
Shaping of Hepatic Ischemia/Reperfusion Events: The Crucial Role of Mitochondria

 ,
,  , ,
, , 


Abstract
:1. Introduction
1.1. The Liver
1.2. Ischemia/Reperfusion
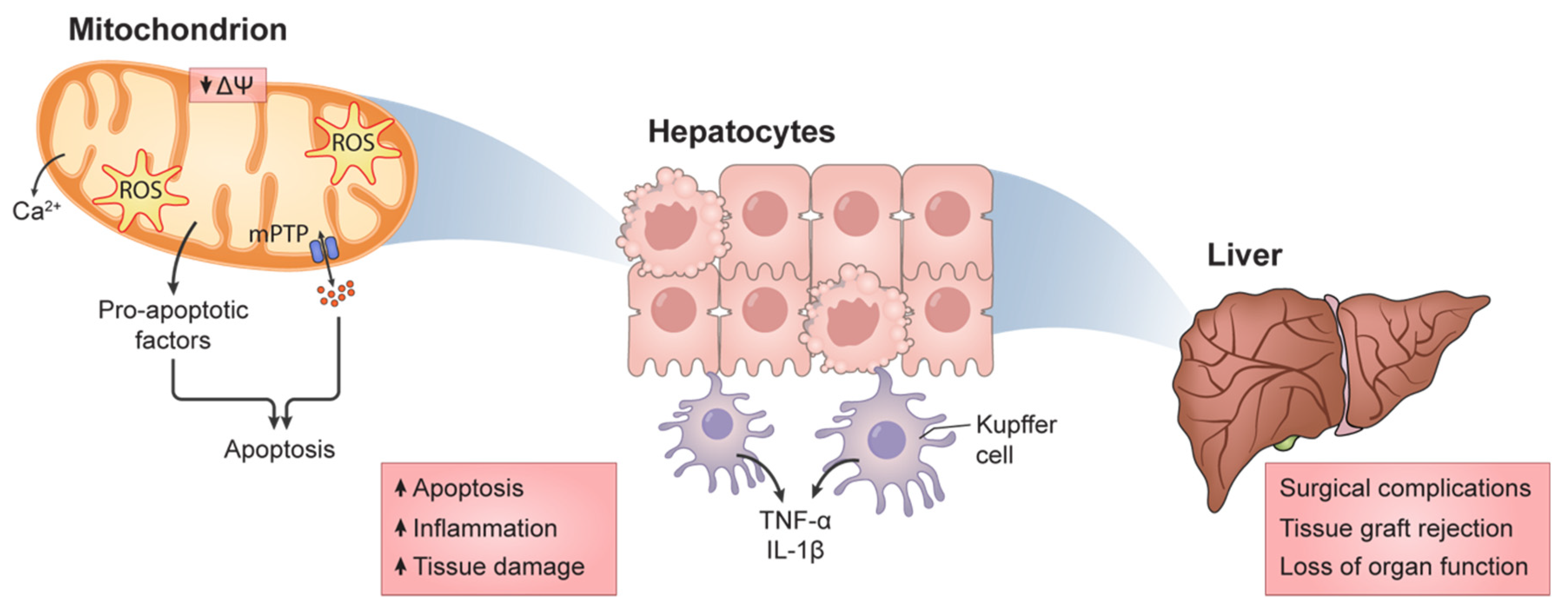
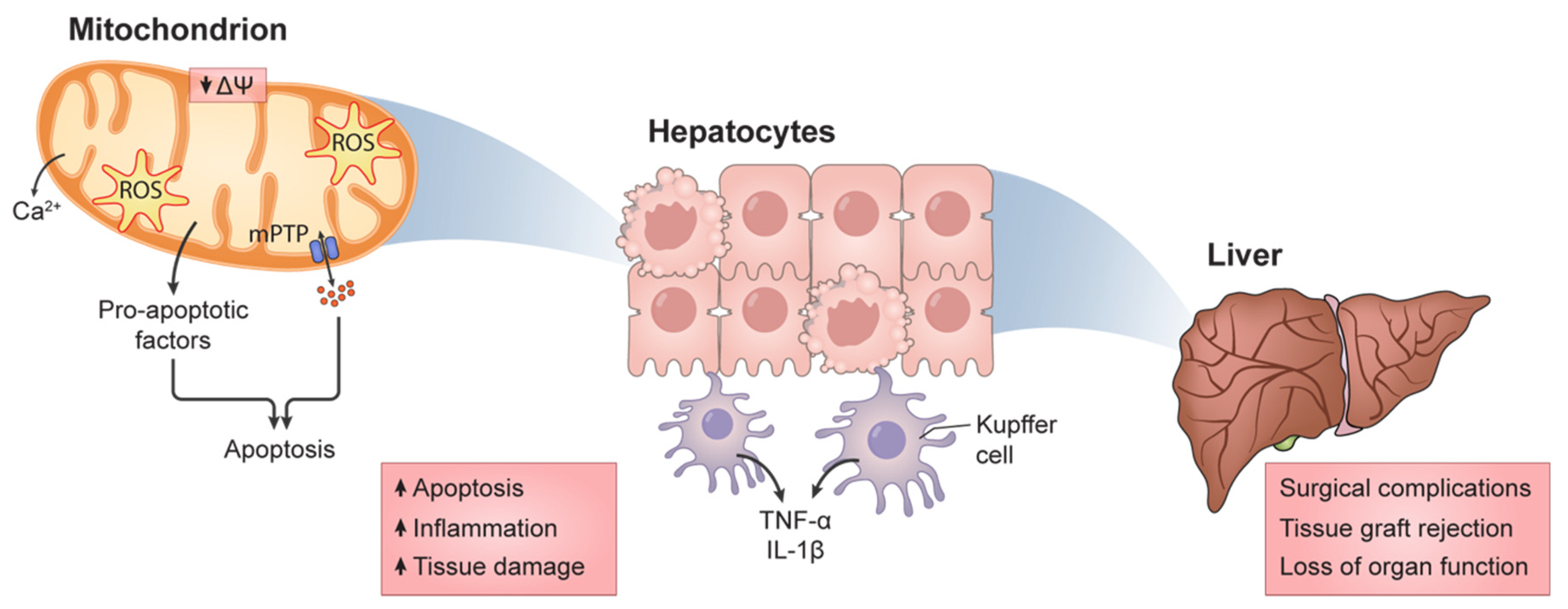
1.3. Mitochondrial Function and HIRI
2. Surgical Approaches in HIRI Mitochondrial Function Compromise
2.1. Ischemic Preconditioning
2.2. Intermittent Clamping
2.3. Remote Ischemic Preconditioning
2.4. Machine Perfusion
2.5. Mitochondrial Transplantation
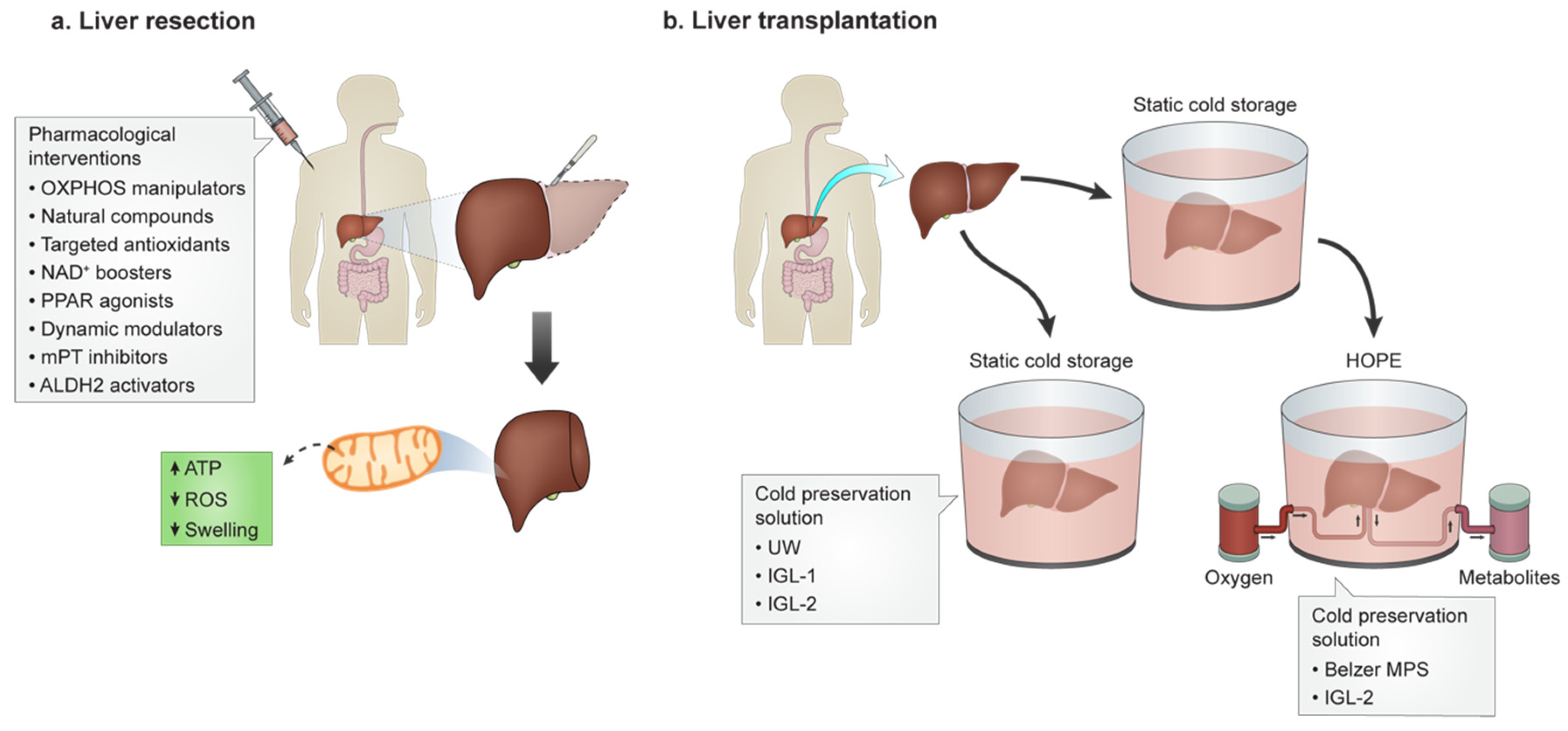
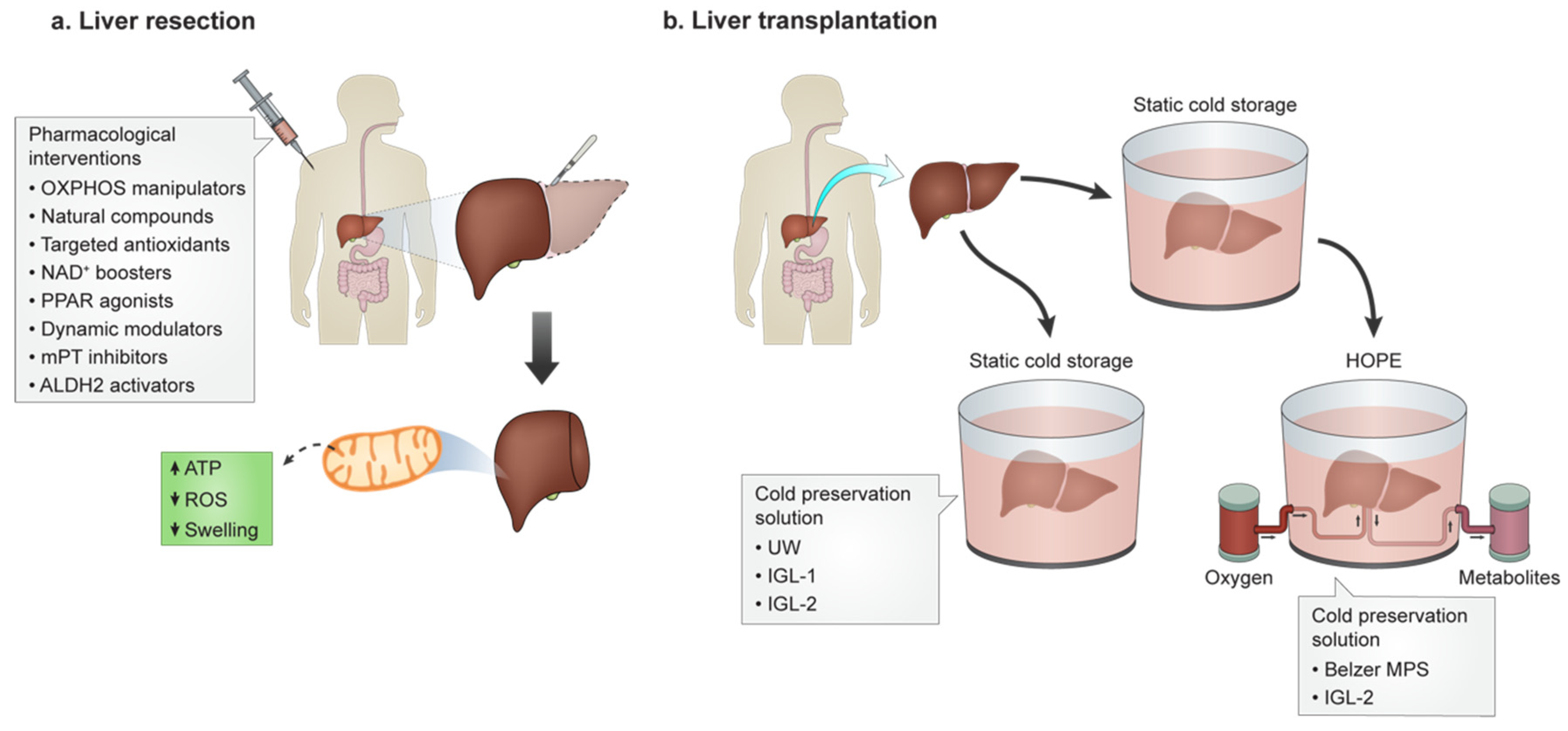
3. Pharmacological Intervention for HIRI
3.1. Mitochondrial Targeting for HIRI
3.1.1. Natural Compounds
3.1.2. Synthetic and Directed Agents
OXPHOS Elements Manipulation
Mitochondrially Targeted Antioxidants
NAD+ Metabolism
PPAR Agonists
Mitochondrial Dynamics Modulators
mPT Inhibiton
3.2. Mitochondrial Aldehyde Dehydrogenase 2 as a Therapeutical Target
3.3. Polyethylene Glycol: A New Promising Approach
4. HIRI and Transplantation
4.1. Static Cold Preservation
4.2. Dynamic PreservationRe
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Kalra, A.; Tuma, F. Physiology, Liver; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Rui, L. Energy Metabolism in the Liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodzin, A.S.; Baker, T.B. Liver Transplantation Today: Where We Are Now and Where We Are Going. Liver Transplant. 2018, 24, 1470–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghinolfi, D.; Melandro, F.; Torri, F.; Martinelli, C.; Cappello, V.; Babboni, S.; Silvestrini, B.; de Simone, P.; Basta, G.; del Turco, S. Extended Criteria Grafts and Emerging Therapeutics Strategy in Liver Transplantation. The Unstable Balance between Damage and Repair. Transplant. Rev. 2021, 35, 100639–100649. [Google Scholar] [CrossRef]
- Lee, S.H.; Culberson, C.; Korneszczuk, K.; Clemens, M.G. Differential Mechanisms of Hepatic Vascular Dysregulation with Mild vs. Moderate Ischemia-Reperfusion. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1219–G1226. [Google Scholar] [CrossRef]
- Sheng, M.; Zhou, Y.; Yu, W.; Weng, Y.; Xu, R.; Du, H. Protective Effect of Berberine Pretreatment in Hepatic Ischemia/Reperfusion Injury of Rat. Transplant. Proc. 2015, 47, 275–282. [Google Scholar] [CrossRef]
- Nardo, B.; Bertelli, R.; Montalti, R.; Beltempo, P.; Puviani, L.; Pacilè, V.; Cavallari, A. Preliminary Results of a Clinical Randomized Study Comparing Celsior and HTK Solutions in Liver Preservation for Transplantation. Transplant. Proc. 2005, 37, 320–322. [Google Scholar] [CrossRef]
- Cannistrà, M.; Ruggiero, M.; Zullo, A.; Gallelli, G.; Serafini, S.; Maria, M.; Naso, A.; Grande, R.; Serra, R.; Nardo, B. Hepatic Ischemia Reperfusion Injury: A Systematic Review of Literature and the Role of Current Drugs and Biomarkers. Int. J. Surg. 2016, 33, S57–S70. [Google Scholar] [CrossRef]
- Zhai, Y.; Petrowsky, H.; Hong, J.C.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Ischaemia-Reperfusion Injury in Liver Transplantation-from Bench to Bedside. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 79–89. [Google Scholar] [CrossRef]
- Zhai, Y.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Liver Ischemia and Reperfusion Injury: New Insights into Mechanisms of Innate-Adaptive Immune-Mediated Tissue Inflammation. Am. J. Transplant. 2011, 11, 1563–1569. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: A Concept in Redox Biology and Medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Weigand, K.; Brost, S.; Steinebrunner, N.; Bchler, M.; Schemmer, P.; Müller, M. Ischemia/Reperfusion Injury in Liver Surgery and Transplantation: Pathophysiology. HPB Surg. 2012, 2012, 176723–176731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalvo-Jave, E.E.; Escalante-Tattersfield, T.; Ortega-Salgado, J.A.; Piña, E.; Geller, D.A. Factors in the Pathophysiology of the Liver Ischemia-Reperfusion Injury. J. Surg. Res. 2008, 147, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siriussawakul, A.; Zaky, A.; Lang, J.D. Role of Nitric Oxide in Hepatic Ischemia-Reperfusion Injury. World J. Gastroenterol. 2010, 16, 6079–6086. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.-Y.; Fu, P.-Y.; Li, P.-D.; Li, Z.-N.; Liu, H.-Y.; Xin, M.-G.; Li, W. Mechanisms of Hepatic Ischemia-Reperfusion Injury and Protective Effects of Nitric Oxide. World J. Gastrointest. Surg. 2014, 6, 122. [Google Scholar] [CrossRef]
- Datta, G.; Fuller, B.J.; Davidson, B.R. Molecular Mechanisms of Liver Ischemia Reperfusion Injury: Insights from Transgenic Knockout Models. World J. Gastroenterol. 2013, 19, 1683–1698. [Google Scholar] [CrossRef]
- Guan, Y.-F. Ischemic Post-Conditioning to Counteract Intestinal Ischemia/Reperfusion Injury. World J. Gastrointest. Pathophysiol. 2010, 1, 137–143. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial Electron Transport Chain: Oxidative Phosphorylation, Oxidant Production, and Methods of Measurement. Redox Biol. 2020, 37, 101674–101683. [Google Scholar] [CrossRef]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef] [Green Version]
- Shields, H.J.; Traa, A.; van Raamsdonk, J.M. Beneficial and Detrimental Effects of Reactive Oxygen Species on Lifespan: A Comprehensive Review of Comparative and Experimental Studies. Front. Cell Dev. Biol. 2021, 9, 628157–628184. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Chen, Q.; Tandler, B.; Hoppel, C.L. Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 535–565. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Paradies, V.; Ruggiero, F.M. Role of Cardiolipin Peroxidation and Ca2+ in Mitochondrial Dysfunction and Disease. Cell Calcium 2009, 45, 643–650. [Google Scholar] [CrossRef]
- Dolder, M.; Wendt, S.; Wallimann, T. Mitochondrial Creatine Kinase in Contact Sites: Interaction with Porin and Adenine Nucleotide Translocase, Role in Permeability Transition and Sensitivity to Oxidative Damage. NeuroSignals 2001, 10, 93–111. [Google Scholar] [CrossRef]
- Wagner, G.R.; Bhatt, D.P.; O’Connell, T.M.; Thompson, J.W.; Dubois, L.G.; Backos, D.S.; Yang, H.; Mitchell, G.A.; Ilkayeva, O.R.; Stevens, R.D.; et al. A Class of Reactive Acyl-CoA Species Reveals the Non-Enzymatic Origins of Protein Acylation. Cell Metab. 2017, 25, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, G.R.; Hirschey, M.D. Nonenzymatic Protein Acylation as a Carbon Stress Regulated by Sirtuin Deacylases. Mol. Cell 2014, 54, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.J.; Choi, J.M.; Kim, L.; Park, S.E.; Rhee, E.J.; Lee, W.Y.; Oh, K.W.; Park, S.W.; Park, C.Y. Nicotinamide Improves Glucose Metabolism and Affects the Hepatic NAD-Sirtuin Pathway in a Rodent Model of Obesity and Type 2 Diabetes. J. Nutr. Biochem. 2014, 25, 66–72. [Google Scholar] [CrossRef]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; de Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxidative Med. Cell. Longev. 2016, 2016, 3907147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Metabolic Regulation of Mitochondrial Dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P. Mitohormesis and Metabolic Health_ The Interplay between ROS, CAMP and Sirtuins. Free Radic. Biol. Med. 2019, 141, 483–491. [Google Scholar] [CrossRef]
- Sastre, J.; Serviddio, G.; Javier, P.; Minana, J.B.; Arduini, A.; Vendemiale, G.; Poli, G.; Pallardo, F.V.; Vina, J. Mitochondrial Function in Liver Disease. Front. Biosci. 2007, 12, 1200–1209. [Google Scholar] [CrossRef] [Green Version]
- Rasola, A.; Bernardi, P. Mitochondrial Permeability Transition in Ca(2+)-Dependent Apoptosis and Necrosis. Cell Calcium 2011, 50, 222–233. [Google Scholar] [CrossRef]
- Ichas, F.; Mazat, J.P. From Calcium Signaling to Cell Death: Two Conformations for the Mitochondrial Permeability Transition Pore. Switching from Low- to High-Conductance State. Biochim. Biophys. Acta Bioenergy 1998, 1366, 33–50. [Google Scholar] [CrossRef] [Green Version]
- Suh, D.H.; Kim, M.K.; Kim, H.S.; Chung, H.H.; Song, Y.S. Mitochondrial Permeability Transition Pore as a Selective Target for Anti-Cancer Therapy. Front. Oncol. 2013, 3, 41. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.W.; Ashcroft, M. Exploring the Molecular Interface between Hypoxia-Inducible Factor Signalling and Mitochondria. Cell. Mol. Life Sci. 2019, 76, 1759–1777. [Google Scholar] [CrossRef] [Green Version]
- Marin, W.; Marin, D.; Ao, X.; Liu, Y. Mitochondria as a Therapeutic Target for Cardiac Ischemia-Reperfusion Injury (Review). Int. J. Mol. Med. 2021, 47, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Y.; Jiao, J.; Wang, J.; Li, Y.; Qin, D.; Li, P. Mitofusin 1 Is Negatively Regulated by MicroRNA 140 in Cardiomyocyte Apoptosis. Mol. Cell. Biol. 2014, 34, 1788–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boengler, K.; Lochnit, G.; Schulz, R. Mitochondria “THE” Target of Myocardial Conditioning. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1215–H1231. [Google Scholar] [CrossRef] [Green Version]
- Go, K.L.; Lee, S.; Zendejas, I.; Behrns, K.E.; Kim, J.S. Mitochondrial Dysfunction and Autophagy in Hepatic Ischemia/Reperfusion Injury. BioMed Res. Int. 2015, 2015, 183469. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Li, L. Pre-Conditions for Eliminating Mitochondrial Dysfunction and Maintaining Liver Function after Hepatic Ischaemia Reperfusion. J. Cell. Mol. Med. 2017, 21, 1719–1731. [Google Scholar] [CrossRef]
- Cancela, J.M.; Petersen, O.H. Regulation of Intracellular Ca2+ Stores by Multiple Ca2+-Releasing Messengers. Diabetes 2002, 51 (Suppl. 3), S349–S357. [Google Scholar] [CrossRef] [Green Version]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.G.; Pathan, N.; Ethell, I.M.; Krajewski, S.; Yamaguchi, Y.; Shibasaki, F.; McKeon, F.; Bobo, T.; Franke, T.F.; Reed, J.C. Ca2+-Induced Apoptosis Through Calcineurin Dephosphorylation of BAD. Science 1999, 284, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Ariyoshi, H.; Sakon, M.; Kambayashi, J.I.; Yoshikawa, N.; Shinoki, N.; Kawasaki, T.; Monden, M. A Role for Local Calcium Gradients upon Hypoxic Injury in Human Umbilical Vein Endothelial Cells (HUVEC). Cell Calcium 1998, 24, 49–57. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Zhang, Y.; Hosch, M.S.; Lang, A.; Zwacka, R.M.; Engelhardt, J.F. Subcellular Site of Superoxide Dismutase Expression Differentially Controls AP-1 Activity and Injury in Mouse Liver Following Ischemia/Reperfusion. Hepatology 2001, 33, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Nagendra, A.R.; Mickelson, J.K.; Smith, C.W. CD18 Integrin and CD54-Dependent Neutrophil Adhesion to Cytokine- Stimulated Human Hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 1997, 272, G408–G416. [Google Scholar] [CrossRef]
- Martins, R.M.; Teodoro, J.S.; Furtado, E.; Rolo, A.P.; Palmeira, C.M.; Tralhão, J.G. Recent Insights into Mitochondrial Targeting Strategies in Liver Transplantation. Int. J. Med. Sci. 2018, 15, 248–256. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and Endoplasmic Reticulum Calcium Homeostasis and Cell Death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Duchen, M.R. Mitochondria and Calcium: From Cell Signalling to Cell Death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef]
- Cervantes-Silva, M.P.; Cox, S.L.; Curtis, A.M. Alterations in Mitochondrial Morphology as a Key Driver of Immunity and Host Defence. EMBO Rep. 2021, 22, e53086–e53104. [Google Scholar] [CrossRef]
- Vringer, E.; Tait, S.W.G. Mitochondria and Inflammation: Cell Death Heats Up. Front. Cell Dev. Biol. 2019, 7, 100. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in Innate Immune Responses and Inflammatory Pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Liang, S.; Zhong, Z. Emerging Role of Mitochondrial DNA as a Major Driver of Inflammation and Disease Progression. Trends Immunol. 2019, 40, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in Inflammation and Immunity. EMBO Rep. 2020, 21, e49799–e49816. [Google Scholar] [CrossRef] [PubMed]
- Little, J.P.; Simtchouk, S.; Schindler, S.M.; Villanueva, E.B.; Gill, N.E.; Walker, D.G.; Wolthers, K.R.; Klegeris, A. Mitochondrial Transcription Factor A (Tfam) Is a pro-Inflammatory Extracellular Signaling Molecule Recognized by Brain Microglia. Mol. Cell. Neurosci. 2014, 60, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Chaung, W.W.; Wu, R.; Ji, Y.; Dong, W.; Wang, P. Mitochondrial Transcription Factor A Is a Proinflammatory Mediator in Hemorrhagic Shock. Int. J. Mol. Med. 2012, 30, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.-M.; Tarkowski, A. Endogenously Oxidized Mitochondrial DNA Induces in Vivo and in Vitro Inflammatory Responses. J. Leukoc. Biol. 2004, 75, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The Role of Mitochondria in NLRP3 Inflammasome Activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New Mitochondrial DNA Synthesis Enables NLRP3 Inflammasome Activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupfer, C.; Thomas, P.G.; Anand, P.K.; Vogel, P.; Milasta, S.; Martinez, J.; Huang, G.; Green, M.; Kundu, M.; Chi, H.; et al. Receptor Interacting Protein Kinase 2-Mediated Mitophagy Regulates Inflammasome Activation during Virus Infection. Nat. Immunol. 2013, 14, 480–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Q.; Wood, C.R.; Cimen, S.; Venkatachalam, A.B.; Alwayn, I.P.J. Mitochondrial Damage-Associated Molecular Patterns (MTDs) Are Released during Hepatic Ischemia Reperfusion and Induce Inflammatory Responses. PLoS ONE 2015, 10, e0140122. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Genovese, I.; Perrone, M.; Vezzani, B.; Vitto, V.A.M.; Giorgi, C. The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders. J. Clin. Med. 2020, 9, 740. [Google Scholar] [CrossRef] [Green Version]
- Selzner, N.; Rudiger, H.; Graf, R.; Clavien, P.A. Protective Strategies against Ischemic Injury of the Liver. Gastroenterology 2003, 125, 917–936. [Google Scholar] [CrossRef]
- Selzner, N.; Selzner, M.; Jochum, W.; Clavien, P.A. Ischemic Preconditioning Protects the Steatotic Mouse Liver against Reperfusion Injury: An ATP Dependent Mechanism. J. Hepatol. 2003, 39, 55–61. [Google Scholar] [CrossRef]
- Martins, R.M.; Teodoro, J.S.; Furtado, E.; Rolo, A.P.; Palmeira, C.M.; Tralhão, J.G. Evaluation of Bioenergetic and Mitochondrial Function in Liver Transplantation. Clin. Mol. Hepatol. 2019, 25, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Alexandrino, H.; Varela, A.T.; Teodoro, J.S.; Martins, M.A.; Rolo, A.P.; Tralhão, J.G.; Palmeira, C.M.; Castro e Sousa, F. Mitochondrial Bioenergetics and Posthepatectomy Liver Dysfunction. Eur. J. Clin. Investig. 2016, 46, 627–635. [Google Scholar] [CrossRef]
- Alexandrino, H.; Rolo, A.; Teodoro, J.S.; Donato, H.; Martins, R.; Serôdio, M.; Martins, M.; Tralhão, J.G.; Caseiro Alves, F.; Palmeira, C.; et al. Bioenergetic Adaptations of the Human Liver in the ALPPS Procedure—How Liver Regeneration Correlates with Mitochondrial Energy Status. HPB 2017, 19, 1091–1103. [Google Scholar] [CrossRef] [Green Version]
- Kong, W.N.; Li, W.; Bai, C.; Dong, Y.; Wu, Y.; An, W. Augmenter of Liver Regeneration-Mediated Mitophagy Protects against Hepatic Ischemia/Reperfusion Injury. Am. J. Transplant. 2021, 22, 130–143. [Google Scholar] [CrossRef]
- Gu, J.; Zhang, T.; Guo, J.; Chen, K.; Li, H.; Wang, J. PINK1 Activation and Translocation to Mitochondria-Associated Membranes Mediates Mitophagy and Protects Against Hepatic Ischemia/Reperfusion Injury. Shock 2020, 54, 783–793. [Google Scholar] [CrossRef]
- Sun, K.; Liu, Z.S.; Sun, Q. Role of Mitochondria in Cell Apoptosis during Hepatic Ischema-Reperfusion Injury and Protective Effect of Ischemic Postconditioning. World J. Gastroenterol. 2004, 10, 1934–1938. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Chen, L.; Lu, T.; Zhang, Y.; Sui, X.; Li, Y.; Huang, X.; He, L.; Cai, J.; Zhou, C.; et al. MSCs Ameliorate Hepatocellular Apoptosis Mediated by PINK1-Dependent Mitophagy in Liver Ischemia/Reperfusion Injury through AMPKα Activation. Cell Death Dis. 2020, 11, 256–275. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Fang, H.; Wei, W.; Dirsch, O.; Dahmen, U. Ischemic Preconditioning Protects against Liver Ischemia/Reperfusion Injury via Heme Oxygenase-1-Mediated Autophagy. Crit. Care Med. 2014, 42, e762–e771. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, J.; Xiong, X.; Xu, Y.; Zhang, H.; Huang, C.; Tian, Y.; Jiao, C.; Wang, X.; Li, X. Remote Ischemic Preconditioning Protects against Liver Ischemia-Reperfusion Injury via Heme Oxygenase-1-Induced Autophagy. PLoS ONE 2014, 9, e98846. [Google Scholar] [CrossRef]
- Clavien, P.A.; Yadav, S.; Sindram, D.; Bentley, R.C. Protective Effects of Ischemic Preconditioning for Liver Resection Performed under Inflow Occlusion in Humans. Ann. Surg. 2000, 232, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Rüdiger, H.A.; Graf, R.; Clavien, P.A. Sub-Lethal Oxidative Stress Triggers the Protective Effects of Ischemic Preconditioning in the Mouse Liver. J. Hepatol. 2003, 39, 972–977. [Google Scholar] [CrossRef]
- Tejima, K.; Arai, M.; Ikeda, H.; Tomiya, T.; Yanase, M.; Inoue, Y.; Nagashima, K.; Nishikawa, T.; Watanabe, N.; Omata, M.; et al. Ischemic Preconditioning Protects Hepatocytes via Reactive Oxygen Species Derived from Kupffer Cells in Rats. Gastroenterology 2004, 127, 1488–1496. [Google Scholar] [CrossRef]
- Petrowsky, H.; McCormack, L.; Trujillo, M.; Selzner, M.; Jochum, W.; Clavien, P.A. A Prospective, Randomized, Controlled Trial Comparing Intermittent Portal Triad Clamping versus Ischemic Preconditioning with Continuous Clamping for Major Liver Resection. Ann. Surg. 2006, 244, 921–928. [Google Scholar] [CrossRef]
- Rüdiger, H.A.; Kang, K.J.; Sindram, D.; Riehle, H.M.; Clavien, P.A. Comparison of Ischemic Preconditioning and Intermittent and Continuous Inflow Occlusion in the Murine Liver. Ann. Surg. 2002, 235, 400–407. [Google Scholar] [CrossRef] [Green Version]
- Ben Mosbah, I.; Duval, H.; Mbatchi, S.F.; Ribault, C.; Grandadam, S.; Pajaud, J.; Morel, F.; Boudjema, K.; Compagnon, P.; Corlu, A. Intermittent Selective Clamping Improves Rat Liver Regeneration by Attenuating Oxidative and Endoplasmic Reticulum Stress. Cell Death Dis. 2014, 5, e1107–e1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricca, L.; Lemoine, A.; Cauchy, F.; Hamelin, J.; Sebagh, M.; Esposti, D.D.; Salloum, C.; Vibert, E.; Balducci, G.; Azoulay, D. Ischemic Postconditioning of the Liver Graft in Adult Liver Transplantation. Transplantation 2015, 99, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Monbaliu, D.; Liu, Q.; Libbrecht, L.; de Vos, R.; Vekemans, K.; Debbaut, C.; Detry, O.; Roskams, T.; van Pelt, J.; Pirenne, J. Preserving the Morphology and Evaluating the Quality of Liver Grafts by Hypothermic Machine Perfusion: A Proof-of-Concept Study Using Discarded Human Livers. Liver Transplant. 2012, 18, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.H.; Lee, J.-H.; Ko, J.S.; Min, J.J.; Gwak, M.S.; Kim, G.S.; Lee, S.K. Effect of Remote Ischemic Postconditioning on Patients Undergoing Living Donor Liver Transplantation. Liver Transplant. 2014, 20, 1383–1392. [Google Scholar] [CrossRef] [Green Version]
- Tara, A.; Dominic, J.L.; Patel, J.N.; Garg, I.; Yeon, J.; Memon, M.S.; Gergal Gopalkrishna Rao, S.R.; Bugazia, S.; Dhandapani, T.P.M.; Kannan, A.; et al. Mitochondrial Targeting Therapy Role in Liver Transplant Preservation Lines: Mechanism and Therapeutic Strategies. Cureus 2021, 13, e16599. [Google Scholar] [CrossRef]
- Tapuria, N.; Junnarkar, S.; Abu-Amara, M.; Fuller, B.; Seifalian, A.M.; Davidson, B.R. Modulation of Microcirculatory Changes in the Late Phase of Hepatic Ischaemia-Reperfusion Injury by Remote Ischaemic Preconditioning. HPB 2012, 14, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Li, L.; Sun, H.; Li, H.; Wu, Y.; Zhang, X.; Zhang, J. Remote Ischemic Preconditioning Attenuates Hepatic Ischemia/Reperfusion Injury after Hemorrhagic Shock by Increasing Autophagy. Int. J. Med. Sci. 2021, 18, 873–882. [Google Scholar] [CrossRef]
- Choi, E.K.; Jung, H.; Jeon, S.; Lim, J.A.; Lee, J.; Kim, H.; Hong, S.W.; Jang, M.H.; Lim, D.G.; Kwak, K.H. Role of Remote Ischemic Preconditioning in Hepatic Ischemic Reperfusion Injury. Dose-Response 2020, 18, 1559325820946923–1559325820946929. [Google Scholar] [CrossRef]
- Kanoria, S.; Robertson, F.P.; Mehta, N.N.; Fusai, G.; Sharma, D.; Davidson, B.R. Effect of Remote Ischaemic Preconditioning on Liver Injury in Patients Undergoing Major Hepatectomy for Colorectal Liver Metastasis: A Pilot Randomised Controlled Feasibility Trial. World J. Surg. 2017, 41, 1322–1330. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Lin, S.; Liu, W.; Ding, H. Hepatic Remote Ischemic Preconditioning (RIPC) Protects Heart Damages Induced by Ischemia Reperfusion Injury in Mice. Front. Physiol. 2021, 12, 713564–713571. [Google Scholar] [CrossRef]
- van Rijn, R.; Schurink, I.J.; de Vries, Y.; van den Berg, A.P.; Cortes Cerisuelo, M.; Darwish Murad, S.; Erdmann, J.I.; Gilbo, N.; de Haas, R.J.; Heaton, N.; et al. Hypothermic Machine Perfusion in Liver Transplantation—A Randomized Trial. N. Engl. J. Med. 2021, 384, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, R.; Jassem, W.; Mergental, H.; Heaton, N.; Mirza, D.; Perera, M.T.P.R.; Quaglia, A.; Holroyd, D.; Vogel, T.; Coussios, C.C.; et al. Liver Transplantation After Ex Vivo Normothermic Machine Preservation: A Phase 1 (First-in-Man) Clinical Trial. Am. J. Transplant. 2016, 16, 1779–1787. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.C.; Liu, S.Y.; Lai, H.S.; Lai, I.R. Isolated Mitochondria Infusion Mitigates Ischemia-Reperfusion Injury of the Liver in Rats. Shock 2013, 39, 304–310. [Google Scholar] [CrossRef]
- Hayashida, K.; Takegawa, R.; Shoaib, M.; Aoki, T.; Choudhary, R.C.; Kuschner, C.E.; Nishikimi, M.; Miyara, S.J.; Rolston, D.M.; Guevara, S.; et al. Mitochondrial Transplantation Therapy for Ischemia Reperfusion Injury: A Systematic Review of Animal and Human Studies. J. Transl. Med. 2021, 19, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Ito, M.; Arai, M.; Hibino, M.; Tsujioka, T.; Harashima, H. Challenges in Promoting Mitochondrial Transplantation Therapy. Int. J. Mol. Sci. 2020, 21, 6365. [Google Scholar] [CrossRef]
- Fu, A.; Shi, X.; Zhang, H.; Fu, B. Mitotherapy for Fatty Liver by Intravenous Administration of Exogenous Mitochondria in Male Mice. Front. Pharmacol. 2017, 8, 239–241. [Google Scholar] [CrossRef] [Green Version]
- Soares, R.O.S.; Losada, D.M.; Jordani, M.C.; Évora, P.; Castro-E-Silva, O. Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies. Int. J. Mol. Sci. 2019, 20, 5034. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Wang, J.; Fan, Z.; Zhang, S.; Qiao, R.; Liu, Y.; Yang, J.; Yang, L.; Wang, H. Cardioprotective Effects of Melatonin against Myocardial Ischaemia/Reperfusion Injury: Activation of AMPK/Nrf2 Pathway. J. Cell. Mol. Med. 2021, 25, 6455–6459. [Google Scholar] [CrossRef]
- Kalimeris, K.; Briassoulis, P.; Ntzouvani, A.; Nomikos, T.; Papaparaskeva, K.; Politi, A.; Batistaki, C.; Kostopanagiotou, G. N-Acetylcysteine Ameliorates Liver Injury in a Rat Model of Intestinal Ischemia Reperfusion. J. Surg. Res. 2016, 206, 263–272. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, J.M.; Huang, H.; Kuznicki, M.; Zheng, S.; Sun, W.; Quan, N.; Wang, L.; Yang, H.; Guo, H.M.; et al. The Protective Effect of Trimetazidine on Myocardial Ischemia/Reperfusion Injury through Activating AMPK and ERK Signaling Pathway. Metab. Clin. Exp. 2016, 65, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Teodoro, J.S.; Varela, A.T.; Duarte, F.V.; Gomes, A.P.; Palmeira, C.M.; Rolo, A.P. Indirubin and NAD+ Prevent Mitochondrial Ischaemia/Reperfusion Damage in Fatty Livers. Eur. J. Clin. Investig. 2018, 48, e12932. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Xu, J.; Tian, F.; Hu, S.; Chen, Y.; Fu, Z. Melatonin Attenuates Myocardial Ischemia-Reperfusion Injury via Improving Mitochondrial Fusion/Mitophagy and Activating the AMPK-OPA1 Signaling Pathways. J. Pineal Res. 2019, 66, e12542. [Google Scholar] [CrossRef] [PubMed]
- Zhai, M.; Li, B.; Duan, W.; Jing, L.; Zhang, B.; Zhang, M.; Yu, L.; Liu, Z.; Yu, B.; Ren, K.; et al. Melatonin Ameliorates Myocardial Ischemia Reperfusion Injury through SIRT3-Dependent Regulation of Oxidative Stress and Apoptosis. J. Pineal Res. 2017, 63, e12419. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Chen, Y.T.; Yang, C.C.; Chen, K.H.; Sung, P.H.; Chiang, H.J.; Chen, C.H.; Chua, S.; Chung, S.Y.; Chen, Y.L.; et al. Melatonin Pretreatment Enhances the Therapeutic Effects of Exogenous Mitochondria against Hepatic Ischemia–Reperfusion Injury in Rats through Suppression of Mitochondrial Permeability Transition. J. Pineal Res. 2016, 61, 52–68. [Google Scholar] [CrossRef]
- Jegatheeswaran, S.; Siriwardena, A.K. Experimental and Clinical Evidence for Modification of Hepatic Ischaemia-Reperfusion Injury by N-Acetylcysteine during Major Liver Surgery. HPB 2011, 13, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Pu, L.Y.; Lu, L.; Wang, X.H.; Zhang, F.; Rao, J.H. N-Acetylcysteine Attenuates Reactive-Oxygen-Speciesmediated Endoplasmic Reticulum Stress during Liver Ischemia-Reperfusion Injury. World J. Gastroenterol. 2014, 20, 15289–15298. [Google Scholar] [CrossRef] [Green Version]
- Cayuela, N.C.; Koike, M.K.; de Fátima Jacysyn, J.; Rasslan, R.; Cerqueira, A.R.A.; Costa, S.K.P.; Diniz-Júnior, J.A.P.; Utiyama, E.M.; de Souza Montero, E.F. N-Acetylcysteine Reduced Ischemia and Reperfusion Damage Associated with Steatohepatitis in Mice. Int. J. Mol. Sci. 2020, 21, 4106. [Google Scholar] [CrossRef]
- Varela, A.T.; Rolo, A.P.; Palmeira, C.M. Fatty Liver and Ischemia/Reperfusion: Are There Drugs Able to Mitigate Injury? Curr. Med. Chem. 2011, 18, 4987–5002. [Google Scholar] [CrossRef]
- Tikhaze, A.K.; Lankin, V.Z.; Zharova, E.A.; Kolycheva, S.V. Trimetazidine as Indirect Antioxidant. Bull. Exp. Biol. Med. 2000, 130, 951–953. [Google Scholar] [CrossRef]
- Pantazi, E.; Zaouali, M.A.; Bejaoui, M.; Folch-Puy, E.; Abdennebi, H.B.; Varela, A.T.; Rolo, A.P.; Palmeira, C.M.; Roselló-Catafau, J. Sirtuin 1 in Rat Orthotopic Liver Transplantation: An IGL-1 Preservation Solution Approach. World J. Gastroenterol. 2015, 21, 1765–1774. [Google Scholar] [CrossRef]
- Varela, A.T.; Simões, A.M.; Teodoro, J.S.; Duarte, F.V.; Gomes, A.P.; Palmeira, C.M.; Rolo, A.P. Indirubin-3’-Oxime Prevents Hepatic I/R Damage by Inhibiting GSK-3β and Mitochondrial Permeability Transition. Mitochondrion 2010, 10, 456–463. [Google Scholar] [CrossRef]
- Hurst, S.; Gonnot, F.; Dia, M.; Crola Da Silva, C.; Gomez, L.; Sheu, S.S. Phosphorylation of Cyclophilin D at Serine 191 Regulates Mitochondrial Permeability Transition Pore Opening and Cell Death after Ischemia-Reperfusion. Cell Death Dis. 2020, 11, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.L.; Costa, A.S.H.; Gruszczyk, A.V.; Beach, T.E.; Allen, F.M.; Prag, H.A.; Hinchy, E.C.; Mahbubani, K.; Hamed, M.; Tronci, L.; et al. Succinate Accumulation Drives Ischaemia-Reperfusion Injury during Organ Transplantation. Nat. Metab. 2019, 1, 966–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, A.; Szczepanek, K.; Hu, Y.; Lesnefsky, E.J.; Chen, Q. Cardioprotection by Modulation of Mitochondrial Respiration during Ischemia-Reperfusion: Role of Apoptosis-Inducing Factor. Biochem. Biophys. Res. Commun. 2013, 435, 627–633. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cochemé, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-Nitrosation of a Cysteine Switch on Mitochondrial Complex i. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portakal, O.; Inal-Erden, M. Effects of Pentoxifylline and Coenzyme Q10 in Hepatic Ischemia/Reperfusion Injury. Clin. Biochem. 1999, 32, 461–466. [Google Scholar] [CrossRef]
- Genova, M.L.; Bonacorsi, E.; D’Aurelio, M.; Formiggini, G.; Nardo, B.; Cuccomarino, S.; Turi, P.; Pich, M.M.; Lenaz, G.; Bovina, C. Protective Effect of Exogenous Coenzyme Q in Rats Subjected to Partial Hepatic Ischemia and Reperfusion. BioFactors 1999, 9, 345–349. [Google Scholar] [CrossRef]
- Cherkashina, D.V.; Sosimchik, I.A.; Semenchenko, O.A.; Volina, V.V.; Petrenko, A.Y. Mitochondria-Targeted Plastoquinone Derivative SkQ 1 Decreases Ischemia-Reperfusion Injury during Liver Hypothermic Storage for Transplantation. Biochemistry 2011, 76, 1022–1029. [Google Scholar] [CrossRef]
- van Golen, R.F.; Reiniers, M.J.; Marsman, G.; Alles, L.K.; van Rooyen, D.M.; Petri, B.; van der Mark, V.A.; van Beek, A.A.; Meijer, B.; Maas, M.A.; et al. The Damage-Associated Molecular Pattern HMGB1 Is Released Early after Clinical Hepatic Ischemia/Reperfusion. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1192–1200. [Google Scholar] [CrossRef]
- Brown, D.A.; Hale, S.L.; Baines, C.P.; Rio, C.L.D.; Hamlin, R.L.; Yueyama, Y.; Kijtawornrat, A.; Yeh, S.T.; Frasier, C.R.; Stewart, L.M.; et al. Reduction of Early Reperfusion Injury with the Mitochondria-Targeting Peptide Bendavia. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Kezic, A.; Spasojevic, I.; Lezaic, V.; Bajcetic, M. Mitochondria-Targeted Antioxidants: Future Perspectives in Kidney Ischemia Reperfusion Injury. Oxidative Med. Cell. Longev. 2016, 2016, 2950503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musleh, W.; Bruce, A.; Malfroy, B.; Baudry, M. Effects of EUK-8, a Synthetic Catalytic Superoxide Scavenger, on Hypoxia- and Acidosis-Induced Damage in Hippocampal Slices. Neuropharmacology 1994, 33, 929–934. [Google Scholar] [CrossRef]
- Davila, A.; Liu, L.; Chellappa, K.; Redpath, P.; Nakamaru-Ogiso, E.; Paolella, L.M.; Zhang, Z.; Migaud, M.E.; Rabinowitz, J.D.; Baur, J.A. Nicotinamide Adenine Dinucleotide Is Transported into Mammalian Mitochondria. eLife 2018, 7, e33246. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Byun, J.; Zhai, P.; Ikeda, Y.; Oka, S.; Sadoshima, J. Nicotinamide Mononucleotide, an Intermediate of NAD+ Synthesis, Protects the Heart from Ischemia and Reperfusion. PLoS ONE 2014, 9, e98972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toropova, Y.G.; Pechnikova, N.A.; Zelinskaya, I.A.; Zhuravsky, S.G.; Kornyushin, O.V.; Gonchar, A.I.; Ivkin, D.Y.; Leonova, Y.V.; Karev, V.E.; Karabak, I.A. Nicotinamide Riboside Has Protective Effects in a Rat Model of Mesenteric Ischaemia-Reperfusion. Int. J. Exp. Pathol. 2018, 99, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Kaur, T.; Sharma, A.K.; Kaur, J.; Yadav, H.N.; Pathak, D.; Singh, A.P. Fenofibrate Attenuates Ischemia Reperfusion-Induced Acute Kidney Injury and Associated Liver Dysfunction in Rats. Drug Dev. Res. 2021, 82, 412–421. [Google Scholar] [CrossRef]
- Somi, M.H.; Hajipour, B.; Asl, N.A.; Estakhri, R.; Azar, A.N.; Zade, M.N.; Haghjou, A.G.; Vatankhah, A.M. Pioglitazone Attenuates Ischemia/Reperfusion-Induced Liver Injury in Rats. Transplant. Proc. 2009, 41, 4105–4109. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, D.; Gong, X.; Ge, P.; Dai, J.; Lin, L.; Zhang, L. Protective Benefits of AMP-Activated Protein Kinase in Hepatic Ischemia-Reperfusion Injury. Am. J. Transl. Res. 2017, 9, 823–829. [Google Scholar]
- Bi, J.; Zhang, J.; Ren, Y.; Du, Z.; Li, Q.; Wang, Y.; Wei, S.; Yang, L.; Zhang, J.; Liu, C.; et al. Irisin Alleviates Liver Ischemia-Reperfusion Injury by Inhibiting Excessive Mitochondrial Fission, Promoting Mitochondrial Biogenesis and Decreasing Oxidative Stress. Redox Biol. 2019, 20, 296–306. [Google Scholar] [CrossRef]
- Dusabimana, T.; Kim, S.R.; Kim, H.J.; Kim, H.; Park, S.W. Nobiletin Ameliorates Hepatic Ischemia and Reperfusion Injury through the Activation of SIRT-1/FOXO3a-Mediated Autophagy and Mitochondrial Biogenesis. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Jia, L.; Yu, W.; Du, H. Dephosphorylation by Calcineurin Regulates Translocation of Dynamin-Related Protein 1 to Mitochondria in Hepatic Ischemia Reperfusion Induced Hippocampus Injury in Young Mice. Brain Res. 2019, 1711, 68–76. [Google Scholar] [CrossRef]
- de la Cruz López, K.G.; Toledo Guzmán, M.E.; Sánchez, E.O.; García Carrancá, A. MTORC1 as a Regulator of Mitochondrial Functions and a Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 1373–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Guo, J.; Gu, J.; Chen, K.; Li, H.; Wang, J. Protective Role of MTOR in Liver Ischemia/Reperfusion Injury: Involvement of Inflammation and Autophagy. Oxidative Med. Cell. Longev. 2019, 2019, 7861290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Hang, H.; Huang, M.; Li, J.; Xu, D.; Jiao, J.; Wang, F.; Wu, H.; Sun, X.; Gu, J.; et al. DJ-1 Deficiency in Hepatocytes Improves Liver Ischemia-Reperfusion Injury by Enhancing Mitophagy. CMGH 2021, 12, 567–584. [Google Scholar] [CrossRef]
- Shi, Q.; Zhao, G.; Wei, S.; Guo, C.; Wu, X.; Zhao, R.C.; Di, G. Pterostilbene Alleviates Liver Ischemia/Reperfusion Injury via PINK1-Mediated Mitophagy. J. Pharmacol. Sci. 2022, 148, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Konukoǧlu, D.; Taşci, I.; Çetinkale, O. Effects of Cyclosporin A and Ibuprofen on Liver Ischemia-Reperfusion Injury in the Rat. Clin. Chim. Acta 1998, 275, 1–8. [Google Scholar] [CrossRef]
- Travis, D.L.; Fabia, R.; Netto, G.G.; Husberg, B.S.; Goldstein, R.M.; Klintmalm, G.B.; Levy, M.F. Protection by Cyclosporine a against Normothermic Liver Ischemia- Reperfusion in Pigs. J. Surg. Res. 1998, 75, 116–126. [Google Scholar] [CrossRef]
- Theruvath, T.P.; Zhong, Z.; Pediaditakis, P.; Ramshesh, V.K.; Currin, R.T.; Tikunov, A.; Holmuhamedov, E.; Lemasters, J.J. Minocycline and N-Methyl-4-Isoleucine Cyclosporin (NIM811) Mitigate Storage/Reperfusion Injury after Rat Liver Transplantation through Suppression of the Mitochondrial Permeability Transition. Hepatology 2008, 47, 236–246. [Google Scholar] [CrossRef] [Green Version]
- Clarke, S.J.; McStay, G.P.; Halestrap, A.P. Sanglifehrin A Acts as a Potent Inhibitor of the Mitochondrial Permeability Transition and Reperfusion Injury of the Heart by Binding to Cyclophilin-D at a Different Site from Cyclosporin A. J. Biol. Chem. 2002, 277, 34793–34799. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.H.; Ferreira, J.C.B.; Gross, E.R.; Mochly-Rosen, D. Targeting Aldehyde Dehydrogenase 2: New Therapeutic Opportunities. Physiol. Rev. 2014, 94, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Zaouali, M.A.; Abdennebi, H.B.; Padrissa-Altés, S.; Alfany-Fernandez, I.; Rimola, A.; Roselló-Catafau, J. How Institut Georges Lopez Preservation Solution Protects Nonsteatotic and Steatotic Livers against Ischemia-Reperfusion Injury. Transplant. Proc. 2011, 43, 77–79. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Zhang, Q.; Luo, Q.; Ying, Y.; Liu, Y.; Li, Y.; Wei, W.; Yan, F.; Zhang, H. Alda-1 Attenuates Lung Ischemia-Reperfusion Injury by Reducing 4-Hydroxy-2-Nonenal in Alveolar Epithelial Cells. Crit. Care Med. 2016, 44, e544. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; He, G.; Wang, J.; Wang, Y.; Chen, W. Pretreatment with the ALDH2 Agonist Alda-1 Reduces Intestinal Injury Induced by Ischaemia and Reperfusion in Mice. Clin. Sci. 2017, 131, 1123–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Xu, M.; Li, J.; Chen, L.; Xu, D.; Tong, Y.; Zhang, J.; Wu, H.; Kong, X.; Xia, Q. Alda-1 Ameliorates Liver Ischemia-Reperfusion Injury by Activating Aldehyde Dehydrogenase 2 and Enhancing Autophagy in Mice. J. Immunol. Res. 2018, 2018, 9807139. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Ye, S.; Zhong, X.; Wang, W.; Lai, C.H.; Yang, W.; Yue, P.; Luo, J.; Huang, X.; Zhong, Z.; et al. Pretreatment with the ALDH2 Activator Alda-1 Protects Rat Livers from Ischemia/Reperfusion Injury by Inducing Autophagy. Mol. Med. Rep. 2020, 22, 2373–2385. [Google Scholar] [CrossRef] [PubMed]
- Puts, C.F.; Berendsen, T.A.; Bruinsma, B.G.; Ozer, S.; Luitje, M.; Usta, O.B.; Yarmush, M.L.; Uygun, K. Polyethylene Glycol Protects Primary Hepatocytes during Supercooling Preservation. Cryobiology 2015, 71, 125–129. [Google Scholar] [CrossRef] [Green Version]
- Zaouali, M.A.; Bejaoui, M.; Calvo, M.; Folch-Puy, E.; Pantazi, E.; Pasut, G.; Rimola, A.; Abdennebi, H.B.; Adam, R.; Roselló-Catafau, J. Polyethylene Glycol Rinse Solution: An Effective Way to Prevent Ischemia-Reperfusion Injury. World J. Gastroenterol. 2014, 20, 16203–16214. [Google Scholar] [CrossRef]
- Ferrero-Andrés, A.; Panisello-Roselló, A.; Serafín, A.; Roselló-Catafau, J.; Folch-Puy, E. Polyethylene Glycol 35 (PEG35) Protects against Inflammation in Experimental Acute Necrotizing Pancreatitis and Associated Lung Injury. Int. J. Mol. Sci. 2020, 21, 917. [Google Scholar] [CrossRef] [Green Version]
- Ackland, G.L.; Gutierrez Del Arroyo, A.; Yao, S.T.; Stephens, R.C.; Dyson, A.; Klein, N.J.; Singer, M.; Gourine, A.V. Low-Molecular-Weight Polyethylene Glycol Improves Survival in Experimental Sepsis. Crit. Care Med. 2010, 38, 629–636. [Google Scholar] [CrossRef]
- Panisello-Roselló, A.; Lopez, A.; Folch-Puy, E.; Carbonell, T.; Rolo, A.; Palmeira, C.; Adam, R.; Net, M.; Roselló-Catafau, J. Role of Aldehyde Dehydrogenase 2 in Ischemia Reperfusion Injury: An Update. World J. Gastroenterol. 2018, 24, 2984–2994. [Google Scholar] [CrossRef]
- Panisello-Roselló, A.; Alva, N.; Flores, M.; Lopez, A.; Benítez, C.C.; Folch-Puy, E.; Rolo, A.; Palmeira, C.; Adam, R.; Carbonell, T.; et al. Aldehyde Dehydrogenase 2 (ALDH2) in Rat Fatty Liver Cold Ischemia Injury. Int. J. Mol. Sci. 2018, 19, 2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardallo, R.G.; da Silva, R.T.; Carbonell, T.; Folch-Puy, E.; Palmeira, C.; Roselló-Catafau, J.; Pirenne, J.; Adam, R.; Panisello-Roselló, A. Role of Peg35, Mitochondrial Aldh2, and Glutathione in Cold Fatty Liver Graft Preservation: An Igl-2 Approach. Int. J. Mol. Sci. 2021, 22, 5332. [Google Scholar] [CrossRef]
- Nehrt, A.; Hamann, K.; Ouyang, H.; Shi, R. Polyethylene Glycol Enhances Axolemmal Resealing Following Transection in Cultured Cells and in Ex Vivo Spinal Cord. J. Neurotrauma 2010, 27, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Shi, R. Polyethylene Glycol Repairs Membrane Damage and Enhances Functional Recovery: A Tissue Engineering Approach to Spinal Cord Injury. Neurosci. Bull. 2013, 29, 460–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Philip, J.L.; Razzaque, M.A.; Lloyd, J.W.; Muller, C.M.; Akhter, S.A. High-Molecular-Weight Polyethylene Glycol Inhibits Myocardial Ischemia-Reperfusion Injury in Vivo. J. Thorac. Cardiovasc. Surg. 2015, 149, 588–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejaoui, M.; Pantazi, E.; Folch-Puy, E.; Panisello, A.; Calvo, M.; Pasut, G.; Rimola, A.; Navasa, M.; Adam, R.; Roselló-Catafau, J. Protective Effect of Intravenous High Molecular Weight Polyethylene Glycol on Fatty Liver Preservation. BioMed Res. Int. 2015, 2015, 79428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejaoui, M.; Pantazi, E.; Calvo, M.; Folch-Puy, E.; Serafín, A.; Pasut, G.; Panisello, A.; Adam, R.; Roselló-Catafau, J. Polyethylene Glycol Preconditioning: An Effective Strategy to Prevent Liver Ischemia Reperfusion Injury. Oxidative Med. Cell. Longev. 2016, 2016, 9096549. [Google Scholar] [CrossRef] [Green Version]
- Rosello, A.P.; da Silva, R.T.; Castro, C.; Bardallo, R.G.; Calvo, M.; Folch-Puy, E.; Carbonell, T.; Palmeira, C.; Catafau, J.R.; Adam, R. Polyethylene Glycol 35 as a Perfusate Additive for Mitochondrial and Glycocalyx Protection in Hope Liver Preservation. Int. J. Mol. Sci. 2020, 21, 5703. [Google Scholar] [CrossRef]
- Kahn, J.; Schemmer, P. Control of Ischemia-Reperfusion Injury in Liver Transplantation: Potentials for Increasing the Donor Pool. Visc. Med. 2018, 34, 444–448. [Google Scholar] [CrossRef]
- Feng, S.; Lai, J.C. Expanded Criteria Donors. Clin. Liver Dis. 2014, 18, 633–649. [Google Scholar] [CrossRef] [Green Version]
- Peralta, C.; Bulbena, O.; Xaus, C.; Prats, N.; Cutrin, J.C.; Poli, G.; Gelpi, E.; Roselló-Catafau, J. Ischemic Preconditioning: A Defense Mechanism against the Reactive Oxygen Species Generated after Hepatic Ischemia Reperfusion. Transplantation 2002, 73, 1203–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.R. Control of Water Content of Respiring Kidney Slices by Sodium Chloride and Polyethylene Glycol. J. Physiol. 1978, 282, 285–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gores, G.J.; Nieminen, A.L.; Wray, B.E.; Herman, B.; Lemasters, J.J. Intracellular PH during “chemical Hypoxia” in Cultured Rat Hepatocytes. Protection by Intracellular Acidosis against the Onset of Cell Death. J. Clin. Investig. 1989, 83, 386–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamieson, N.V.; Lindell, S.; Sundberg, R.; Southard, J.H.; Belzer, F.O. An Analysis of the Components in Uw Solution Using the Isolated Perfused Rabbit Liver. Transplantation 1988, 46, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Mangino, M.J. Preservation Methods for Kidney and Liver. Organogenesis 2009, 5, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Hu, X.; Xia, Z.; Liu, Z.; Zhong, Z.; Lu, Z.; Liu, A.; Ye, S.; Cao, Q.; Wang, Y.; et al. Mild Hypothermia Attenuates Hepatic Ischemia-Reperfusion Injury through Regulating the JAK2/STAT3-CPT1a-Dependent Fatty Acid β -Oxidation. Oxidative Med. Cell. Longev. 2020, 2020, 5849794. [Google Scholar] [CrossRef]
- Czigany, Z.; Lurje, I.; Schmelzle, M.; Schöning, W.; Öllinger, R.; Raschzok, N.; Sauer, I.M.; Tacke, F.; Strnad, P.; Trautwein, C.; et al. Ischemia-Reperfusion Injury in Marginal Liver Grafts and the Role of Hypothermic Machine Perfusion: Molecular Mechanisms and Clinical Implications. J. Clin. Med. 2020, 9, 846. [Google Scholar] [CrossRef] [Green Version]
- Brüggenwirth, I.M.A.; van Leeuwen, O.B.; de Vries, Y.; Bodewes, S.B.; Adelmeijer, J.; Wiersema-Buist, J.; Lisman, T.; Martins, P.N.; de Meijer, V.E.; Porte, R.J. Extended Hypothermic Oxygenated Machine Perfusion Enables Ex Situ Preservation of Porcine Livers for up to 24 Hours. JHEP Rep. 2020, 2, 100092. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.J.; Baicu, S.C. Current State of Hypothermic Machine Perfusion Preservation of Organs: The Clinical Perspective. Cryobiology 2010, 60, S20–S35. [Google Scholar] [CrossRef] [Green Version]
- Dutkowski, P.; Guarrera, J.V.; de Jonge, J.; Martins, P.N.; Porte, R.J.; Clavien, P.A. Evolving Trends in Machine Perfusion for Liver Transplantation. Gastroenterology 2019, 156, 1542–1547. [Google Scholar] [CrossRef] [Green Version]
- Collins, G.M.; Bravo-Shugarman, M.; Terasaki, P.I. Kidney Preservation for Transportation. Initial Perfusion and 30 Hours’ Ice Storage. Lancet 1969, 2, 1219–1222. [Google Scholar] [CrossRef]
- Giraud, S.; Thuillier, R.; Codas, R.; Manguy, E.; Barrou, B.; Valagier, A.; Puichaud, A.; Badet, L.; Nicolas, E.; Eugene, M.; et al. The Optimal Peg for Kidney Preservation: A Preclinical Porcine Study. Int. J. Mol. Sci. 2018, 19, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salahudeen, A.K. Cold Ischemic Injury of Transplanted Kidneys: New Insights from Experimental Studies. Am. J. Physiol. Ren. Physiol. 2004, 287, F181–F187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hüter, L.; Simon, T.P.; Weinmann, L.; Schuerholz, T.; Reinhart, K.; Wolf, G.; Amann, K.U.; Marx, G. Hydroxyethylstarch Impairs Renal Function and Induces Interstitial Proliferation, Macrophage Infiltration and Tubular Damage in an Isolated Renal Perfusion Model. Crit. Care 2009, 13, R23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicomb, W.N.; Hill, J.D.; Avery, J.; Collins, G.M. Optimal Cardioplegia and 24-Hour Heart Storage with Simplified UW Solution Containing Polyethylene Glycol. Transplantation 1990, 49, 261–264. [Google Scholar] [CrossRef]
- Daniel, M.R.; Wakerley, C.L. Factors Influencing the Survival of Cell Monolayers during Storage at 4°. Br. J. Exp. Pathol. 1976, 57, 95–118. [Google Scholar]
- Ganote, C.E.; Worstell, J.; Iannotti, J.P.; Kaltenbach, J.P. Cellular Swelling and Irreversible Myocardial Injury. Effects of Polyethylene Glycol and Mannitol in Perfused Rat Hearts. Am. J. Pathol. 1977, 88, 95–118. [Google Scholar]
- Adam, R.; Delvart, V.; Karam, V.; Ducerf, C.; Navarro, F.; Letoublon, C.; Belghiti, J.; Pezet, D.; Castaing, D.; le Treut, Y.P.; et al. Compared Efficacy of Preservation Solutions in Liver Transplantation: A Long-Term Graft Outcome Study from the European Liver Transplant Registry. Am. J. Transplant. 2015, 15, 395–406. [Google Scholar] [CrossRef]
- Franco-Gou, R.; Mosbah, I.B.; Serafin, A.; Abdennebi, H.B.; Roselló-Catafau, J.; Peralta, C. New Preservation Strategies for Preventing Liver Grafts against Cold Ischemia Reperfusion Injury. J. Gastroenterol. Hepatol. 2007, 22, 1120–1126. [Google Scholar] [CrossRef]
- Codas, R.; Petruzzo, P.; Morelon, E.; Lefrançois, N.; Danjou, F.; Berthillot, C.; Contu, P.; Espa, M.; Martin, X.; Badet, L. IGL-1 Solution in Kidney Transplantation: First Multi-Center Study. Clin. Transplant. 2009, 23, 337–342. [Google Scholar] [CrossRef]
- Dondéro, F.; Paugam-Burtz, C.; Danjou, F.; Stocco, J.; Durand, F.; Belghiti, J. A Randomized Study Comparing IGL-1 to the University of Wisconsin Preservation Solution in Liver Transplantation. Ann. Transplant. 2010, 15, 1–7. [Google Scholar] [PubMed]
- Hauet, T.; Eugene, M. A New Approach in Organ Preservation: Potential Role of New Polymers. Kidney Int. 2008, 74, 998–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valuckaite, V.; Seal, J.; Zaborina, O.; Tretiakova, M.; Testa, G.; Alverdy, J.C. High Molecular Weight Polyethylene Glycol (PEG 15-20) Maintains Mucosal Microbial Barrier Function during Intestinal Graft Preservation. J. Surg. Res. 2013, 183, 869–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaouali, M.A.; Mosbah, I.B.; Boncompagni, E.; Abdennebi, H.B.; Mitjavila, M.T.; Bartrons, R.; Freitas, I.; Rimola, A.; Roselló-Catafau, J. Hypoxia Inducible Factor-1α Accumulation in Steatotic Liver Preservation: Role of Nitric Oxide. World J. Gastroenterol. 2010, 16, 3499–3509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, A.; Muller, X.; Mueller, M.; Stepanova, A.; Kron, P.; de Rougemont, O.; Muiesan, P.; Clavien, P.A.; Galkin, A.; Meierhofer, D.; et al. Hypothermic Oxygenated Perfusion Protects from Mitochondrial Injury before Liver Transplantation. EBioMedicine 2020, 60, 103014–103028. [Google Scholar] [CrossRef]
- Czigany, Z.; Lurje, I.; Tolba, R.H.; Neumann, U.P.; Tacke, F.; Lurje, G. Machine Perfusion for Liver Transplantation in the Era of Marginal Organs—New Kids on the Block. Liver Int. 2019, 39, 228–249. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, A.; Kron, P.; Graf, R.; Dutkowski, P.; Clavien, P.A. Warm vs. Cold Perfusion Techniques to Rescue Rodent Liver Grafts. J. Hepatol. 2014, 61, 1267–1275. [Google Scholar] [CrossRef]
- Stegemann, J.; Minor, T. Energy Charge Restoration, Mitochondrial Protection and Reversal of Preservation Induced Liver Injury by Hypothermic Oxygenation Prior to Reperfusion. Cryobiology 2009, 58, 331–336. [Google Scholar] [CrossRef]
- Schlegel, A.; Muller, X.; Dutkowski, P. Hypothermic Machine Preservation of the Liver: State of the Art. Curr. Transplant. Rep. 2018, 5, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic Accumulation of Succinate Controls Reperfusion Injury through Mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Kron, P.; Schlegel, A.; Mancina, L.; Clavien, P.A.; Dutkowski, P. Hypothermic Oxygenated Perfusion (HOPE) for Fatty Liver Grafts in Rats and Humans. J. Hepatol. 2018, 68, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Dutkowski, P.; Furrer, K.; Tian, Y.; Graf, R.; Clavien, P.A. Novel Short-Term Hypothermic Oxygenated Perfusion (HOPE) System Prevents Injury in Rat Liver Graft from Non-Heart Beating Donor. Ann. Surg. 2006, 244, 968–976. [Google Scholar] [CrossRef]
- Zeng, Y.; Zhang, X.F.; Fu, B.M.; Tarbell, J.M. The Role of Endothelial Surface Glycocalyx in Mechanosensing and Transduction. Adv. Exp. Med. Biol. 2018, 1097, 1–27. [Google Scholar] [PubMed]
- Schiefer, J.; Faybik, P.; Koch, S.; Tudor, B.; Kollmann, D.; Kuessel, L.; Krenn, C.G.; Berlakovich, G.; Baron, D.M.; Baron-Stefaniak, J. Glycocalyx Damage within Human Liver Grafts Correlates with Graft Injury and Postoperative Graft Function after Orthotopic Liver Transplantation. Transplantation 2019, 104, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Lopez, A.; Panisello-Rosello, A.; Castro-Benitez, C.; Adam, R. Glycocalyx Preservation and NO Production in Fatty Livers—The Protective Role of High Molecular Polyethylene Glycol in Cold Ischemia Injury. Int. J. Mol. Sci. 2018, 19, 2375. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Components | UW | IGL-1 | IGL-2 | Celsior | Belzer-MPS |
|---|---|---|---|---|---|
| K+ (mmol/L) | 125 | 25 | 25 | 15 | 25 |
| Na+ (mmol/L) | 27 | 125 | 125 | 100 | 120 |
| Mg2+ (mmol/L) | 5 | 5 | 5 | 13 | 5 |
| SO42− (mmol/L) | 4 | 5 | 5 | - | 5 |
| Ca2+ (mmol/L) | - | 0.5 | - | 0.25 | 0.5 |
| Cl− (mmol/L) | - | - | - | 40 | - |
| Zn2+ (mmol/L) | - | - | 0.091 | - | - |
| Diphosphate (mmol/L) | 25 | 25 | 25 | - | 25 |
| HEPES (mmol/L) | - | - | - | - | 10 |
| Histidine (mmol/L) | - | - | 30 | 30 | - |
| Raffinose | - | 30 | - | - | - |
| Mannitol (mmol/L) | - | - | 60 | 60 | 30 |
| Lactobionic acid (mmol/L) | 105 | 100 | 80 | 80 | - |
| Dextrose (mmol/L) | - | - | - | - | 10 |
| Ribose (mmol/L) | - | - | - | - | 5 |
| Gluconate (mmol/L) | - | - | - | - | 85 |
| Hydroxyethyl starch (g/L) | 50 | - | - | - | 50 |
| Polyethylene glycol 35 (g/L) | - | 1 | 5 | - | - |
| Glutathione (mmol/L) | 3 | 3 | 9 | 3 | 3 |
| Allopurinol | - | 1 | - | - | - |
| Adenosine (mmol/L) | 5 | 5 | 5 | - | - |
| Glutamic acid (mmol/L) | - | - | - | 20 | - |
| Adenine (mmol/L) | - | - | - | - | 5 |
| NaNO2 (nmol/L) | - | - | 50 | - | - |
| pH | 7.4 | 7.4 | 7.4 | 7.4 | 7.4 |
| Osmolarity (mosmol/L) | 320 | 320 | 320 | 320 | 320 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teodoro, J.S.; Da Silva, R.T.; Machado, I.F.; Panisello-Roselló, A.; Roselló-Catafau, J.; Rolo, A.P.; Palmeira, C.M. Shaping of Hepatic Ischemia/Reperfusion Events: The Crucial Role of Mitochondria. Cells 2022, 11, 688. https://doi.org/10.3390/cells11040688
Teodoro JS, Da Silva RT, Machado IF, Panisello-Roselló A, Roselló-Catafau J, Rolo AP, Palmeira CM. Shaping of Hepatic Ischemia/Reperfusion Events: The Crucial Role of Mitochondria. Cells. 2022; 11(4):688. https://doi.org/10.3390/cells11040688
Chicago/Turabian StyleTeodoro, João S., Rui T. Da Silva, Ivo F. Machado, Arnau Panisello-Roselló, Joan Roselló-Catafau, Anabela P. Rolo, and Carlos M. Palmeira. 2022. "Shaping of Hepatic Ischemia/Reperfusion Events: The Crucial Role of Mitochondria" Cells 11, no. 4: 688. https://doi.org/10.3390/cells11040688
APA StyleTeodoro, J. S., Da Silva, R. T., Machado, I. F., Panisello-Roselló, A., Roselló-Catafau, J., Rolo, A. P., & Palmeira, C. M. (2022). Shaping of Hepatic Ischemia/Reperfusion Events: The Crucial Role of Mitochondria. Cells, 11(4), 688. https://doi.org/10.3390/cells11040688