
Ca2+ Transportome and the Interorganelle Communication in Hepatocellular Carcinoma

 ,
,  and
and
Abstract
:1. Introduction
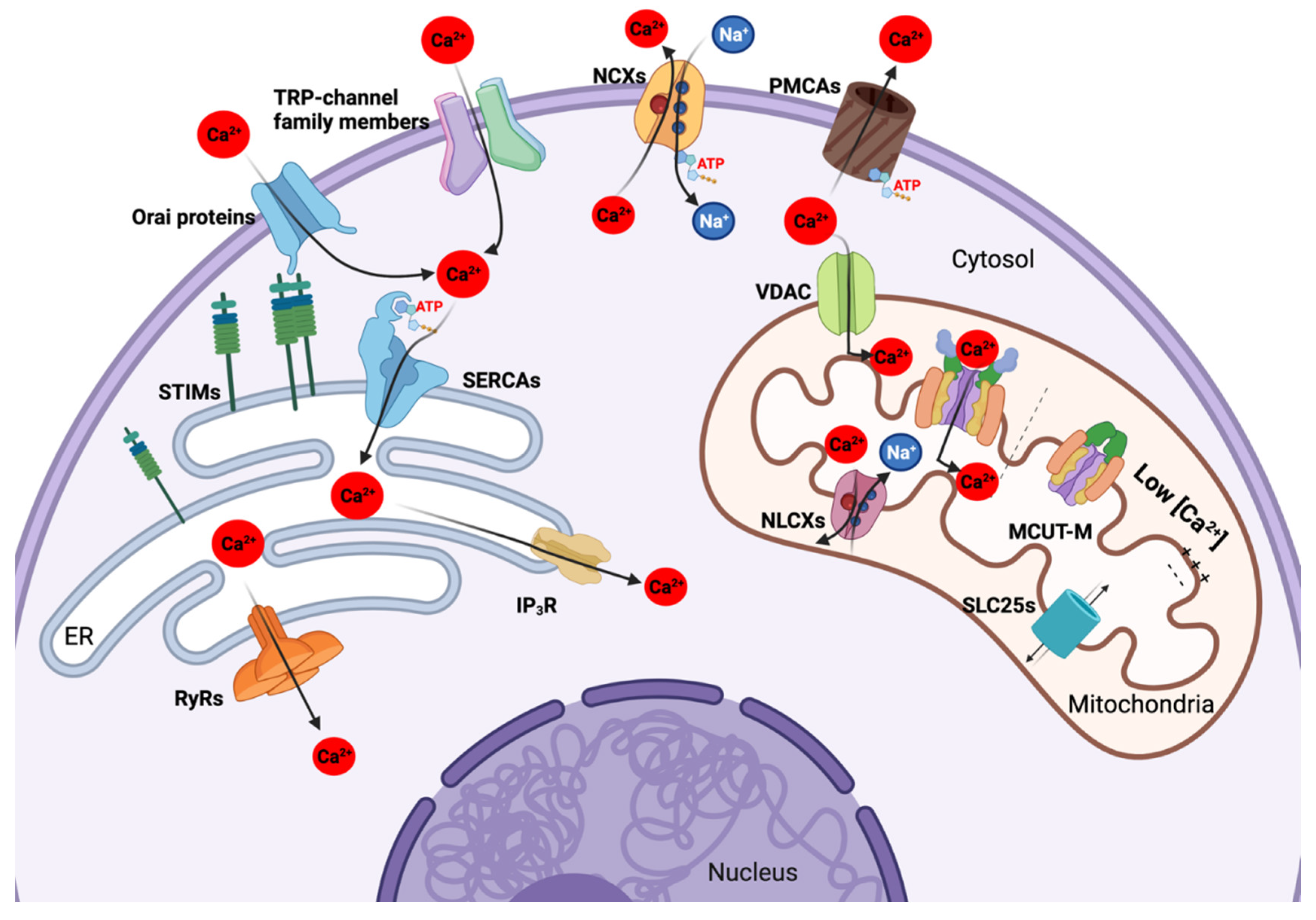
2. Ca2+ Transportome Composition and Function
2.1. Plasma Membrane Ca2+ Transportome
2.1.1. Ca2+-Permeable Ion Channels of the Plasma Membrane
2.1.2. Energy-Dependent Ca2+ Channels and Ca2+ Extrusion Systems of the Plasma Membrane
2.2. The Ca2+ Transportome of the Endoplasmic Reticulum
2.2.1. Ca2+ Permeable Efflux Transporters in the ER
2.2.2. Energy-Dependent ER Ca2+ Influx Transporters
2.2.3. Stromal Interaction Molecule 1 (STIM1)-Mediated Store-Operated Ca2+ Entry (SOCE) Mechanism
2.3. Mitochondrial Ca2+ Transportome
2.3.1. Mitochondrial Ca2+ Uptake Machinery
2.3.2. Other Transporters in Mitochondria
3. Deregulation of the Ca2+ Transportome and Consequences on HCC
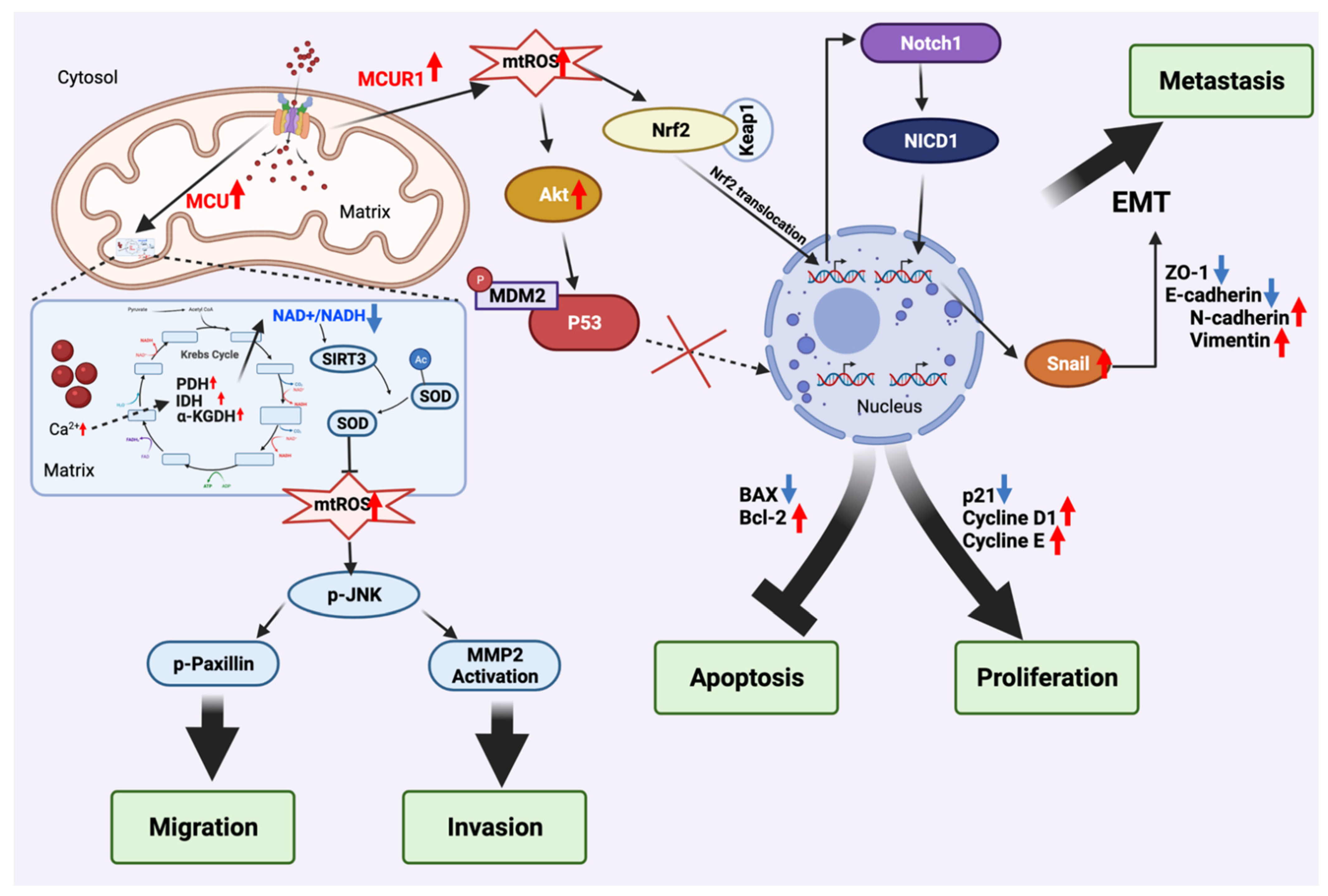
3.1. Mitochondria Ca2+ Uptake Machinery (MCUT-M)
3.1.1. Mitochondrial Ca2+ Uniporter (MCU)
3.1.2. Mitochondrial Ca2+ Uniporter Regulator 1 (MCUR1)
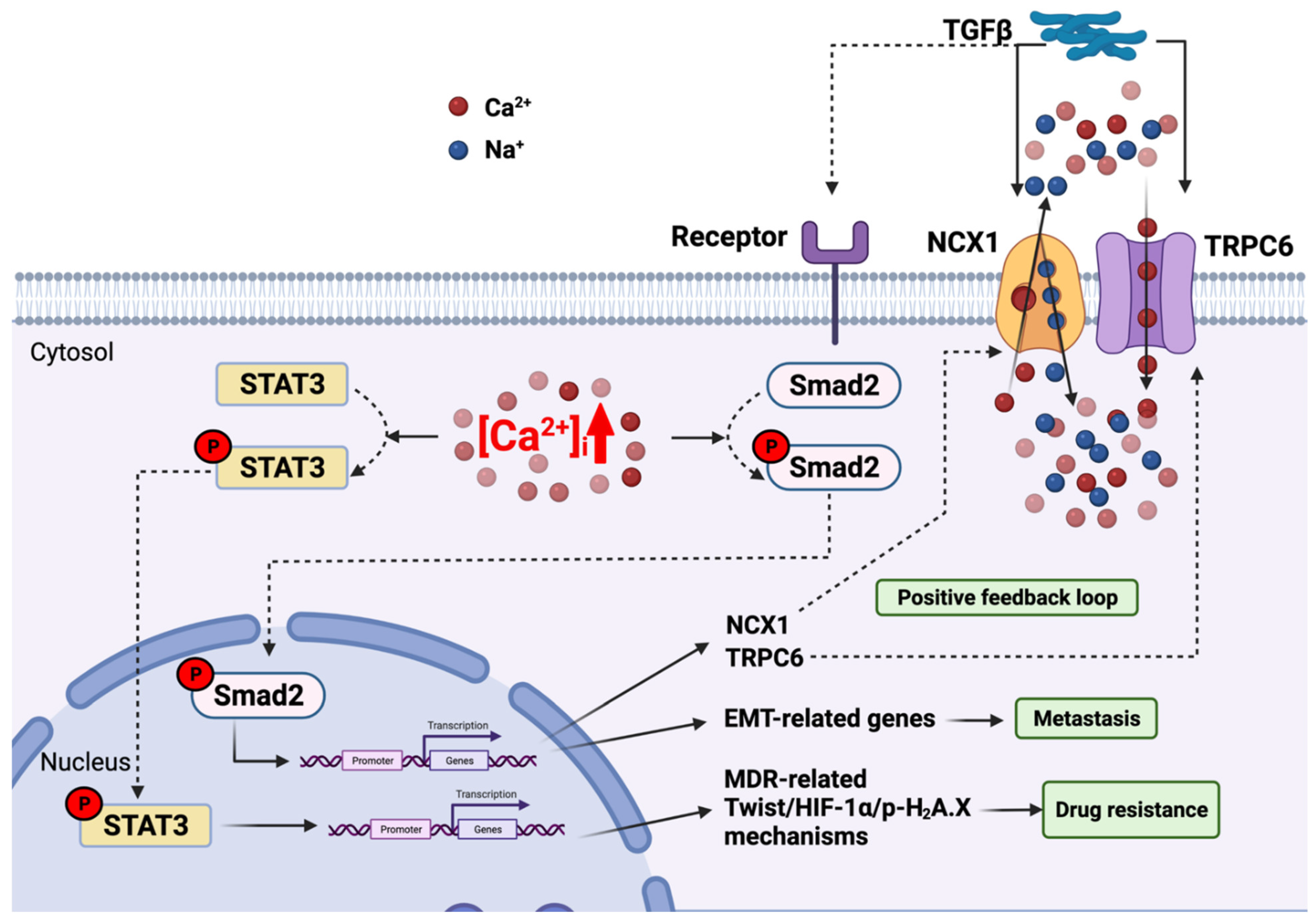
3.2. Plasma Membrane Channel—Transient Receptor Potential Cation Channel Subfamily C Member 6 (TRPC6)—Link with the TGFβ Pathway
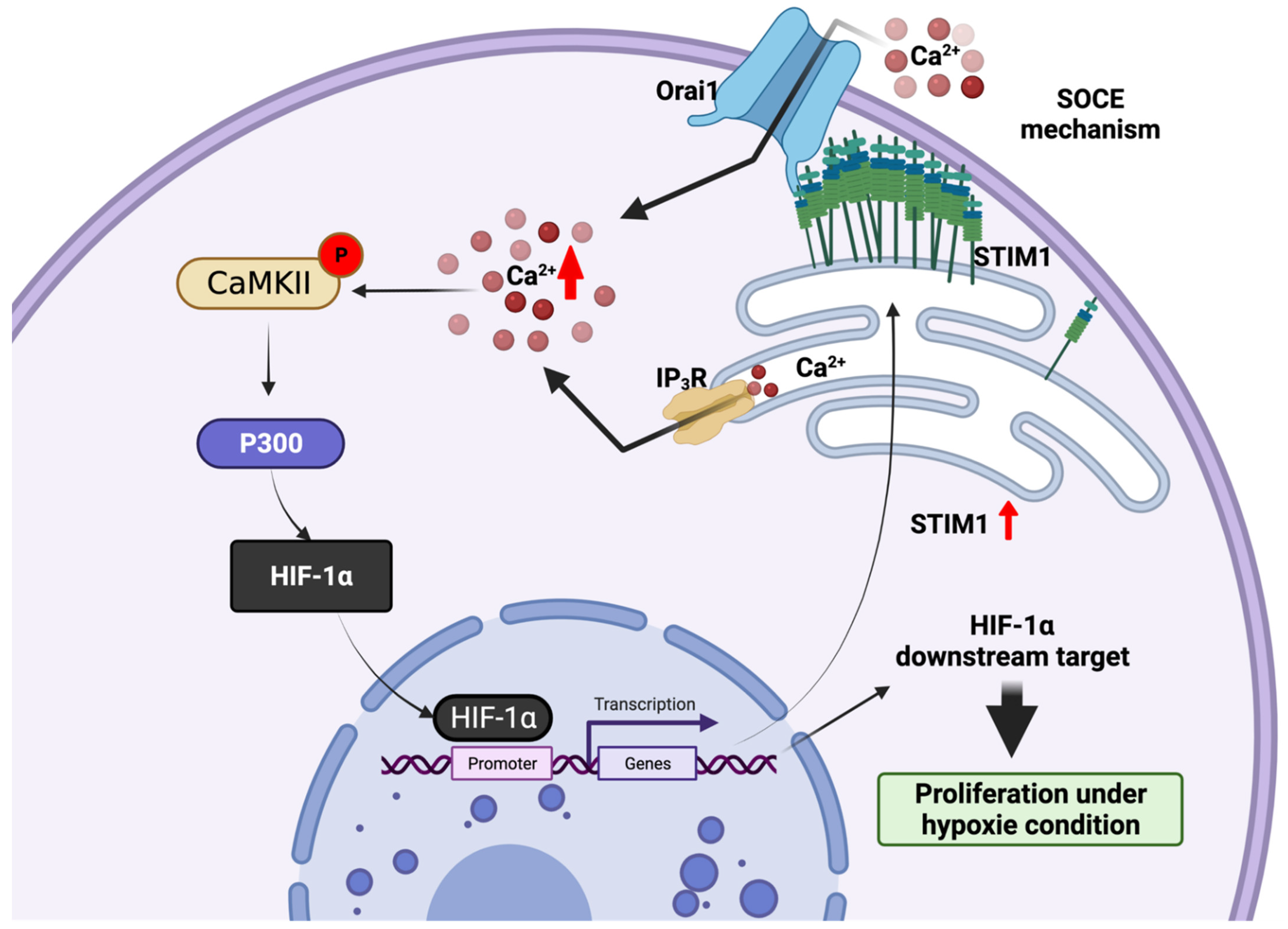
3.3. Endoplasmic Reticulum STIM1-a Metabolic Checkpoint Pathway in HCC
4. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424, Erratum in 2020, 70, 313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J. Hepatol. 2020, 72, 250–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wands, J. Hepatocellular Carcinoma and Sex. New Engl. J. Med. 2007, 357, 1974–1976. [Google Scholar] [CrossRef] [Green Version]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef]
- Lin, L.; Yan, L.; Liu, Y.; Qu, C.; Ni, J.; Li, H. The Burden and Trends of Primary Liver Cancer Caused by Specific Etiologies from 1990 to 2017 at the Global, Regional, National, Age, and Sex Level Results from the Global Burden of Disease Study 2017. Liver Cancer 2020, 9, 563–582. [Google Scholar] [CrossRef]
- Llovet, J.M.; Brú, C.; Bruix, J. Prognosis of hepatocellular carcinoma: The BCLC staging classification. In Seminars in Liver Disease; Thieme Medical Publishers, Inc.: New York, NY, USA, 1999; Volume 19, pp. 329–338. [Google Scholar]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.D.; Heimbach, J.K. New advances in the diagnosis and management of hepatocellular carcinoma. BMJ 2020, 371, m3544. [Google Scholar] [CrossRef]
- Minemura, M.; Tanimura, H.; Tabor, E. Overexpression of multidrug resistance genes MDR1 and cMOAT in human hepatocellular carcinoma and hepatoblastoma cell lines. Int. J. Oncol. 1999, 15, 559–563. [Google Scholar] [CrossRef]
- Chenivesse, X.; Franco, D.; Bréchot, C. MDR1 (multidrug resistance) gene expression in human primary liver cancer and cirrhosis. J. Hepatol. 1993, 18, 168–172. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Garcia, S.; Prado-Garcia, H. Mitochondrial calcium: Transport and modulation of cellular processes in homeostasis and cancer (Review). Int. J. Oncol. 2019, 54, 1155–1167. [Google Scholar] [CrossRef] [Green Version]
- Missiroli, S.; Perrone, M.; Genovese, I.; Pinton, P.; Giorgi, C. Cancer metabolism and mitochondria: Finding novel mechanisms to fight tumours. eBioMedicine 2020, 59, 102943. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [Green Version]
- Dubois, C.; Van den Abeele, F.; Lehen’Kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of Channel-Forming ORAI Proteins Determines an Oncogenic Switch in Prostate Cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Faouzi, M.; Kischel, P.; Hague, F.; Ahidouch, A.; Benzerdjeb, N.; Sevestre, H.; Penner, R.; Ouadid-Ahidouch, H. ORAI3 silencing alters cell proliferation and cell cycle progression via c-myc pathway in breast cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 752–760. [Google Scholar] [CrossRef] [Green Version]
- Motiani, R.K.; Hyzinski-García, M.C.; Zhang, X.; Henkel, M.M.; Abdullaev, I.F.; Kuo, Y.-H.; Matrougui, K.; Mongin, A.A.; Trebak, M. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflügers Arch.-Eur. J. Physiol. 2013, 465, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Lu, F.; He, H.; Shen, J.; Messina, J.; Mathew, R.; Wang, D.; Sarnaik, A.A.; Chang, W.-C.; Kim, M.; et al. STIM1- and Orai1-mediated Ca2+ oscillation orchestrates invadopodium formation and melanoma invasion. J. Cell Biol. 2014, 207, 535–548. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Lkhagvadorj, S.; Lee, M.-R.; Hwang, K.-H.; Chung, H.C.; Jung, J.H.; Cha, S.-K.; Eom, M. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochem. Biophys. Res. Commun. 2014, 448, 76–82. [Google Scholar] [CrossRef]
- Thebault, S.; Flourakis, M.; Vanoverberghe, K.; Vandermoere, F.; Roudbaraki, M.; Lehen’Kyi, V.; Slomianny, C.; Beck, B.; Mariot, P.; Bonnal, J.-L.; et al. Differential Role of Transient Receptor Potential Channels in Ca2+ Entry and Proliferation of Prostate Cancer Epithelial Cells. Cancer Res. 2006, 66, 2038–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raphaël, M.; Lehen’Kyi, V.; Vandenberghe, M.; Beck, B.; Khalimonchyk, S.; Abeele, F.V.; Farsetti, L.; Germain, E.; Bokhobza, A.; Mihalache, A.; et al. TRPV6 calcium channel translocates to the plasma membrane via Orai1-mediated mechanism and controls cancer cell survival. Proc. Natl. Acad. Sci. USA 2014, 111, E3870–E3879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajeddine, N.; Gailly, P. TRPC1 Protein Channel Is Major Regulator of Epidermal Growth Factor Receptor Signaling. J. Biol. Chem. 2012, 287, 16146–16157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szado, T.; Vanderheyden, V.; Parys, J.B.; De Smedt, H.; Rietdorf, K.; Kotelevets, L.; Chastre, E.; Khan, F.; Landegren, U.; Söderberg, O.; et al. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2427–2432. [Google Scholar] [CrossRef] [Green Version]
- Tosatto, A.; Sommaggio, R.; Kummerow, C.; Bentham, R.B.; Blacker, T.S.; Berecz, T.; Duchen, M.R.; Rosato, A.; Bogeski, I.; Szabadkai, G.; et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1alpha. EMBO Mol. Med. 2016, 8, 569–585. [Google Scholar] [CrossRef]
- Liu, Y.; Jin, M.; Wang, Y.; Zhu, J.; Tan, R.; Zhao, J.; Ji, X.; Jin, C.; Jia, Y.; Ren, T.; et al. MCU-induced mitochondrial calcium uptake promotes mitochondrial biogenesis and colorectal cancer growth. Signal. Transduct. Target. Ther. 2020, 5, 59. [Google Scholar] [CrossRef]
- Chakraborty, P.K.; Mustafi, S.B.; Xiong, X.; Dwivedi, S.K.D.; Nesin, V.; Saha, S.; Zhang, M.; Dhanasekaran, D.; Jayaraman, M.; Mannel, R.; et al. MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat. Commun. 2017, 8, 14634. [Google Scholar] [CrossRef]
- Singer, S.J.; Nicolson, G.L. The Fluid Mosaic Model of the Structure of Cell Membranes. Science 1972, 175, 720–731. [Google Scholar] [CrossRef]
- Kalappurakkal, J.M.; Sil, P.; Mayor, S. Toward a new picture of the living plasma membrane. Protein Sci. 2020, 29, 1355–1365. [Google Scholar] [CrossRef]
- Dewenter, M.; Von Der Lieth, A.; Katus, H.A.; Backs, J. Calcium Signaling and Transcriptional Regulation in Cardiomyocytes. Circ. Res. 2017, 121, 1000–1020. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.; Montell, C. TRP Channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapovalov, G.; Ritaine, A.; Skryma, R.; Prevarskaya, N. Role of TRP ion channels in cancer and tumorigenesis. Semin. Immunopathol. 2016, 38, 357–369. [Google Scholar] [CrossRef]
- Wen, L.; Liang, C.; Chen, E.; Chen, W.; Liang, F.; Zhi, X.; Wei, T.; Xue, F.; Li, G.; Yang, Q.; et al. Regulation of Multi-drug Resistance in hepatocellular carcinoma cells is TRPC6/Calcium Dependent. Sci. Rep. 2016, 6, 23269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Yang, Y.; Xie, R.; Liu, J.; Nie, X.; An, J.; Wen, G.; Liu, X.; Jin, H.; Tuo, B. The NCX1/TRPC6 Complex Mediates TGFβ-Driven Migration and Invasion of Human Hepatocellular Carcinoma Cells. Cancer Res. 2018, 78, 2564–2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.-F.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362. [Google Scholar] [CrossRef] [Green Version]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal Structure of the Calcium Release-Activated Calcium Channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [Green Version]
- Liou, J.; Kim, M.L.; Do Heo, W.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM Is a Ca2+ Sensor Essential for Ca2+-Store-Depletion-Triggered Ca2+ Influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [Green Version]
- Roos, J.; Digregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Boustany, C.; Bidaux, G.; Enfissi, A.; Delcourt, P.; Prevarskaya, N.; Capiod, T. Capacitative calcium entry and transient receptor potential canonical 6 expression control human hepatoma cell proliferation. Hepatology 2008, 47, 2068–2077. [Google Scholar] [CrossRef] [PubMed]
- Fiorio Pla, A.; Kondratska, K.; Prevarskaya, N. STIM and ORAI proteins: Crucial roles in hallmarks of cancer. Am. J. Physiol. Cell Physiol. 2016, 310, C509–C519. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International Union of Pharmacology. XLVIII. Nomenclature and Structure-Function Relationships of Voltage-Gated Calcium Channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Auld, A.; Chen, J.; Brereton, H.M.; Wang, Y.-J.; Gregory, R.B.; Barritt, G.J. Store-operated Ca2+ inflow in Reuber hepatoma cells is inhibited by voltage-operated Ca2+ channel antagonists and, in contrast to freshly isolated hepatocytes, does not require a pertussis toxin-sensitive trimeric GTP-binding protein. Biochim. et Biophys. Acta 2000, 1497, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Brereton, H.M.; Harland, M.; Froscio, M.; Petronijevic, T.; Barritt, G.J. Novel variants of voltage-operated calcium channel α1-subunit transcripts in a rat liver-derived cell line: Deletion in the IVS4 voltage sensing region. Cell Calcium 1997, 22, 39–52. [Google Scholar] [CrossRef]
- Sana, A.B.; Maryan, W.F.; Manal, M.N.; Mamdouh, M.E.-S. Calcium channel α2δ1 subunit as a novel biomarker for diagnosis of hepatocellular carcinoma. Cancer Biol. Med. 2018, 15, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Wang, L.; Han, H.; Jin, K.; Lin, N.; Guo, T.; Chen, Y.; Cheng, H.; Lu, F.; Fang, W.; et al. 1B50-1, a mAb Raised against Recurrent Tumor Cells, Targets Liver Tumor-Initiating Cells by Binding to the Calcium Channel α2δ1 Subunit. Cancer Cell 2013, 23, 541–556. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Carafoli, E. The Plasma Membrane Ca2+ ATPase and the Plasma Membrane Sodium Calcium Exchanger Cooperate in the Regulation of Cell Calcium. Cold Spring Harb. Perspect. Biol. 2010, 3, a004168. [Google Scholar] [CrossRef]
- Schatzmann, H.J. ATP-dependent Ca++-Extrusion from human red cells. Experientia 1966, 22, 364–365. [Google Scholar] [CrossRef]
- Reuter, H.; Seitz, N. The dependence of calcium efflux from cardiac muscle on temperature and external ion composition. J. Physiol. 1968, 195, 451–470. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.F.; Blaustein, M.P.; Hodgkin, A.L.; Steinhardt, R.A. The influence of calcium on sodium efflux in squid axons. J. Physiol. 1969, 200, 431–458. [Google Scholar] [CrossRef] [PubMed]
- Rimessi, A.; Coletto, L.; Pinton, P.; Rizzuto, R.; Brini, M.; Carafoli, E. Inhibitory Interaction of the 14-3-3ϵ Protein with Isoform 4 of the Plasma Membrane Ca2+-ATPase Pump. J. Biol. Chem. 2005, 280, 37195–37203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, J.I. Metabolic regulation of the PMCA: Role in cell death and survival. Cell Calcium 2017, 69, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Greeb, J.; Shull, G.E. Molecular Cloning of a Third Isoform of the Calmodulin-sensitive Plasma Membrane Ca2+-Transporting ATPase That Is Expressed Predominantly in Brain and Skeletal Muscle. J. Biol. Chem. 1989, 264, 18569–18576. [Google Scholar] [CrossRef]
- Sritangos, P.; Alarcon, E.P.; James, A.; Sultan, A.; Richardson, D.A.; Bruce, J.I.E. Plasma Membrane Ca2+ ATPase Isoform 4 (PMCA4) Has an Important Role in Numerous Hallmarks of Pancreatic Cancer. Cancers 2020, 12, 218. [Google Scholar] [CrossRef] [Green Version]
- Naffa, R.; Padányi, R.; Ignácz, A.; Hegyi, Z.; Jezsó, B.; Tóth, S.; Varga, K.; Homolya, L.; Hegedűs, L.; Schlett, K.; et al. The Plasma Membrane Ca2+ Pump PMCA4b Regulates Melanoma Cell Migration through Remodeling of the Actin Cytoskeleton. Cancers 2021, 13, 1354. [Google Scholar] [CrossRef]
- Stafford, N.; Wilson, C.; Oceandy, D.; Neyses, L.; Cartwright, E.J. The Plasma Membrane Calcium ATPasesand Their Role as Major New Players in Human Disease. Physiol. Rev. 2017, 97, 1089–1125. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.J.; Roberts-Thomson, S.J.; Holman, N.A.; May, F.J.; Lehrbach, G.M.; Monteith, G.R. Expression of plasma membrane calcium pump isoform mRNAs in breast cancer cell lines. Cell. Signal. 2002, 14, 1015–1022. [Google Scholar] [CrossRef]
- Delgado-Coello, B.; Santiago-García, J.; Zarain-Herzberg, A.; Mas-Oliva, J. Plasma membrane Ca2+-ATPase mRNA expression in murine hepatocarcinoma and regenerating liver cells. Mol. Cell. Biochem. 2003, 247, 177–184. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Lederer, W.J. Sodium/Calcium Exchange: Its Physiological Implications. Physiol. Rev. 1999, 79, 763–854. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, D.A.; Sawaya, M.R.; Kwon, S.; Cascio, D.; Philipson, K.D.; Abramson, J. The Crystal Structure of the Primary Ca2+ Sensor of the Na+/Ca2+ Exchanger Reveals a Novel Ca2+ Binding Motif. J. Biol. Chem. 2006, 281, 21577–21581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytton, J. Na+/Ca2+ exchangers: Three mammalian gene families control Ca2+ transport. Biochem. J. 2007, 406, 365–382. [Google Scholar] [CrossRef]
- Gerkau, N.J.; Rakers, C.; Durry, S.; Petzold, G.C.; Rose, C.R. Reverse NCX Attenuates Cellular Sodium Loading in Metabolically Compromised Cortex. Cereb. Cortex 2017, 28, 4264–4280. [Google Scholar] [CrossRef]
- Linck, B.; Qiu, Z.; He, Z.; Tong, Q.; Hilgemann, D.W.; Philipson, K.D. Functional comparison of the three isoforms of the Na+/Ca2+ exchanger (NCX1, NCX2, NCX3). Am. J. Physiol. Physiol. 1998, 274, C415–C423. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.R.; Claude, A.; Fullam, E.F. A study of tissue culture cells by electron microscopy. J. Exp. Med. 1945, 81, 233–246. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Somlyo, A.P.; Bond, M.; Somlyo, A.V. Calcium content of mitochondria and endoplasmic reticulum in liver frozen rapidly in vivo. Nature 1985, 314, 622–625. [Google Scholar] [CrossRef]
- Copeland, D.E.; Dalton, A.J. An Association between Mitochondria and the Endoplasmic Reticulum in Cells of the Pseudobranch Gland of a Teleost. J. Cell Biol. 1959, 5, 393–396. [Google Scholar] [CrossRef]
- Liou, J.; Fivaz, M.; Inoue, T.; Meyer, T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 9301–9306. [Google Scholar] [CrossRef] [Green Version]
- Perkins, H.T.; Allan, V. Intertwined and Finely Balanced: Endoplasmic Reticulum Morphology, Dynamics, Function, and Diseases. Cells 2021, 10, 2341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Konja, D.; Zhang, Y.; Wang, Y. Communications between Mitochondria and Endoplasmic Reticulum in the Regulation of Metabolic Homeostasis. Cells 2021, 10, 2195. [Google Scholar] [CrossRef] [PubMed]
- Burdakov, D.; Petersen, O.; Verkhratsky, A. Intraluminal calcium as a primary regulator of endoplasmic reticulum function. Cell Calcium 2005, 38, 303–310. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Muik, M.; Frischauf, I.; Derler, I.; Fahrner, M.; Bergsmann, J.; Eder, P.; Schindl, R.; Hesch, C.; Polzinger, B.; Fritsch, R.; et al. Dynamic Coupling of the Putative Coiled-coil Domain of ORAI1 with STIM1 Mediates ORAI1 Channel Activation. J. Biol. Chem. 2008, 283, 8014–8022. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Pozzan, T. Microdomains of Intracellular Ca2+: Molecular Determinants and Functional Consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine Receptors: Structure, Expression, Molecular Details, and Function in Calcium Release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soeller, C.; Crossman, D.; Gilbert, R.; Cannell, M.B. Analysis of ryanodine receptor clusters in rat and human cardiac myocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 14958–14963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierobon, N.; Renard-Rooney, D.C.; Gaspers, L.D.; Thomas, A. Ryanodine Receptors in Liver. J. Biol. Chem. 2006, 281, 34086–34095. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Teoh, N.; Xu, L.; Pok, S.; Li, X.; Chu, E.S.H.; Chiu, J.; Dong, L.; Arfianti, E.; Haigh, W.G.; et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Parys, J.B.; Decuypere, J.-P.; Bultynck, G. Role of the inositol 1,4,5-trisphosphate receptor/Ca2+-release channel in autophagy. Cell Commun. Signal. 2012, 10, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, M.T.; Florentino, R.M.; Franca, A.; Filho, A.C.L.; Dos Santos, M.L.; Fonseca, R.; Lemos, F.; Fonseca, M.; Kruglov, E.; Mennone, A.; et al. Expression of the type 3 InsP3receptor is a final common event in the development of hepatocellular carcinoma. Gut 2019, 68, 1676–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandecaetsbeek, I.; Vangheluwe, P.; Raeymaekers, L.; Wuytack, F.; Vanoevelen, J. The Ca2+ Pumps of the Endoplasmic Reticulum and Golgi Apparatus. Cold Spring Harb. Perspect. Biol. 2011, 3, a004184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoshima, C.; Sasabe, H.; Stokes, D.L. Three-dimensional cryo-electron microscopy of the calcium ion pump in the sarcoplasmic reticulum membrane. Nature 1993, 362, 469–471. [Google Scholar] [CrossRef]
- Pagliaro, L.; Marchesini, M.; Roti, G. Targeting oncogenic Notch signaling with SERCA inhibitors. J. Hematol. Oncol. 2021, 14, 1–17. [Google Scholar] [CrossRef]
- Papp, B.; Brouland, J.-P.; Arbabian, A.; Gélébart, P.; Kovács, T.; Bobe, R.; Enouf, J.; Varin-Blank, N.; Apáti, Á. Endoplasmic Reticulum Calcium Pumps and Cancer Cell Differentiation. Biomolecules 2012, 2, 165–186. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Qiao, S.; Xiang, Y.; Cui, M.; Yao, X.; Lin, R.; Zhang, X. Endoplasmic reticulum stress: Multiple regulatory roles in hepatocellular carcinoma. Biomed. Pharmacother. 2021, 142, 112005. [Google Scholar] [CrossRef]
- Fan, L.; Li, A.; Li, W.; Cai, P.; Yang, B.; Zhang, M.; Gu, Y.; Shu, Y.; Sun, Y.; Shen, Y.; et al. Novel role of Sarco/endoplasmic reticulum calcium ATPase 2 in development of colorectal cancer and its regulation by F36, a curcumin analog. Biomed. Pharmacother. 2014, 68, 1141–1148. [Google Scholar] [CrossRef]
- Xia, S.; Wu, J.; Zhou, W.; Zhang, M.; Zhao, K.; Tian, D.; Liu, J.; Liao, J. HRC promotes anoikis resistance and metastasis by suppressing endoplasmic reticulum stress in hepatocellular carcinoma. Int. J. Med. Sci. 2021, 18, 3112–3124. [Google Scholar] [CrossRef]
- Hammad, A.; Machaca, K. Store Operated Calcium Entry in Cell Migration and Cancer Metastasis. Cells 2021, 10, 1246. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, S.; Meyer, T. STIM Proteins and the Endoplasmic Reticulum-Plasma Membrane Junctions. Annu. Rev. Biochem. 2011, 80, 973–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernster, L.; Schatz, G. Mitochondria: A historical review. J. Cell Biol. 1981, 91, 227s–255s. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Han, Y.; Li, M.; Qiu, F.; Zhang, M.; Zhang, Y.-H. Cell-permeable organic fluorescent probes for live-cell long-term super-resolution imaging reveal lysosome-mitochondrion interactions. Nat. Commun. 2017, 8, 1307. [Google Scholar] [CrossRef]
- Desai, R.; East, D.A.; Hardy, L.; Faccenda, D.; Rigon, M.; Crosby, J.; Alvarez, M.S.; Singh, A.; Mainenti, M.; Hussey, L.K.; et al. Mitochondria form contact sites with the nucleus to couple prosurvival retrograde response. Sci. Adv. 2020, 6, eabc9955. [Google Scholar] [CrossRef]
- Gil-Hernández, A.; Arroyo-Campuzano, M.; Simoni-Nieves, A.; Zazueta, C.; Gomez-Quiroz, L.E.; Silva-Palacios, A. Relevance of Membrane Contact Sites in Cancer Progression. Front. Cell Dev. Biol. 2021, 8, 622215. [Google Scholar] [CrossRef]
- Qi, H.; Li, L.; Shuai, J. Optimal microdomain crosstalk between endoplasmic reticulum and mitochondria for Ca2+ oscillations. Sci. Rep. 2015, 5, 7984. [Google Scholar] [CrossRef] [Green Version]
- Kamer, K.J.; Mootha, V.K. The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Biol. 2015, 16, 545–553. [Google Scholar] [CrossRef]
- Nemani, N.; Shanmughapriya, S.; Madesh, M. Molecular regulation of MCU: Implications in physiology and disease. Cell Calcium 2018, 74, 86–93. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallafacchina, G.; Zanin, S.; Rizzuto, R. From the Identification to the Dissection of the Physiological Role of the Mitochondrial Calcium Uniporter: An Ongoing Story. Biomolecules 2021, 11, 786. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Zhang, J.; Tsai, C.-W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.-F.; Feng, L. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 2020, 582, 129–133. [Google Scholar] [CrossRef]
- Wang, C.; Jacewicz, A.; Delgado, B.; Baradaran, R.; Long, S.B. Structures reveal gatekeeping of the mitochondrial Ca2+ uniporter by MICU1-MICU2. eLife 2020, 9, e59991. [Google Scholar] [CrossRef]
- Huang, G.; Vercesi, A.E.; Docampo, R. Essential regulation of cell bioenergetics in Trypanosoma brucei by the mitochondrial calcium uniporter. Nat. Commun. 2013, 4, 2865. [Google Scholar] [CrossRef] [Green Version]
- Langenbacher, A.D.; Shimizu, H.; Hsu, W.; Zhao, Y.; Borges, A.; Koehler, C.; Chen, J.-N. Mitochondrial Calcium Uniporter Deficiency in Zebrafish Causes Cardiomyopathy with Arrhythmia. Front. Physiol. 2020, 11, 617492. [Google Scholar] [CrossRef]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabo, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [Green Version]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 2010, 467, 291–296. [Google Scholar] [CrossRef] [Green Version]
- Mallilankaraman, K.; Doonan, P.; Cardenas, C.; Chandramoorthy, H.C.; Muller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgo, J.; Birnbaum, M.J.; et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liu, J.; Holmström, K.; Menazza, S.; Parks, R.J.; Fergusson, M.M.; Yu, Z.-X.; Springer, D.A.; Halsey, C.; Liu, C.; et al. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep. 2016, 16, 1561–1573. [Google Scholar] [CrossRef] [Green Version]
- Antony, A.N.; Paillard, M.; Moffat, C.; Juskeviciute, E.; Correnti, J.; Bolon, B.; Rubin, E.; Csordás, G.; Seifert, E.L.; Hoek, J.B.; et al. MICU1 regulation of mitochondrial Ca2+ uptake dictates survival and tissue regeneration. Nat. Commun. 2016, 7, 10955. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Suzuki, J.; Paranjpe, I.; Unsulangi, T.; Boyman, L.; Milescu, L.S.; Lederer, W.J.; Kirichok, Y. The mechanism of MICU-dependent gating of the mitochondrial Ca2+ uniporter. eLife 2021, 10, e69312. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Grabarek, Z.; Mootha, V.K. High-affinity cooperative Ca2+ binding by MICU 1– MICU 2 serves as an on–off switch for the uniporter. EMBO Rep. 2017, 18, 1397–1411. [Google Scholar] [CrossRef]
- Payne, R.; Hoff, H.; Roskowski, A.; Foskett, J.K. MICU2 Restricts Spatial Crosstalk between InsP 3 R and MCU Channels by Regulating Threshold and Gain of MICU1-Mediated Inhibition and Activation of MCU. Cell Rep. 2017, 21, 3141–3154. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovács-Bogdán, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE Is an Essential Component of the Mitochondrial Calcium Uniporter Complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Nguyen, N.X.; She, J.; Zeng, W.; Yang, Y.; Bai, X.-C.; Jiang, Y. Structural Mechanism of EMRE-Dependent Gating of the Human Mitochondrial Calcium Uniporter. Cell 2019, 177, 1252–1261. [Google Scholar] [CrossRef]
- Tsai, M.-F.; Phillips, C.B.; Ranaghan, M.; Tsai, C.-W.; Wu, Y.; Willliams, C.; Miller, C. Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. eLife 2016, 5, e15545. [Google Scholar] [CrossRef]
- Mallilankaraman, K.; Cárdenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenár, T.; Csordás, G.; Madireddi, P.; Yang, J.; Müller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nature 2012, 14, 1336–1343. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, D.; Artiga, D.; Abiria, S.A.; Clapham, D.E. Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc. Natl. Acad. Sci. USA 2016, 113, E1872–E1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paupe, V.; Prudent, J.; Dassa, E.P.; Rendon, O.Z.; Shoubridge, E.A. CCDC90A (MCUR1) Is a Cytochrome c Oxidase Assembly Factor and Not a Regulator of the Mitochondrial Calcium Uniporter. Cell Metab. 2015, 21, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Sander, P.; Gudermann, T.; Schredelseker, J. A Calcium Guard in the Outer Membrane: Is VDAC a Regulated Gatekeeper of Mitochondrial Calcium Uptake? Int. J. Mol. Sci. 2021, 22, 946. [Google Scholar] [CrossRef] [PubMed]
- Varughese, J.; Buchanan, S.; Pitt, A. The Role of Voltage-Dependent Anion Channel in Mitochondrial Dysfunction and Human Disease. Cells 2021, 10, 1737. [Google Scholar] [CrossRef]
- Heslop, K.A.; Milesi, V.; Maldonado, E.N. VDAC Modulation of Cancer Metabolism: Advances and Therapeutic Challenges. Front. Physiol. 2021, 12, 742839. [Google Scholar] [CrossRef] [PubMed]
- Pittala, S.; Krelin, Y.; Shoshan-Barmatz, V. Targeting Liver Cancer and Associated Pathologies in Mice with a Mitochondrial VDAC1-Based Peptide. Neoplasia 2018, 20, 594–609. [Google Scholar] [CrossRef] [PubMed]
- Sohlang, M.N.; Majaw, S. Altered VDAC-HK association and apoptosis in mouse peripheral blood lymphocytes exposed to diabetic condition: An in vitro and in vivo study. Arch. Physiol. Biochem. 2021, 1–11. [Google Scholar] [CrossRef]
- Rochette, L.; Meloux, A.; Zeller, M.; Malka, G.; Cottin, Y.; Vergely, C. Mitochondrial SLC25 Carriers: Novel Targets for Cancer Therapy. Molecules 2020, 25, 2417. [Google Scholar] [CrossRef]
- Hoffman, N.E.; Chandramoorthy, H.C.; Shanmughapriya, S.; Zhang, X.Q.; Vallem, S.; Doonan, P.J.; Malliankaraman, K.; Guo, S.; Rajan, S.; Elrod, J.; et al. SLC25A23 augments mitochondrial Ca2+ uptake, interacts with MCU, and induces oxidative stress–mediated cell death. Mol. Biol. Cell 2014, 25, 936–947. [Google Scholar] [CrossRef]
- Roy, S.; Dey, K.; Hershfinkel, M.; Ohana, E.; Sekler, I. Identification of residues that control Li+ versus Na+ dependent Ca2+ exchange at the transport site of the mitochondrial NCLX. Biochim. et Biophys. Acta 2017, 1864, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, U.; Domingo, J.S.; Castelbou, C.; Sekler, I.; Wiederkehr, A.; Demaurex, N. NCLX Protein, but Not LETM1, Mediates Mitochondrial Ca2+ Extrusion, Thereby Limiting Ca2+-induced NAD(P)H Production and Modulating Matrix Redox State. J. Biol. Chem. 2014, 289, 20377–20385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, T.; Gueguinou, M.; Walter, V.; Delierneux, C.; Johnson, M.T.; Zhang, X.; Xin, P.; Yoast, R.; Emrich, S.M.; Yochum, G.S.; et al. Dichotomous role of the human mitochondrial Na+/Ca2+/Li+ exchanger NCLX in colorectal cancer growth and metastasis. eLife 2020, 9, e59686. [Google Scholar] [CrossRef] [PubMed]
- Peruzzo, R.; Costa, R.; Bachmann, M.; Leanza, L.; Szabò, I. Mitochondrial Metabolism, Contact Sites and Cellular Calcium Signaling: Implications for Tumorigenesis. Cancers 2020, 12, 2574. [Google Scholar] [CrossRef]
- Jimenez, H.; Wang, M.; Zimmerman, J.W.; Pennison, M.J.; Sharma, S.; Surratt, T.; Xu, Z.-X.; Brezovich, I.; Absher, D.; Myers, R.M.; et al. Tumour-specific amplitude-modulated radiofrequency electromagnetic fields induce differentiation of hepatocellular carcinoma via targeting Cav3.2 T-type voltage-gated calcium channels and Ca2+ influx. eBioMedicine 2019, 44, 209–224. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.R.; Chun, J.N.; So, I.; Kim, H.J.; Baek, S.; Jeon, J.-H.; Shin, S.-Y. Data-driven Analysis of TRP Channels in Cancer: Linking Variation in Gene Expression to Clinical Significance. Cancer Genom. Proteom. 2016, 13, 83–90. [Google Scholar]
- Vriens, J.; Janssens, A.; Prenen, J.; Nilius, B.; Wondergem, R. TRPV channels and modulation by hepatocyte growth factor/scatter factor in human hepatoblastoma (HepG2) cells. Cell Calcium 2004, 36, 19–28. [Google Scholar] [CrossRef]
- Voringer, S.; Schreyer, L.; Nadolni, W.; Meier, M.A.; Woerther, K.; Mittermeier, C.; Ferioli, S.; Singer, S.; Holzer, K.; Zierler, S.; et al. Inhibition of TRPM7 blocks MRTF/SRF-dependent transcriptional and tumorigenic activity. Oncogene 2019, 39, 2328–2344. [Google Scholar] [CrossRef]
- Tang, B.-D.; Xia, X.; Lv, X.-F.; Yu, B.-X.; Yuan, J.-N.; Mai, X.-Y.; Shang, J.-Y.; Zhou, J.-G.; Liang, S.-J.; Pang, R.-P. Inhibition of Orai1-mediated Ca2+ entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J. Cell. Mol. Med. 2016, 21, 904–915. [Google Scholar] [CrossRef]
- Karacicek, B.; Erac, Y.; Tosun, M. Functional consequences of enhanced expression of STIM1 and Orai1 in Huh-7 hepatocellular carcinoma tumor-initiating cells. BMC Cancer 2019, 19, 751. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yan, G.; Zheng, L.; Zhou, Y.; Sheng, H.; Wu, L.; Zhang, Q.; Lei, J.; Zhang, J.; Xin, R.; et al. STIM1 is a metabolic checkpoint regulating the invasion and metastasis of hepatocellular carcinoma. Theranostics 2020, 10, 6483–6499. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.-Q.; Chen, Y.-F.; Chen, J.-R.; Jou, Y.-S.; Wu, P.-C.; Kao, C.-H.; Wang, C.-H.; Huang, Y.-L.; Chen, C.-F.; Huang, T.-S.; et al. CISD2 Haploinsufficiency Disrupts Calcium Homeostasis, Causes Nonalcoholic Fatty Liver Disease, and Promotes Hepatocellular Carcinoma. Cell Rep. 2017, 21, 2198–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Oliveras, A.; Izquierdo-Torres, E.; Meneses-Morales, I.; Rodríguez, G.; Zarain-Herzberg, Á.; Santiago-García, J. Histone deacetylase inhibitors promote ATP2A3 gene expression in hepatocellular carcinoma cells: p300 as a transcriptional regulator. Int. J. Biochem. Cell Biol. 2019, 113, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Zhang, H.; Wang, J.; Zhu, J.; Jin, M.; Wu, Y.; Guo, X.; Ji, L.; Huang, Q.; Yang, H.; et al. MCU-dependent mitochondrial Ca2+ inhibits NAD+/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 2017, 36, 5897–5909. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Wang, J.; Zhang, H.; Yuan, P.; Zhu, J.; Wu, Y.; Huang, Q.; Guo, X.; Zhang, J.; Jiaojiao, W.; et al. MCUR1-Mediated Mitochondrial Calcium Signaling Facilitates Cell Survival of Hepatocellular Carcinoma via Reactive Oxygen Species-Dependent P53 Degradation. Antioxidants Redox Signal. 2018, 28, 1120–1136. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Wang, J.; Ji, X.; Cao, H.; Zhu, J.; Chen, Y.; Yang, J.; Zhao, Z.; Ren, T.; Xing, J. MCUR1 facilitates epithelial-mesenchymal transition and metastasis via the mitochondrial calcium dependent ROS/Nrf2/Notch pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 136. [Google Scholar] [CrossRef] [Green Version]
- Etchegaray, J.-P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Min, L.; He, B.; Hui, L. Mitogen-activated protein kinases in hepatocellular carcinoma development. Semin. Cancer Biol. 2011, 21, 10–20. [Google Scholar] [CrossRef]
- López-Colomé, A.M.; Lee-Rivera, I.; Benavides-Hidalgo, R.; López, E. Paxillin: A crossroad in pathological cell migration. J. Hematol. Oncol. 2017, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Geervliet, E.; Bansal, R. Matrix Metalloproteinases as Potential Biomarkers and Therapeutic Targets in Liver Diseases. Cells 2020, 9, 1212. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Koudelkova, P.; Dituri, F.; Mikulits, W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J. Hepatol. 2016, 65, 798–808. [Google Scholar] [CrossRef] [Green Version]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakao, A.; Imamura, T.; Souchelnytskyi, S.; Kawabata, M.; Ishisaki, A.; Oeda, E.; Tamaki, K.; Hanai, J.; Heldin, C.; Miyazono, K.; et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997, 16, 5353–5362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, F.M.; Azimi, I.; Faville, R.A.; Peters, A.A.; Jalink, K.; Putney, J.W.; Goodhill, G.J.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Induction of epithelial–mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene 2014, 33, 2307–2316. [Google Scholar] [CrossRef] [Green Version]
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002, 62, 3387–3394. [Google Scholar]
- Wang, L.; Mosel, A.J.; Oakley, G.; Peng, A. Deficient DNA Damage Signaling Leads to Chemoresistance to Cisplatin in Oral Cancer. Mol. Cancer Ther. 2012, 11, 2401–2409. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Bao, M.; Fang, Y.; Yu, X.; Ji, J.; Ding, X. STAT3/HIF-1α signaling activation mediates peritoneal fibrosis induced by high glucose. J. Transl. Med. 2021, 19, 283. [Google Scholar] [CrossRef]
- Li, Y.; Guo, B.; Xie, Q.; Ye, D.; Zhang, D.; Zhu, Y.; Chen, H.; Zhu, B. STIM1 Mediates Hypoxia-Driven Hepatocarcinogenesis via Interaction with HIF-1. Cell Rep. 2015, 12, 388–395. [Google Scholar] [CrossRef] [Green Version]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation, and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [Green Version]
- Herst, P.M.; Grasso, C.; Berridge, M.V. Metabolic reprogramming of mitochondrial respiration in metastatic cancer. Cancer Metastasis Rev. 2018, 37, 643–653. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Localization | Protein | Expression in HCC | References |
|---|---|---|---|
| Plasma membrane | CACNA1H | Upregulated | [136] |
| TRPM2 | Upregulated | [137] | |
| TRPV2 | Upregulated | [138] | |
| TRPV4 | Downregulated | [137] | |
| TRPC6 | Upregulated | [35,36] | |
| TRPM7 | Upregulated | [139] | |
| Orai1 | Upregulated | [140] | |
| Endoplasmic Reticulum | STIM1 | Upregulated | [141,142] |
| SERCA2 | Downregulated | [143] | |
| SERCA3 | Downregulated | [144] | |
| IP3R | Upregulated | [83] | |
| Mitochondria | MICU1 | Downregulated | [145] |
| MCU | Upregulated | [145] | |
| MCUR1 | Upregulated | [146,147] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, H.-T.; Canoy, R.J.; Campanella, M.; Vassetzky, Y.; Brenner, C. Ca2+ Transportome and the Interorganelle Communication in Hepatocellular Carcinoma. Cells 2022, 11, 815. https://doi.org/10.3390/cells11050815
Lai H-T, Canoy RJ, Campanella M, Vassetzky Y, Brenner C. Ca2+ Transportome and the Interorganelle Communication in Hepatocellular Carcinoma. Cells. 2022; 11(5):815. https://doi.org/10.3390/cells11050815
Chicago/Turabian StyleLai, Hong-Toan, Reynand Jay Canoy, Michelangelo Campanella, Yegor Vassetzky, and Catherine Brenner. 2022. "Ca2+ Transportome and the Interorganelle Communication in Hepatocellular Carcinoma" Cells 11, no. 5: 815. https://doi.org/10.3390/cells11050815
APA StyleLai, H.-T., Canoy, R. J., Campanella, M., Vassetzky, Y., & Brenner, C. (2022). Ca2+ Transportome and the Interorganelle Communication in Hepatocellular Carcinoma. Cells, 11(5), 815. https://doi.org/10.3390/cells11050815