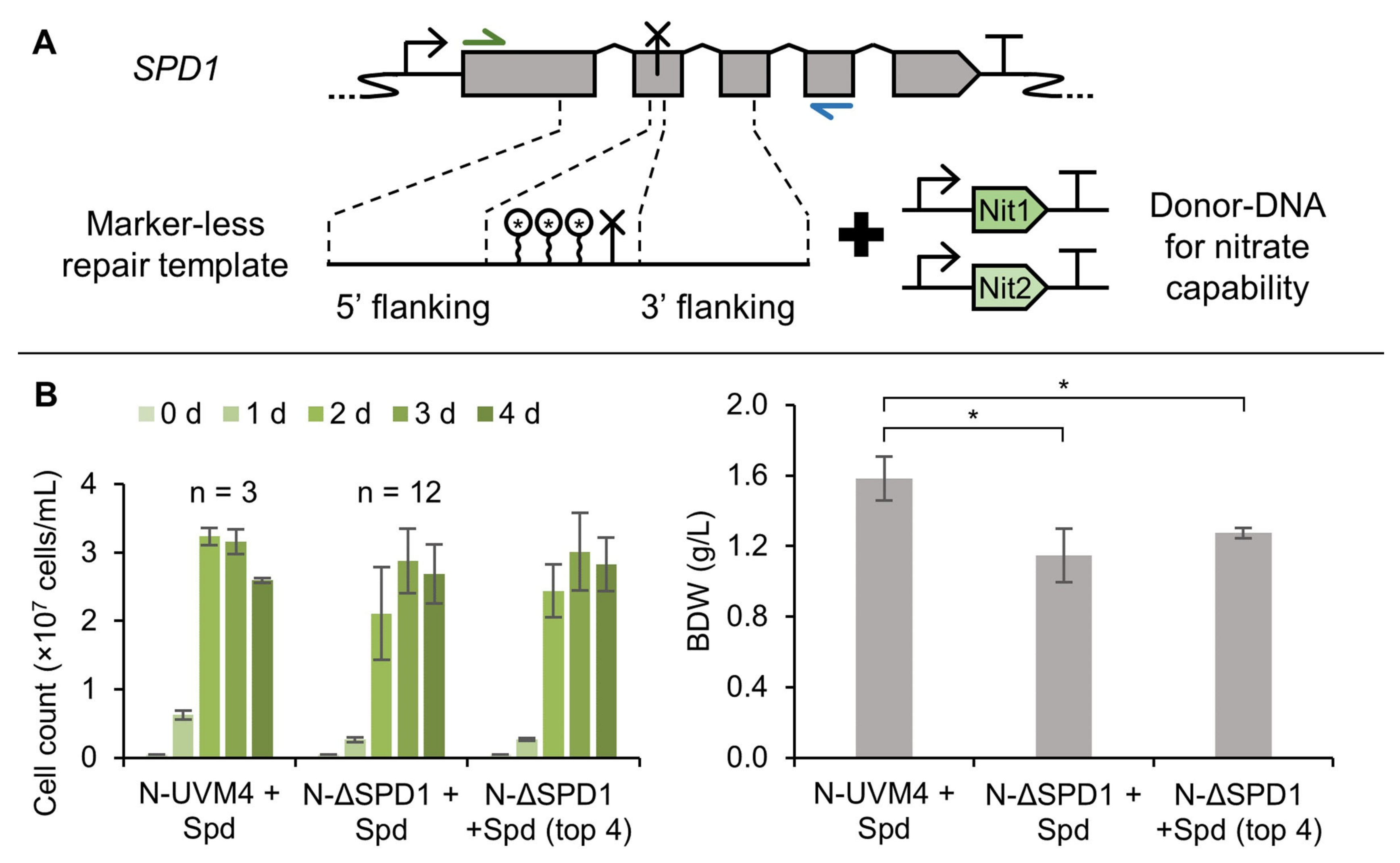
3.5. A Marker-Less ΔSPD1 Mutant Strain
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salomé, P.A.; Merchant, S.S. A Series of Fortunate Events: Introducing Chlamydomonas as a Reference Organism. Plant Cell 2019, 31, 1682–1707. [Google Scholar] [CrossRef] [Green Version]
- Baier, T.; Kros, D.; Feiner, R.C.; Lauersen, K.J.; Müller, K.M.; Kruse, O. Engineered Fusion Proteins for Efficient Protein Secretion and Purification of a Human Growth Factor from the Green Microalga Chlamydomonas reinhardtii. ACS Synth. Biol. 2018, 7, 2547–2557. [Google Scholar] [CrossRef] [PubMed]
- Specht, E.; Miyake-Stoner, S.; Mayfield, S. Micro-algae come of age as a platform for recombinant protein production. Biotechnol. Lett. 2010, 32, 1373–1383. [Google Scholar] [CrossRef] [Green Version]
- Dyo, Y.M.; Purton, S. The algal chloroplast as a synthetic biology platform for production of therapeutic proteins. Microbiol. 2018, 164, 113–121. [Google Scholar] [CrossRef]
- Perozeni, F.; Cazzaniga, S.; Baier, T.; Zanoni, F.; Zoccatelli, G.; Lauersen, K.J.; Wobbe, L.; Ballottari, M. Turning a green alga red: Engineering astaxanthin biosynthesis by intragenic pseudogene revival in Chlamydomonas reinhardtii. Plant Biotechnol. J. 2020, 18, 2053–2067. [Google Scholar] [CrossRef] [Green Version]
- Lauersen, K.J.; Wichmann, J.; Baier, T.; Kampranis, S.C.; Pateraki, I.; Møller, B.L.; Kruse, O. Phototrophic production of heterologous diterpenoids and a hydroxy-functionalized derivative from Chlamydomonas reinhardtii. Metab. Eng. 2018, 49, 116–127. [Google Scholar] [CrossRef]
- Freudenberg, R.A.; Baier, T.; Einhaus, A.; Wobbe, L.; Kruse, O. High cell density cultivation enables efficient and sustainable recombinant polyamine production in the microalga Chlamydomonas reinhardtii. Bioresour. Technol. 2021, 323, 124542. [Google Scholar] [CrossRef] [PubMed]
- Einhaus, A.; Baier, T.; Rosenstengel, M.; Freudenberg, R.A.; Kruse, O. Rational Promoter Engineering Enables Robust Terpene Production in Microalgae. ACS Synth. Biol. 2021, 10, 847–856. [Google Scholar] [CrossRef]
- Baier, T.; Jacobebbinghaus, N.; Einhaus, A.; Lauersen, K.J.; Kruse, O. Introns mediate post-transcriptional enhancement of nuclear gene expression in the green microalga Chlamydomonas reinhardtii. PLoS Genet. 2020, 16, e1008944. [Google Scholar] [CrossRef] [PubMed]
- Crozet, P.; Navarro, F.J.; Willmund, F.; Mehrshahi, P.; Bakowski, K.; Lauersen, K.J.; Pérez-Pérez, M.E.; Auroy, P.; Gorchs Rovira, A.; Sauret-Gueto, S.; et al. Birth of a Photosynthetic Chassis: A MoClo Toolkit Enabling Synthetic Biology in the Microalga Chlamydomonas reinhardtii. ACS Synth. Biol. 2018, 7, 2074–2086. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.S.; Muff, T.J.; Georgianna, D.R.; Mayfield, S.P. Towards a synthetic nuclear transcription system in green algae: Characterization of Chlamydomonas reinhardtii nuclear transcription factors and identification of targeted promoters. Algal Res. 2017, 22, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Scaife, M.A.; Smith, A.G. Towards developing algal synthetic biology. Biochem. Soc. Trans. 2016, 44, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Ghribi, M.; Nouemssi, S.B.; Meddeb-Mouelhi, F.; Desgagné-Penix, I. Genome editing by CRISPR-Cas: A game change in the genetic manipulation of chlamydomonas. Life 2020, 10, 295. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.T.; Kaldenhoff, R. Achievements and challenges of genetic engineering of the model green alga Chlamydomonas reinhardtii. Algal Res. 2020, 50, 101986. [Google Scholar] [CrossRef]
- Liao, H.; Li, X.; Yang, Q.; Bai, Y.; Cui, P.; Wen, C.; Liu, C.; Chen, Z.; Tang, J.; Che, J.; et al. Herbicide Selection Promotes Antibiotic Resistance in Soil Microbiomes. Mol. Biol. Evol. 2021, 38, 2337–2350. [Google Scholar] [CrossRef]
- Wang, W.; Arshad, M.I.; Khurshid, M.; Rasool, M.H.; Nisar, M.A.; Aslam, M.A.; Qamar, M.U. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar]
- Wang, F.; Zhang, W. Synthetic biology: Recent progress, biosafety and biosecurity concerns, and possible solutions. J. Biosaf. Biosecurity 2019, 1, 22–30. [Google Scholar] [CrossRef]
- Lee, J.W.; Chan, C.T.Y.; Slomovic, S.; Collins, J.J. Next-generation biocontainment systems for engineered organisms. Nat. Chem. Biol. 2018, 14, 530–537. [Google Scholar] [CrossRef]
- Murbach, T.S.; Glávits, R.; Endres, J.R.; Hirka, G.; Vértesi, A.; Béres, E.; Szakonyiné, I.P. A Toxicological Evaluation of Chlamydomonas reinhardtii, a Green Algae. Int. J. Toxicol. 2018, 37, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.-E.E.; Lim, J.-M.M.; Koh, H.G.; Kim, E.K.; Kang, N.K.; Jeon, S.; Kwon, S.; Shin, W.-S.S.; Lee, B.; Hwangbo, K.; et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Sci. Rep. 2016, 6, 27810. [Google Scholar] [CrossRef]
- Baek, K.; Kim, D.H.; Jeong, J.; Sim, S.J.; Melis, A.; Kim, J.-S.; Jin, E.; Bae, S. DNA-free two-gene knockout in Chlamydomonas reinhardtii via CRISPR-Cas9 ribonucleoproteins. Sci. Rep. 2016, 6, 30620. [Google Scholar] [CrossRef]
- Slaninová, M.; Hroššová, D.; Vlček, D.; Mages, W. Is it possible to improve homologous recombination in Chlamydomonas reinhardtii? Biologia 2008, 63, 941–946. [Google Scholar] [CrossRef]
- Nour-Eldin, H.H.; Specht, E.A.; Ostrand, J.; Hoang, K.T.D.; Karunanithi, P.S.; Mayfield, S.P. High-throughput system for quantifying and characterizing homologous recombination in Chlamydomonas reinhardtii. Algal Res. 2018, 31, 167–172. [Google Scholar] [CrossRef]
- Picariello, T.; Hou, Y.; Kubo, T.; McNeill, N.A.; Yanagisawa, H.A.; Oda, T.; Witman, G.B. TIM, a targeted insertional mutagenesis method utilizing CRISPR/Cas9 in Chlamydomonas reinhardtii. PLoS ONE 2020, 15, e0232594. [Google Scholar] [CrossRef] [PubMed]
- Dhokane, D.; Bhadra, B.; Dasgupta, S. CRISPR based targeted genome editing of Chlamydomonas reinhardtii using programmed Cas9-gRNA ribonucleoprotein. Mol. Biol. Rep. 2020, 47, 8747–8755. [Google Scholar] [CrossRef] [PubMed]
- Greiner, A.; Kelterborn, S.; Evers, H.; Kreimer, G.; Sizova, I.; Hegemann, P. Targeting of photoreceptor genes in Chlamydomonas reinhardtii via zinc-finger nucleases and CRISPR/Cas9. Plant Cell 2017, 29, 2498–2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angstenberger, M.; De Signori, F.; Vecchi, V.; Dall’Osto, L.; Bassi, R. Cell Synchronization Enhances Nuclear Transformation and Genome Editing via Cas9 Enabling Homologous Recombination in Chlamydomonas reinhardtii. ACS Synth. Biol. 2020, 9, 2840–2850. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.H.; Stern, D.; Witman, G. The Chlamydomonas Sourcebook; Academic Press: Oxford, UK, 2009; Volume 1, ISBN 9780123708731. [Google Scholar]
- Dutcher, S.K.; Galloway, R.E.; Barclay, W.R.; Poortinga, G. Tryptophan analog resistance mutations in Chlamydomonas reinhardtii. Genetics 1992, 131, 593–607. [Google Scholar] [CrossRef]
- Eversole, R.A. Biochemical Mutants of Chlamydomonas reinhardi. Am. J. Bot. 1956, 43, 404. [Google Scholar] [CrossRef]
- Matagne, R.F.; Remacle, C.; Dinant, M. Cytoduction in Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 1991, 88, 7447–7450. [Google Scholar] [CrossRef] [Green Version]
- Helliwell, K.E.; Collins, S.; Kazamia, E.; Purton, S.; Wheeler, G.L.; Smith, A.G. Fundamental shift in vitamin B12 eco-physiology of a model alga demonstrated by experimental evolution. ISME J. 2015, 9, 1446–1455. [Google Scholar] [CrossRef] [Green Version]
- Bunbury, F.; Helliwell, K.E.; Mehrshahi, P.; Davey, M.P.; Salmon, D.L.; Holzer, A.; Smirnoff, N.; Smitha, A.G. Responses of a newly evolved auxotroph of chlamydomonas to B12 deprivation. Plant Physiol. 2020, 183, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.Y.; Lin, H.J. Polyamines in microalgae: Something borrowed, something new. Mar. Drugs 2019, 17, 1. [Google Scholar] [CrossRef] [Green Version]
- Miller-Fleming, L.; Olin-Sandoval, V.; Campbell, K.; Ralser, M. Remaining Mysteries of Molecular Biology: The Role of Polyamines in the Cell. J. Mol. Biol. 2015, 427, 3389–3406. [Google Scholar] [CrossRef]
- Michael, A.J. Polyamines in eukaryotes, bacteria, and archaea. J. Biol. Chem. 2016, 291, 14896–14903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theiss, C.; Bohley, P.; Bisswanger, H.; Voigt, J. Uptake of polyamines by the unicellular green alga Chlamydomonas reinhardtii and their effect on ornithine decarboxylase activity. J. Plant Physiol. 2004, 161, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Hamana, K.; Aizaki, T.; Arai, E.; Saito, A.; Uchikata, K.; Ohnishi, H. Distribution of norspermidine as a cellular polyamine within micro green algae including non-photosynthetic achlorophyllous Polytoma, Polytomella, Prototheca and Helicosporidium. J. Gen. Appl. Microbiol. 2004, 50, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Tassoni, A.; Awad, N.; Griffiths, G. Effect of ornithine decarboxylase and norspermidine in modulating cell division in the green alga Chlamydomonas reinhardtii. Plant Physiol. Biochem. 2018, 123, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solé-Gil, A.; Hernández-García, J.; López-Gresa, M.P.; Blázquez, M.A.; Agustí, J. Conservation of thermospermine synthase activity in vascular and non-vascular plants. Front. Plant Sci. 2019, 10, 663. [Google Scholar] [CrossRef]
- Serif, M.; Dubois, G.; Finoux, A.L.; Teste, M.A.; Jallet, D.; Daboussi, F. One-step generation of multiple gene knock-outs in the diatom Phaeodactylum tricornutum by DNA-free genome editing. Nat. Commun. 2018, 9, 3924. [Google Scholar] [CrossRef] [Green Version]
- Neupert, J.; Karcher, D.; Bock, R. Generation of Chlamydomonas strains that efficiently express nuclear transgenes. Plant J. 2009, 57, 1140–1150. [Google Scholar] [CrossRef]
- Gorman, D.S.; Levine, R.P. Cytochrome f and plastocyanin: Their sequence in the photosynthetic electron transport chain of Chlamydomonas reinhardi. Proc. Natl. Acad. Sci. USA 1965, 54, 1665–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kropat, J.; Hong-Hermesdorf, A.; Casero, D.; Ent, P.; Castruita, M.; Pellegrini, M.; Merchant, S.S.; Malasarn, D. A revised mineral nutrient supplement increases biomass and growth rate in Chlamydomonas reinhardtii. Plant J. 2011, 66, 770–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauersen, K.J.; Baier, T.; Wichmann, J.; Wördenweber, R.; Mussgnug, J.H.; Hübner, W.; Huser, T.; Kruse, O. Efficient phototrophic production of a high-value sesquiterpenoid from the eukaryotic microalga Chlamydomonas reinhardtii. Metab. Eng. 2016, 38, 331–343. [Google Scholar] [CrossRef]
- Kindle, K.L. High-frequency nuclear transformation of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 1990, 87, 1228–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Lee, S.; Baek, K.; Jin, E. Site-Specific Gene Knock-Out and On-Site Heterologous Gene Overexpression in Chlamydomonas reinhardtii via a CRISPR-Cas9-Mediated Knock-in Method. Front. Plant Sci. 2020, 11, 306. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Bae, S.; Kim, J.S. Cas-Designer: A web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics 2015, 31, 4014–4016. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yang, L.; Wen, X.; Chen, Z.; Liang, Q.; Li, J.; Wang, W. Rapid and high efficiency transformation of Chlamydomonas reinhardtii by square-wave electroporation. Biosci. Rep. 2019, 39, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Cao, M.; Fu, Y.; Guo, Y.; Pan, J. Chlamydomonas (Chlorophyceae) colony PCR. Protoplasma 2009, 235, 107–110. [Google Scholar] [CrossRef]
- Fischer, N.; Rochaix, J.-D. The flanking regions of PsaD drive efficient gene expression in the nucleus of the green alga Chlamydomonas reinhardtii. Mol. Genet. Genom. 2001, 265, 888–894. [Google Scholar] [CrossRef]
- López-Paz, C.; Liu, D.; Geng, S.; Umen, J.G. Identification of Chlamydomonas reinhardtii endogenous genic flanking sequences for improved transgene expression. Plant J. 2017, 92, 1232–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baier, T.; Wichmann, J.; Kruse, O.; Lauersen, K.J. Intron-containing algal transgenes mediate efficient recombinant gene expression in the green microalga Chlamydomonas reinhardtii. Nucleic Acids Res. 2018, 46, 6909–6919. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, D.; Baier, T.; Lauersen, K.J. Intronserter, an advanced online tool for design of intron containing transgenes. Algal Res. 2019, 42, 101588. [Google Scholar] [CrossRef]
- Wichmann, J.; Baier, T.; Wentnagel, E.; Lauersen, K.J.; Kruse, O. Tailored carbon partitioning for phototrophic production of (E)-α-bisabolene from the green microalga Chlamydomonas reinhardtii. Metab. Eng. 2018, 45, 211–222. [Google Scholar] [CrossRef]
- Ferenczi, A.; Pyott, D.E.; Xipnitou, A.; Molnar, A.; Merchant, S.S. Efficient targeted DNA editing and replacement in Chlamydomonas reinhardtii using Cpf1 ribonucleoproteins and single-stranded DNA. Proc. Natl. Acad. Sci. USA 2017, 114, 13567–13572. [Google Scholar] [CrossRef] [Green Version]
- Zabawinski, C.; Van den Koornhuyse, N.; D’Hulst, C.; Schlichting, R.; Giersch, C.; Delrue, B.; Lacroix, J.M.; Preiss, J.; Ball, S. Starchless mutants of Chlamydomonas reinhardtii lack the small subunit of a heterotetrameric ADP-glucose pyrophosphorylase. J. Bacteriol. 2001, 183, 1069–1077. [Google Scholar] [CrossRef] [Green Version]
- Park, J.J.; Wang, H.; Gargouri, M.; Deshpande, R.R.; Skepper, J.N.; Holguin, F.O.; Juergens, M.T.; Shachar-Hill, Y.; Hicks, L.M.; Gang, D.R. The response of Chlamydomonas reinhardtii to nitrogen deprivation: A systems biology analysis. Plant J. 2015, 81, 611–624. [Google Scholar] [CrossRef]
- Martin, N.C.; Goodenough, U.W. Gametic differentiation in Chlamydomonas reinhardtii. I. Production of gametes and their fine structure. J. Cell Biol. 1975, 67, 587–605. [Google Scholar] [CrossRef]
- Sathe, S. Temporal and metabolic overlap between lipid acucmulation and programmed cell death due to nitrogen starvation in the unicellular chlorophyte Chlamydomonoas reinhardtii. Phycol. Res. 2019, 67, 173–183. [Google Scholar] [CrossRef]
- Won, M.; Dawid, I.B. PCR artifact in testing for homologous recombination in genomic editing in zebrafish. PLoS ONE 2017, 12, e0172802. [Google Scholar] [CrossRef] [Green Version]
- Theiss, C.; Bohley, P. Regulation by Polyamines of Ornithine Decarboxylase Activity and Cell Division in the Unicellular Green Alga Chlamydomonas reinhardtii. Plant Physiol. 2002, 128, 1470–1479. [Google Scholar] [CrossRef] [Green Version]
- Alcázar, R.; Altabella, T.; Marco, F.; Bortolotti, C.; Reymond, M.; Koncz, C.; Carrasco, P.; Tiburcio, A.F. Polyamines: Molecules with regulatory functions in plant abiotic stress tolerance. Planta 2010, 231, 1237–1249. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Shao, Q.; Yin, L.; Younis, A.; Zheng, B. Polyamine Function in Plants: Metabolism, Regulation on Development, and Roles in Abiotic Stress Responses. Front. Plant Sci. 2019, 9, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Nishimura, K.; Zanelli, C.F.; Valentini, S.R. Functional significance of eIF5A and its hypusine modification in eukaryotes. Amino Acids 2010, 38, 491–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuller, A.P.; Wu, C.C.C.; Dever, T.E.; Buskirk, A.R.; Green, R. eIF5A Functions Globally in Translation Elongation and Termination. Mol. Cell 2017, 66, 194.e5–205.e5. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, M.K.; Myung, H.P.; Tabor, H. Hypusine modification for growth is the major function of spermidine in Saccharomyces cerevisiae polyamine auxotrophs grown in limiting spermidine. Proc. Natl. Acad. Sci. USA 2008, 105, 6554–6559. [Google Scholar] [CrossRef] [Green Version]
- Poidevin, L.; Unal, D.; Belda-Palazón, B.; Ferrando, A. Polyamines as quality control metabolites operating at the post-transcriptional level. Plants 2019, 8, 109. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Russel, D.W. Molecular Cloning. A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; Volume 1, ISBN 0-87969-576-5. [Google Scholar]
- Tardif, M.; Atteia, A.; Specht, M.; Cogne, G.; Rolland, N.; Brugière, S.; Hippler, M.; Ferro, M.; Bruley, C.; Peltier, G.; et al. Predalgo: A new subcellular localization prediction tool dedicated to green algae. Mol. Biol. Evol. 2012, 29, 3625–3639. [Google Scholar] [CrossRef] [Green Version]
- Shu, S.; Guo, S.-R.; Yu, L.-Y. A Review: Polyamines and Photosynthesis. In Advances in Photosynthesis—Fundamental Aspects; InTech: London, UK, 2012; pp. 439–464. [Google Scholar]
- Fernández, E.; Schnell, R.; Ranum, L.P.; Hussey, S.C.; Silflow, C.D.; Lefebvre, P.A. Isolation and characterization of the nitrate reductase structural gene of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 1989, 86, 6449–6453. [Google Scholar] [CrossRef] [Green Version]
- Schnell, R.A.; Lefebvre, P.A. Isolation of the chlamydomonas regulatory gene NIT2 by transposon tagging. Genetics 1993, 134, 737–747. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freudenberg, R.A.; Wittemeier, L.; Einhaus, A.; Baier, T.; Kruse, O. The Spermidine Synthase Gene SPD1: A Novel Auxotrophic Marker for Chlamydomonas reinhardtii Designed by Enhanced CRISPR/Cas9 Gene Editing. Cells 2022, 11, 837. https://doi.org/10.3390/cells11050837
Freudenberg RA, Wittemeier L, Einhaus A, Baier T, Kruse O. The Spermidine Synthase Gene SPD1: A Novel Auxotrophic Marker for Chlamydomonas reinhardtii Designed by Enhanced CRISPR/Cas9 Gene Editing. Cells. 2022; 11(5):837. https://doi.org/10.3390/cells11050837
Chicago/Turabian StyleFreudenberg, Robert A., Luisa Wittemeier, Alexander Einhaus, Thomas Baier, and Olaf Kruse. 2022. "The Spermidine Synthase Gene SPD1: A Novel Auxotrophic Marker for Chlamydomonas reinhardtii Designed by Enhanced CRISPR/Cas9 Gene Editing" Cells 11, no. 5: 837. https://doi.org/10.3390/cells11050837
APA StyleFreudenberg, R. A., Wittemeier, L., Einhaus, A., Baier, T., & Kruse, O. (2022). The Spermidine Synthase Gene SPD1: A Novel Auxotrophic Marker for Chlamydomonas reinhardtii Designed by Enhanced CRISPR/Cas9 Gene Editing. Cells, 11(5), 837. https://doi.org/10.3390/cells11050837