
RNAi Screening Uncovers a Synthetic Sick Interaction between CtIP and the BARD1 Tumor Suppressor

 , , , , ,
, , , , , 
 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNAi Image-Based Screening and Data Analysis
2.3. Antibodies
2.4. siRNA Transfections and Sequences
2.5. Plasmids and Cloning
2.6. Immunoblotting and Immunoprecipitation Assays
2.7. Flow Cytometry Analysis
2.8. Immunofluorescence Microscopy
2.9. Quantitative Image-Based Cytometry (QIBC)
2.10. Cell Proliferation and Viability Assays
2.11. Colony Formation Assay
2.12. HDR Reporter Assay
2.13. DNA Fiber Spreading
2.14. Pulsed Field Gel Electrophoresis (PFGE)
2.15. Metaphase Spreads
2.16. Statistical Analysis
3. Results
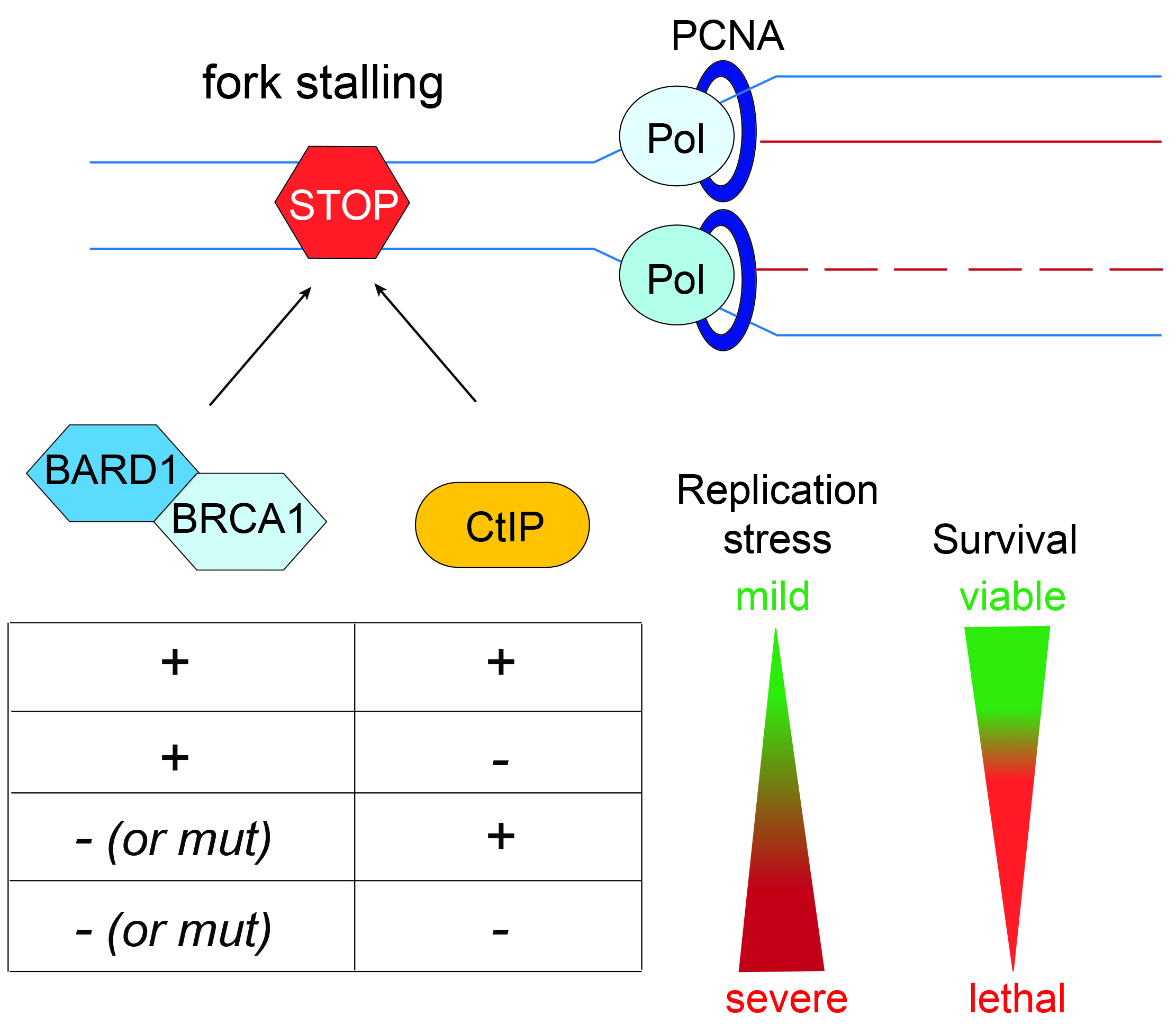
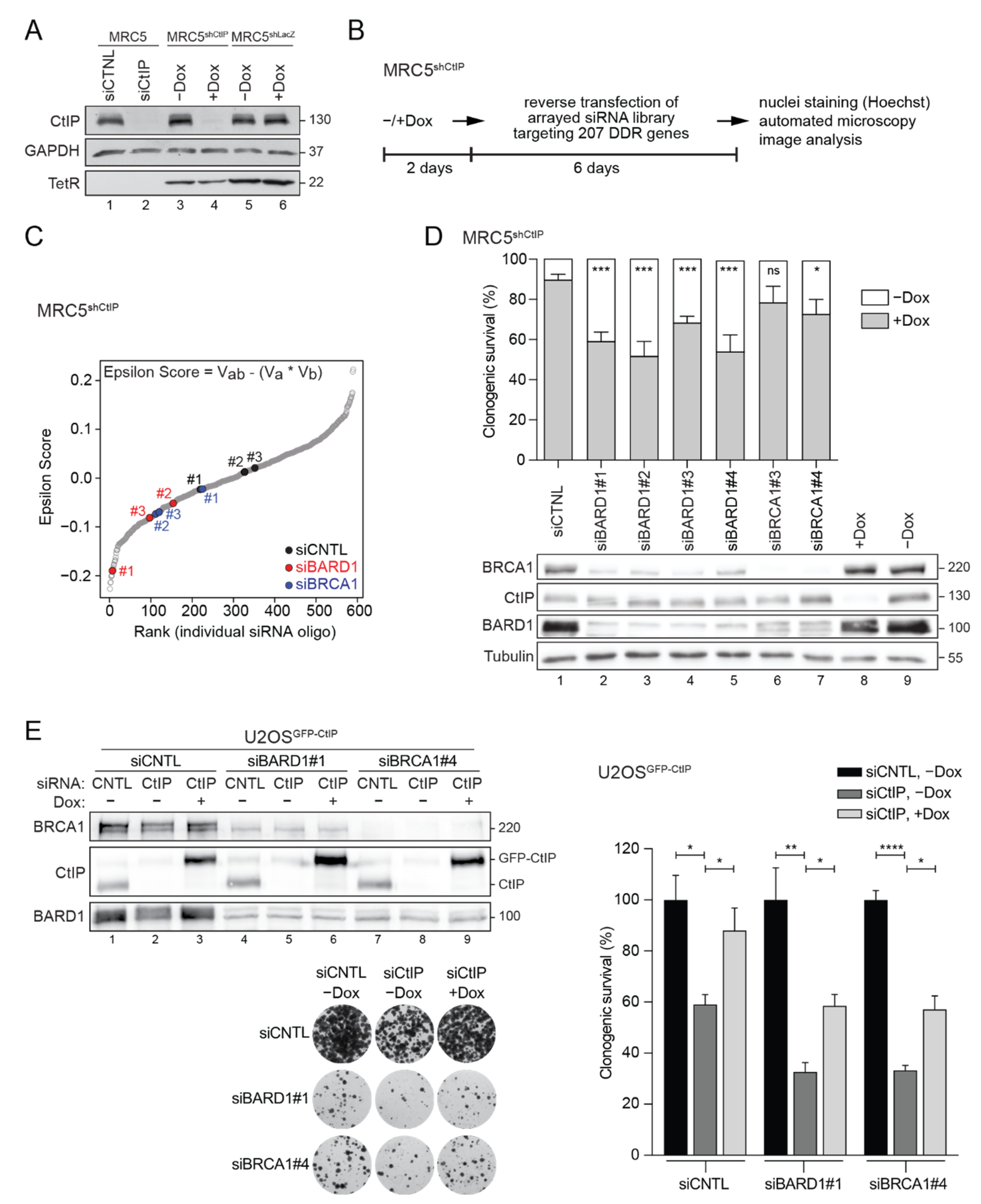
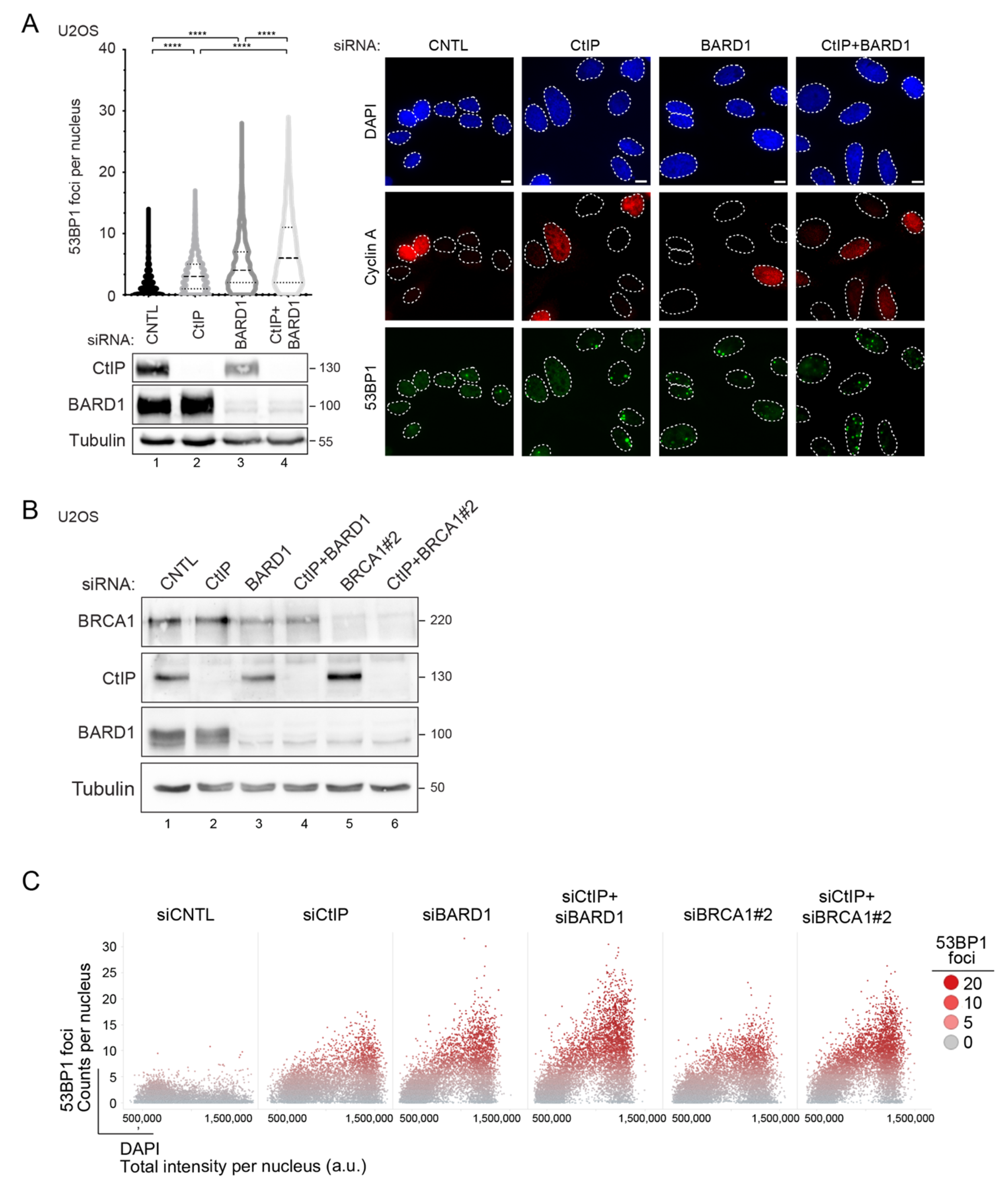
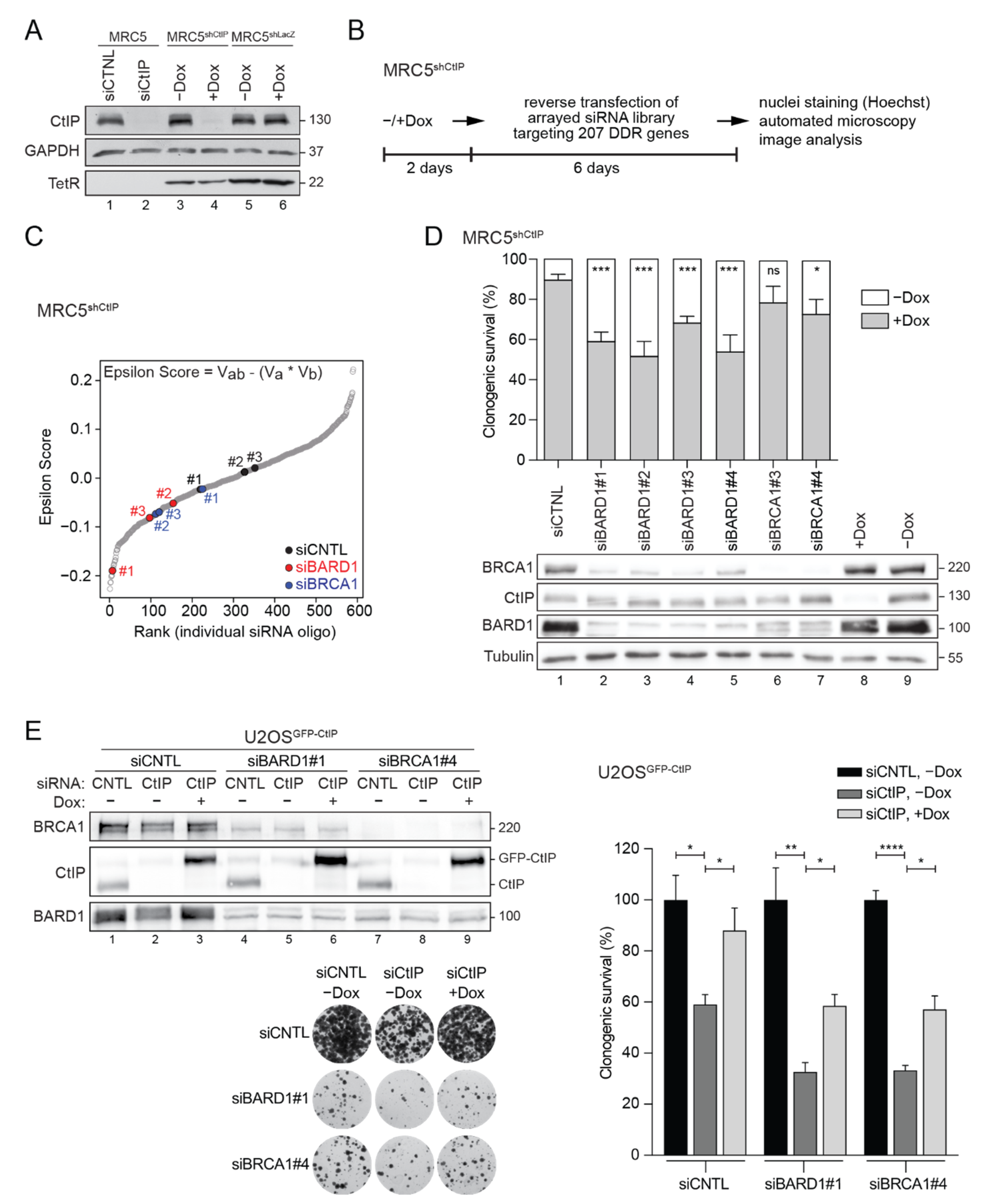
3.1. RNAi Screening Unveils a Negative Genetic Interaction between CtIP and BARD1
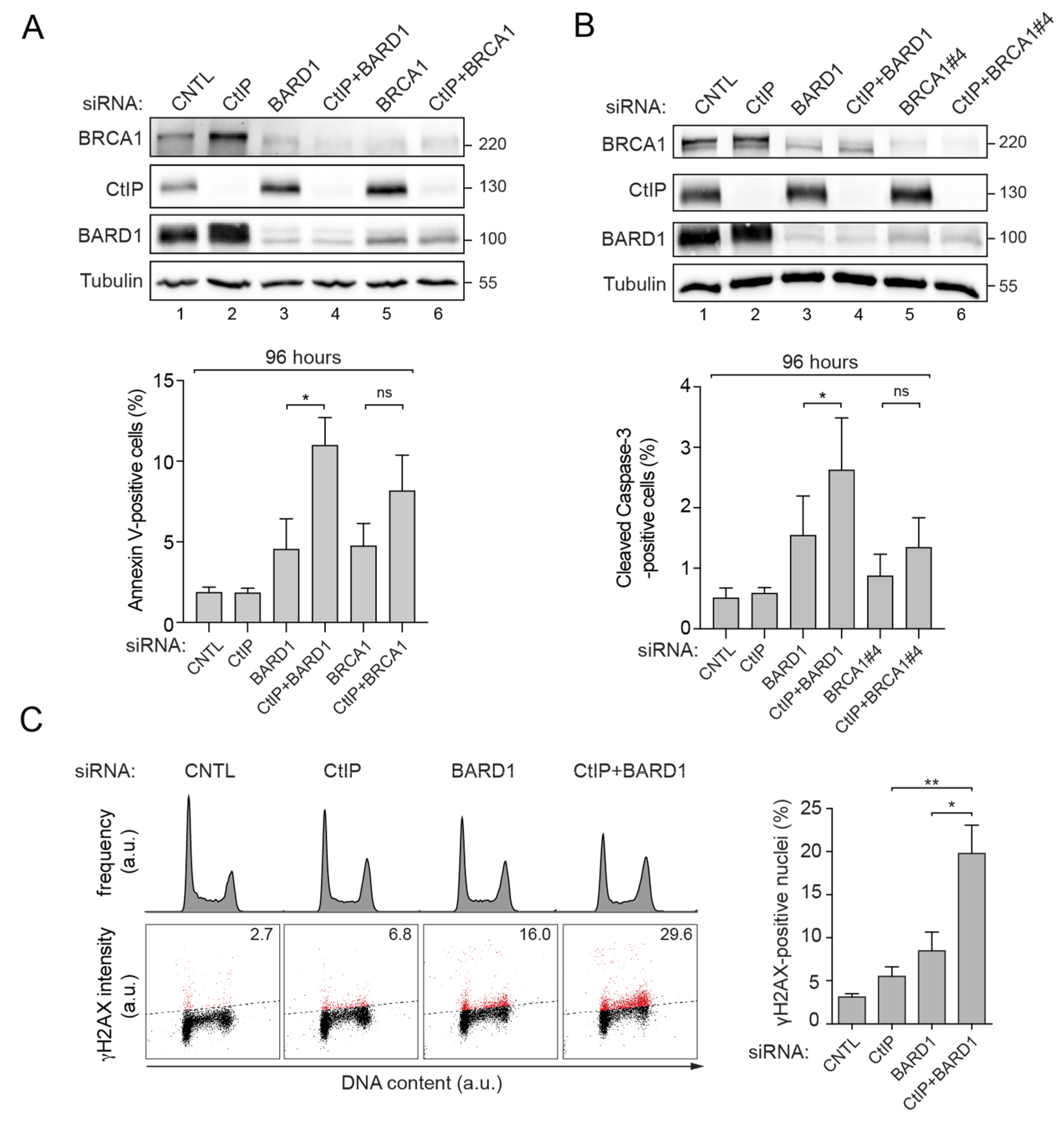
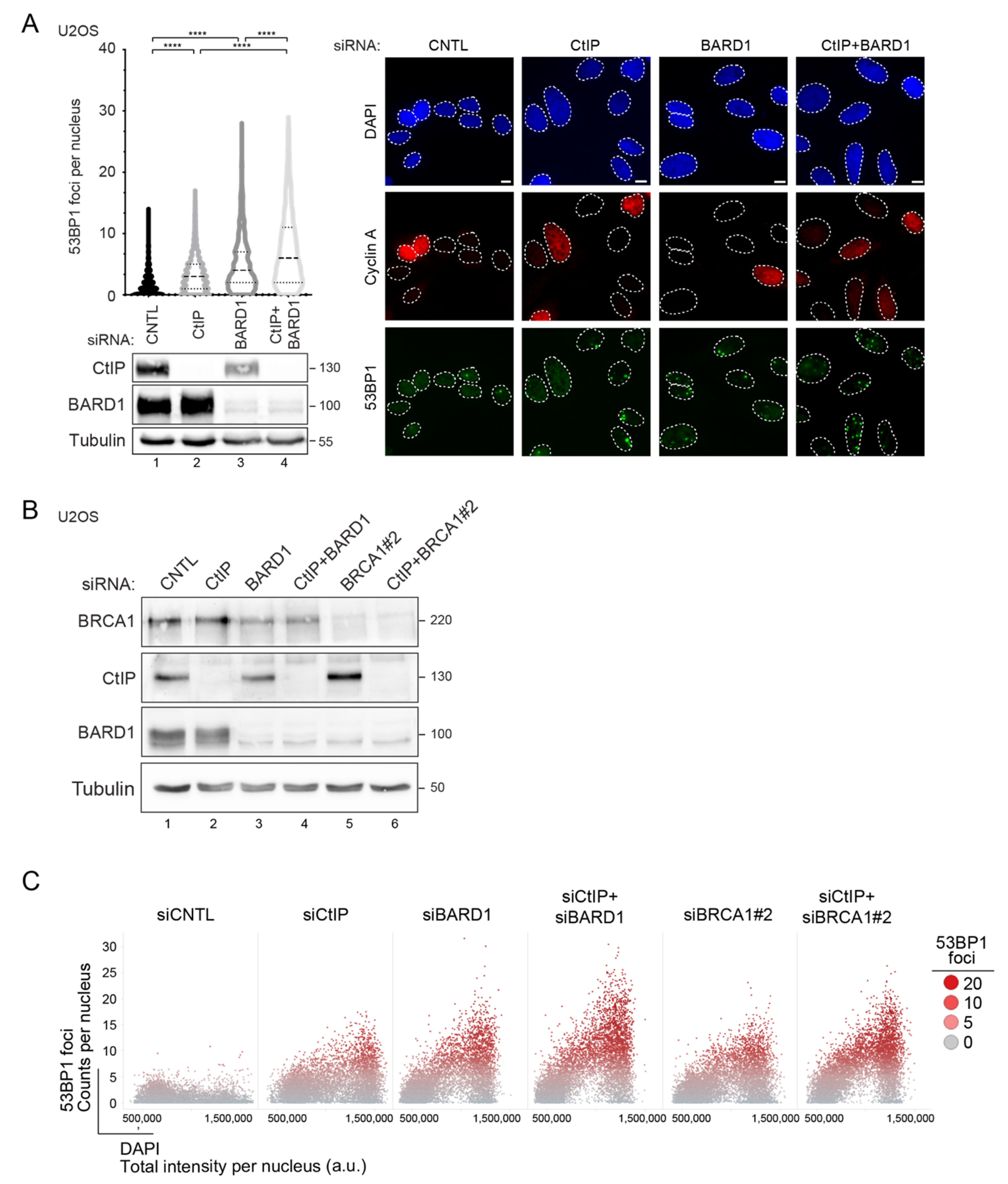
3.2. Prolonged Downregulation of CtIP and BARD1 Results in DNA Damage-Induced Apoptosis
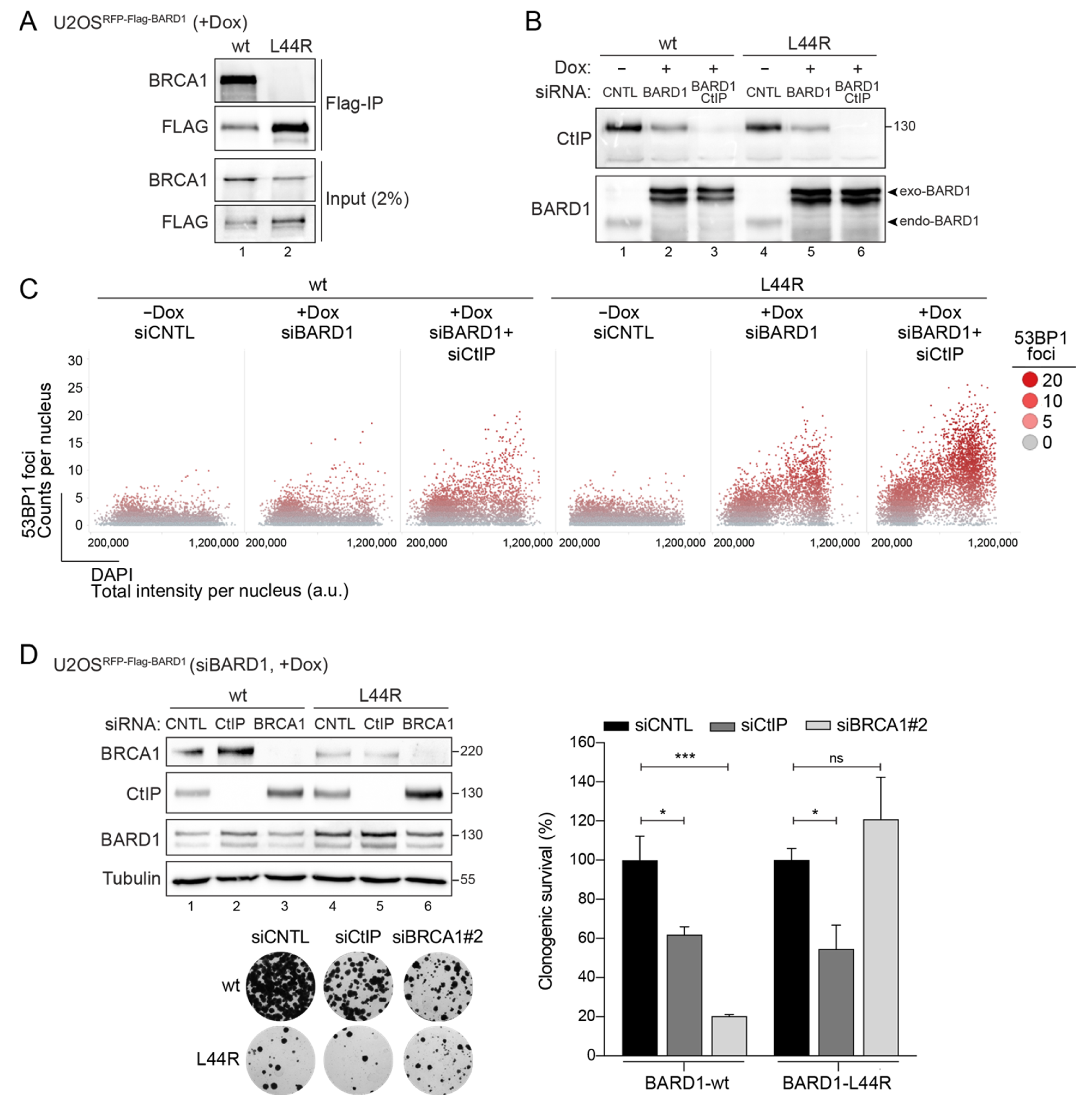
3.3. BARD1-L44R Mutant Defective in BRCA1 Binding Recapitulates SSL Phenotype Observed between CtIP and BARD1
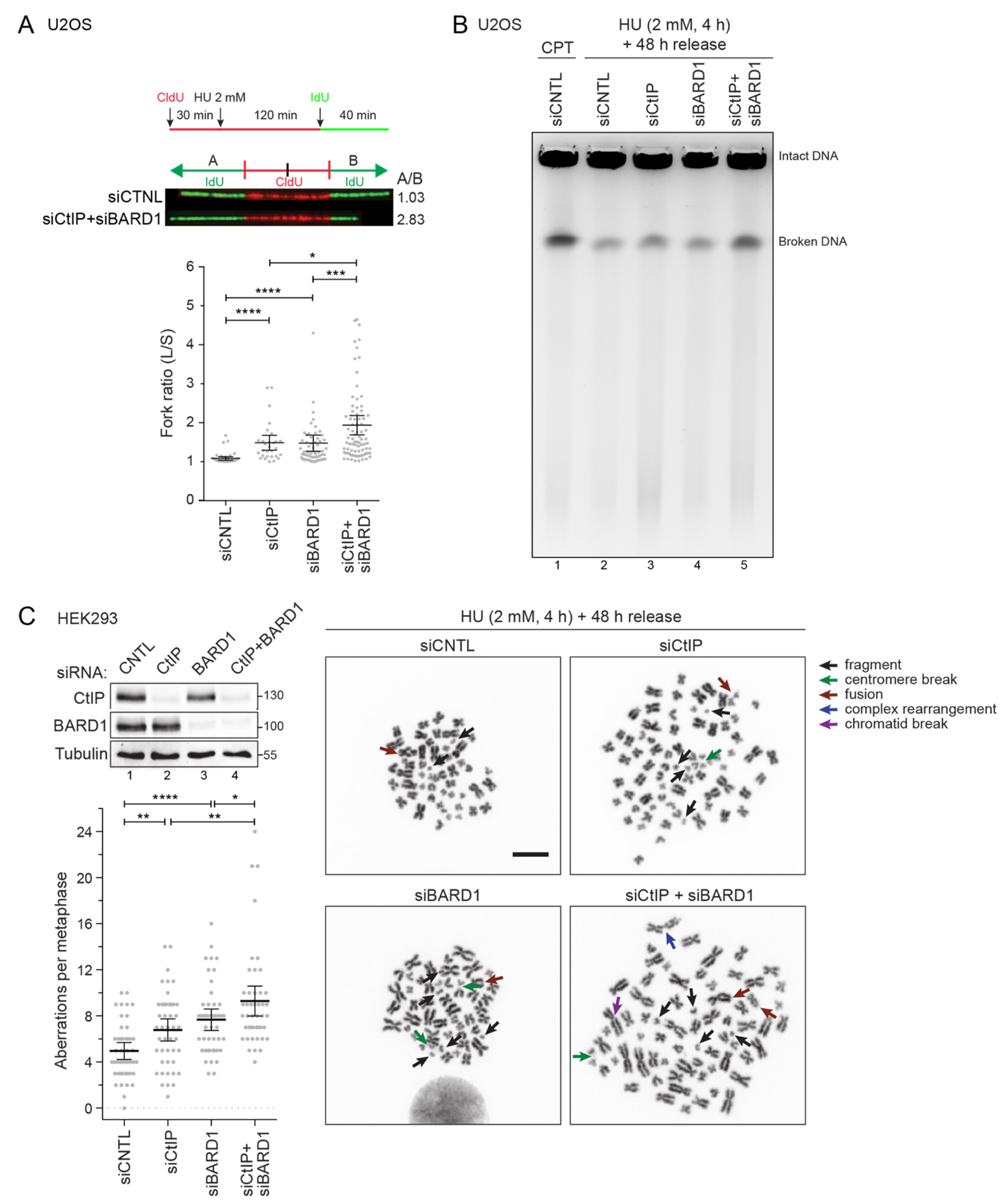
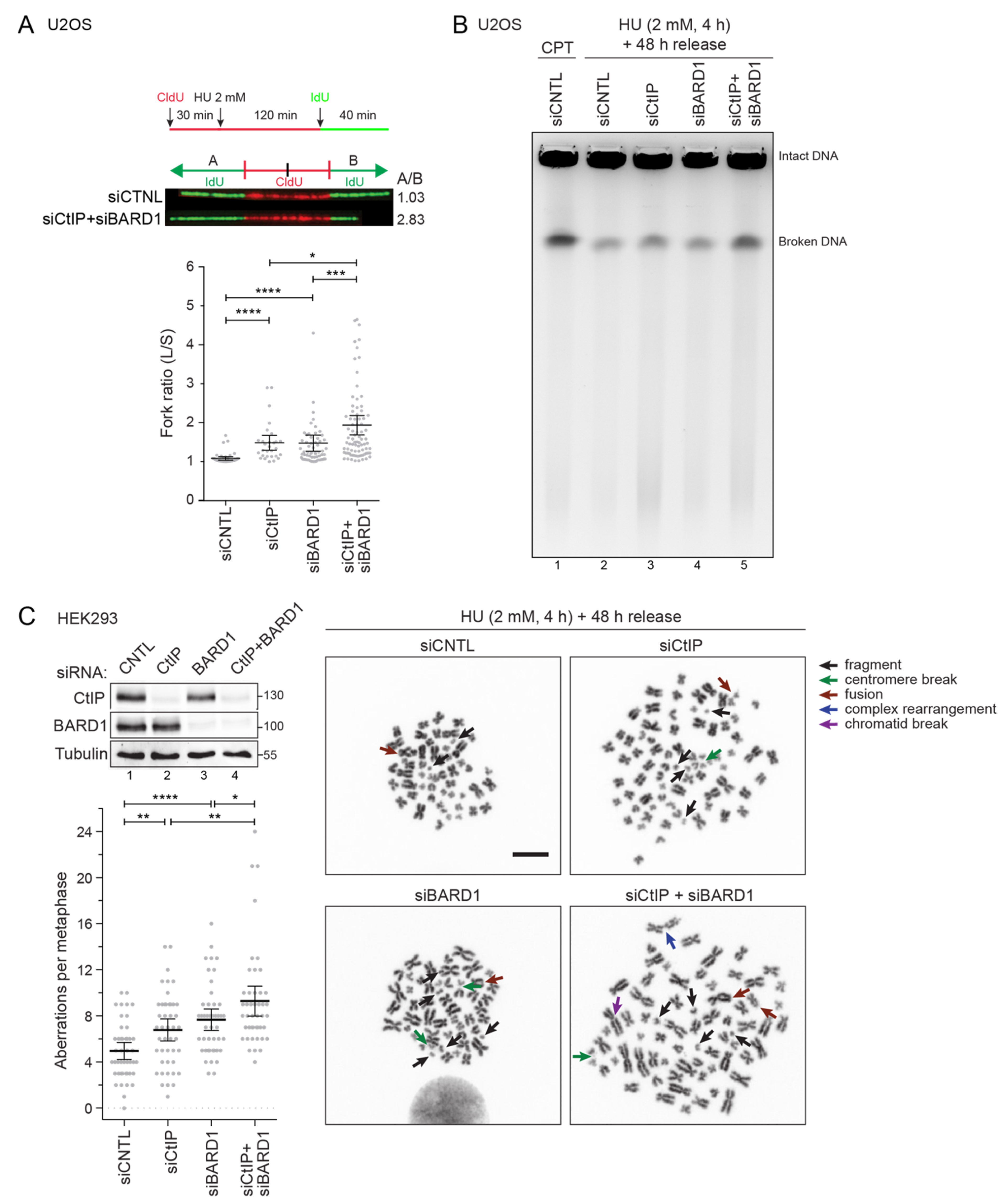
3.4. Combined Depletion of CtIP and BARD1 Fuels Replication Stress-Induced Genomic Instability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Petropoulos, M.; Champeris Tsaniras, S.; Taraviras, S.; Lygerou, Z. Replication Licensing Aberrations, Replication Stress, and Genomic Instability. Trends Biochem. Sci. 2019, 44, 752–764. [Google Scholar] [CrossRef]
- Rickman, K.; Smogorzewska, A. Advances in understanding DNA processing and protection at stalled replication forks. J. Cell Biol. 2019, 218, 1096–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol. 2018, 2, 313–336. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, J.E.; Lee, E.H.; Hendrickson, E.A.; Sobeck, A. CtIP mediates replication fork recovery in a FANCD2-regulated manner. Hum. Mol. Genet. 2014, 23, 3695–3705. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, Y.; Truong, L.N.; Shi, L.Z.; Hwang, P.Y.H.; He, J.; Do, J.; Cho, M.J.; Li, H.; Negrete, A.; et al. CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol. Cell 2014, 54, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Dungrawala, H.; Rose, K.L.; Bhat, K.P.; Mohni, K.N.; Glick, G.G.; Couch, F.B.; Cortez, D. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 2015, 59, 998–1010. [Google Scholar] [CrossRef] [Green Version]
- Przetocka, S.; Porro, A.; Bolck, H.A.; Walker, C.; Lezaja, A.; Trenner, A.; von Aesch, C.; Himmels, S.F.; D’Andrea, A.D.; Ceccaldi, R.; et al. CtIP-Mediated Fork Protection Synergizes with BRCA1 to Suppress Genomic Instability upon DNA Replication Stress. Mol. Cell 2018, 72, 568–582.e6. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Chen, J. DNA Damage-Induced Cell Cycle Checkpoint Control Requires CtIP, a Phosphorylation-Dependent Binding Partner of BRCA1 C-Terminal Domains. Mol. Cell. Biol. 2004, 24, 9478–9486. [Google Scholar] [CrossRef] [Green Version]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.F.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [Green Version]
- Yun, M.H.; Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009, 459, 460–463. [Google Scholar] [CrossRef]
- Chen, L.; Nievera, C.J.; Lee, A.Y.L.; Wu, X. Cell cycle-dependent complex formation of BRCA1·CtIP·MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Kogame, T.; Oshiumi, H.; Shinohara, A.; Sumitomo, Y.; Agama, K.; Pommier, Y.; Tsutsui, K.M.; Tsutsui, K.; Hartsuiker, E.; et al. Collaborative action of Brca1 and CtIP in elimination of covalent modifications from double-strand breaks to facilitate subsequent break repair. PLoS Genet. 2010, 6, e1000828. [Google Scholar] [CrossRef] [Green Version]
- Reczek, C.R.; Shakya, R.; Miteva, Y.; Szabolcs, M.; Ludwig, T.; Baer, R. The DNA resection protein CtIP promotes mammary tumorigenesis. Oncotarget 2016, 7, 32172–32183. [Google Scholar] [CrossRef] [Green Version]
- Polato, F.; Callen, E.; Wong, N.; Faryabi, R.; Bunting, S.; Chen, H.T.; Kozak, M.; Kruhlak, M.J.; Reczek, C.R.; Lee, W.H.; et al. CtIP-mediated resection is essential for viability and can operate independently of BRCA1. J. Exp. Med. 2014, 211, 1027–1036. [Google Scholar] [CrossRef]
- Kaelin, W.G. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Murina, O.; von Aesch, C.; Karakus, U.; Ferretti, L.P.; Bolck, H.A.; Hänggi, K.; Sartori, A.A. FANCD2 and CtIP cooperate to repair DNA interstrand crosslinks. Cell Rep. 2014, 7, 1030–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafranchi, L.; Boer, H.R.; Vries, E.G.; Ong, S.; Sartori, A.A.; Vugt, M.A. APC/CC dh1 controls Ct IP stability during the cell cycle and in response to DNA damage. EMBO J. 2014, 33, 2860–2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daza-Martin, M.; Starowicz, K.; Jamshad, M.; Tye, S.; Ronson, G.E.; MacKay, H.L.; Chauhan, A.S.; Walker, A.K.; Stone, H.R.; Beesley, J.F.J.; et al. Isomerization of BRCA1–BARD1 promotes replication fork protection. Nature 2019, 571, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.D. A pair of new statistical parameters for quality control in RNA interference high-throughput screening assays. Genomics 2007, 89, 552–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, S.R.; Schuldiner, M.; Krogan, N.J.; Weissman, J.S. A strategy for extracting and analyzing large-scale quantitative epistatic interaction data. Genome Biol. 2006, 7, R63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- König, R.; Chiang, C.Y.; Tu, B.P.; Yan, S.F.; DeJesus, P.D.; Romero, A.; Bergauer, T.; Orth, A.; Krueger, U.; Zhou, Y.; et al. A probability-based approach for the analysis of large-scale RNAi screens. Nat. Methods 2007, 4, 847–849. [Google Scholar] [CrossRef]
- Li, M.; Yu, X. Function of BRCA1 in the DNA Damage Response Is Mediated by ADP-Ribosylation. Cancer Cell 2013, 23, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Laufer, M.; Nandula, S.V.; Modi, A.P.; Wang, S.; Jasin, M.; Murty, V.V.V.S.; Ludwig, T.; Baer, R. Structural requirements for the BARD1 tumor suppressor in chromosomal stability and homology-directed DNA repair. J. Biol. Chem. 2007, 282, 34325–34333. [Google Scholar] [CrossRef] [Green Version]
- Forment, J.V.; Jackson, S.P. A flow cytometry-based method to simplify the analysis and quantification of protein association to chromatin in mammalian cells. Nat. Protoc. 2015, 10, 1297–1307. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, M.; Murina, O.; Reijns, M.A.M.; Agathanggelou, A.; Challis, R.; Tarnauskaite, Ž.; Muir, M.; Fluteau, A.; Aregger, M.; McEwan, A.; et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 2018, 559, 285–289. [Google Scholar] [CrossRef]
- Pellegrino, S.; Michelena, J.; Teloni, F.; Imhof, R.; Altmeyer, M. Replication-Coupled Dilution of H4K20me2 Guides 53BP1 to Pre-replicative Chromatin. Cell Rep. 2017, 19, 1819–1831. [Google Scholar] [CrossRef] [Green Version]
- Guzmán, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef]
- Davies, O.R.; Forment, J.V.; Sun, M.; Belotserkovskaya, R.; Coates, J.; Galanty, Y.; Demir, M.; Morton, C.R.; Rzechorzek, N.J.; Jackson, S.P.; et al. CtIP tetramer assembly is required for DNA-end resection and repair. Nat. Struct. Mol. Biol. 2015, 9, e92444. [Google Scholar] [CrossRef] [Green Version]
- Godau, J.; Ferretti, L.P.; Trenner, A.; Dubois, E.; von Aesch, C.; Marmignon, A.; Simon, L.; Kapusta, A.; Guérois, R.; Bétermier, M.; et al. Identification of a miniature Sae2/Ctp1/CtIP ortholog from Paramecium tetraurelia required for sexual reproduction and DNA double-strand break repair. DNA Repair 2019, 77, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Merrick, C.J.; Jackson, D.; Diffley, J.F.X. Visualization of Altered Replication Dynamics after DNA Damage in Human Cells. J. Biol. Chem. 2004, 279, 20067–20075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanada, K.; Budzowska, M.; Davies, S.L.; Van Drunen, E.; Onizawa, H.; Beverloo, H.B.; Maas, A.; Essers, J.; Hickson, I.D.; Kanaar, R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 2007, 14, 1096–1104. [Google Scholar] [CrossRef]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Mehta, M.; Kuo, D.; Sung, M.K.; Chuang, R.; Jaehnig, E.J.; Bodenmiller, B.; Licon, K.; Copeland, W.; Shales, M.; et al. Rewiring of genetic networks in response to DNA damage. Science 2010, 330, 1385–1389. [Google Scholar] [CrossRef] [Green Version]
- Guénolé, A.; Srivas, R.; Vreeken, K.; Wang, Z.Z.; Wang, S.; Krogan, N.J.; Ideker, T.; van Attikum, H. Dissection of DNA Damage Responses Using Multiconditional Genetic Interaction Maps. Mol. Cell 2013, 49, 346–358. [Google Scholar] [CrossRef] [Green Version]
- Bassik, M.C.; Kampmann, M.; Lebbink, R.J.; Wang, S.; Hein, M.Y.; Poser, I.; Weibezahn, J.; Horlbeck, M.A.; Chen, S.; Mann, M.; et al. A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell 2013, 152, 909–922. [Google Scholar] [CrossRef] [Green Version]
- Matos-Rodrigues, G.; Martini, E.; Lopez, B.S. Mouse models for deciphering the impact of homologous recombination on tumorigenesis. Cancers 2021, 13, 2083. [Google Scholar] [CrossRef]
- McCarthy, E.E.; Celebi, J.T.; Baer, R.; Ludwig, T. Loss of Bard1, the Heterodimeric Partner of the Brca1 Tumor Suppressor, Results in Early Embryonic Lethality and Chromosomal Instability. Mol. Cell. Biol. 2003, 152, 909–922. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T. Mechanisms of ATM activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grøhfte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.; Ideker, T. Systematic interpretation of genetic interactions using protein networks. Nat. Biotechnol. 2005, 23, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; Baryshnikova, A.; Myers, C.L.; Andrews, B.; Boone, C. Charting the genetic interaction map of a cell. Curr. Opin. Biotechnol. 2011, 22, 66–74. [Google Scholar] [CrossRef]
- Zinovyev, A.; Kuperstein, I.; Barillot, E.; Heyer, W.D. Synthetic Lethality between Gene Defects Affecting a Single Non-essential Molecular Pathway with Reversible Steps. PLoS Comput. Biol. 2013, 9, e1003016. [Google Scholar] [CrossRef] [Green Version]
- Roguev, A.; Talbot, D.; Negri, G.L.; Shales, M.; Cagney, G.; Bandyopadhyay, S.; Panning, B.; Krogan, N.J. Quantitative genetic-interaction mapping in mammalian cells. Nat. Methods 2013, 10, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, R.A.; Sobhian, B.; Pathania, S.; Cantor, S.B.; Nakatani, Y.; Livingston, D.M. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006, 20, 34–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B. BRCA1 tumor suppressor network: Focusing on its tail. Cell Biosci. 2012, 2, 1–10. [Google Scholar] [CrossRef]
- Morris, J.R.; Keep, N.H.; Solomon, E. Identification of residues required for the interaction of BARD1 with BRCA1. J. Biol. Chem. 2002, 277, 9382–9386. [Google Scholar] [CrossRef] [Green Version]
- Densham, R.M.; Garvin, A.J.; Stone, H.R.; Strachan, J.; Baldock, R.A.; Daza-Martin, M.; Fletcher, A.; Blair-Reid, S.; Beesley, J.; Johal, B.; et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol. 2016, 23, 647–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Zarrizi, R.; Higgs, M.R.; Voßgröne, K.; Rossing, M.; Bertelsen, B.; Bose, M.; Kousholt, A.N.; Rösner, H.; Ejlertsen, B.; Stewart, G.S.; et al. Germline RBBP8 variants associated with early-onset breast cancer compromise replication fork stability. J. Clin. Investig. 2020, 130, 4069–4080. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-L.; Liu, F.; Cai, S.; Lin, X.; Li, A.; Chen, Y.; Gu, B.; Lee, E.Y.-H.P.; Lee, W.-H. Inactivation of CtIP Leads to Early Embryonic Lethality Mediated by G 1 Restraint and to Tumorigenesis by Haploid Insufficiency. Mol. Cell. Biol. 2005, 25, 3535–3542. [Google Scholar] [CrossRef] [Green Version]
- Sallmyr, A.; Tomkinson, A.E. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 2018, 293, 10536–10546. [Google Scholar] [CrossRef] [Green Version]
- Mozaffari, N.L.; Pagliarulo, F.; Sartori, A.A. Human CtIP: A ‘double agent’ in DNA repair and tumorigenesis. Semin. Cell Dev. Biol. 2021, 113, 47–56. [Google Scholar] [CrossRef]
- Suszynska, M.; Kluzniak, W.; Wokolorczyk, D.; Jakubowska, A.; Huzarski, T.; Gronwald, J.; Debniak, T.; Szwiec, M.; Ratajska, M.; Klonowska, K.; et al. Bard1 is a low/moderate breast cancer risk gene: Evidence based on an association study of the central european p.q564x recurrent mutation. Cancers 2019, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Chen, P.L.; Subramanian, T.; Chinnadurai, G.; Tomlinson, G.; Osborne, C.K.; Sharp, Z.D.; Lee, W.H. Binding of CtIP to the BRCT repeats of BRCA1 involved in the transcription regulation of p21 is disrupted upon DNA damage. J. Biol. Chem. 1999, 274, 11334–11338. [Google Scholar] [CrossRef] [Green Version]
- Yasuhara, T.; Kato, R.; Hagiwara, Y.; Shiotani, B.; Yamauchi, M.; Nakada, S.; Shibata, A.; Miyagawa, K. Human Rad52 Promotes XPG-Mediated R-loop Processing to Initiate Transcription-Associated Homologous Recombination Repair. Cell 2018, 175, 558–570.e11. [Google Scholar] [CrossRef] [Green Version]
- Cruz-García, A.; López-Saavedra, A.; Huertas, P. BRCA1 accelerates CtIP-ediated DNA-end resection. Cell Rep. 2014, 9, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartek, J.; Lukas, J.; Bartkova, J. DNA damage response as an anti-cancer barrier: Damage threshold and the concept of “conditional haploinsufficiency”. Cell Cycle 2007, 6, 2344–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathania, S.; Bade, S.; Le Guillou, M.; Burke, K.; Reed, R.; Bowman-Colin, C.; Su, Y.; Ting, D.T.; Polyak, K.; Richardson, A.L.; et al. BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nat. Commun. 2014, 5, 5496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedic, M.; Skibinski, A.; Brown, N.; Gallardo, M.; Mulligan, P.; Martinez, P.; Keller, P.J.; Glover, E.; Richardson, A.L.; Cowan, J.; et al. Haploinsufficiency for BRCA1 leads to cell-type-specific genomic instability and premature senescence. Nat. Commun. 2015, 6, 7505. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Krais, J.J.; Bernhardy, A.J.; Nicolas, E.; Cai, K.Q.; Harrell, M.I.; Kim, H.H.; George, E.; Swisher, E.M.; Simpkins, F.; et al. RING domain-deficient BRCA1 promotes PARP inhibitor and platinum resistance. J. Clin. Investig. 2016, 126, 3145–3157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drost, R.; Dhillon, K.K.; Van Der Gulden, H.; Van Der Heijden, I.; Brandsma, I.; Cruz, C.; Chondronasiou, D.; Castroviejo-Bermejo, M.; Boon, U.; Schut, E.; et al. BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. J. Clin. Investig. 2016, 126, 2903–2918. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolck, H.A.; Przetocka, S.; Meier, R.; von Aesch, C.; Zurfluh, C.; Hänggi, K.; Spegg, V.; Altmeyer, M.; Stebler, M.; Nørrelykke, S.F.; et al. RNAi Screening Uncovers a Synthetic Sick Interaction between CtIP and the BARD1 Tumor Suppressor. Cells 2022, 11, 643. https://doi.org/10.3390/cells11040643
Bolck HA, Przetocka S, Meier R, von Aesch C, Zurfluh C, Hänggi K, Spegg V, Altmeyer M, Stebler M, Nørrelykke SF, et al. RNAi Screening Uncovers a Synthetic Sick Interaction between CtIP and the BARD1 Tumor Suppressor. Cells. 2022; 11(4):643. https://doi.org/10.3390/cells11040643
Chicago/Turabian StyleBolck, Hella A., Sara Przetocka, Roger Meier, Christine von Aesch, Christina Zurfluh, Kay Hänggi, Vincent Spegg, Matthias Altmeyer, Michael Stebler, Simon F. Nørrelykke, and et al. 2022. "RNAi Screening Uncovers a Synthetic Sick Interaction between CtIP and the BARD1 Tumor Suppressor" Cells 11, no. 4: 643. https://doi.org/10.3390/cells11040643
APA StyleBolck, H. A., Przetocka, S., Meier, R., von Aesch, C., Zurfluh, C., Hänggi, K., Spegg, V., Altmeyer, M., Stebler, M., Nørrelykke, S. F., Horvath, P., Sartori, A. A., & Porro, A. (2022). RNAi Screening Uncovers a Synthetic Sick Interaction between CtIP and the BARD1 Tumor Suppressor. Cells, 11(4), 643. https://doi.org/10.3390/cells11040643