
Neurodegenerative Disease-Associated TDP-43 Fragments Are Extracellularly Secreted with CASA Complex Proteins

 , , ,
, , ,  ,
,  ,
,  ,
,  , , , ,
, , , ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
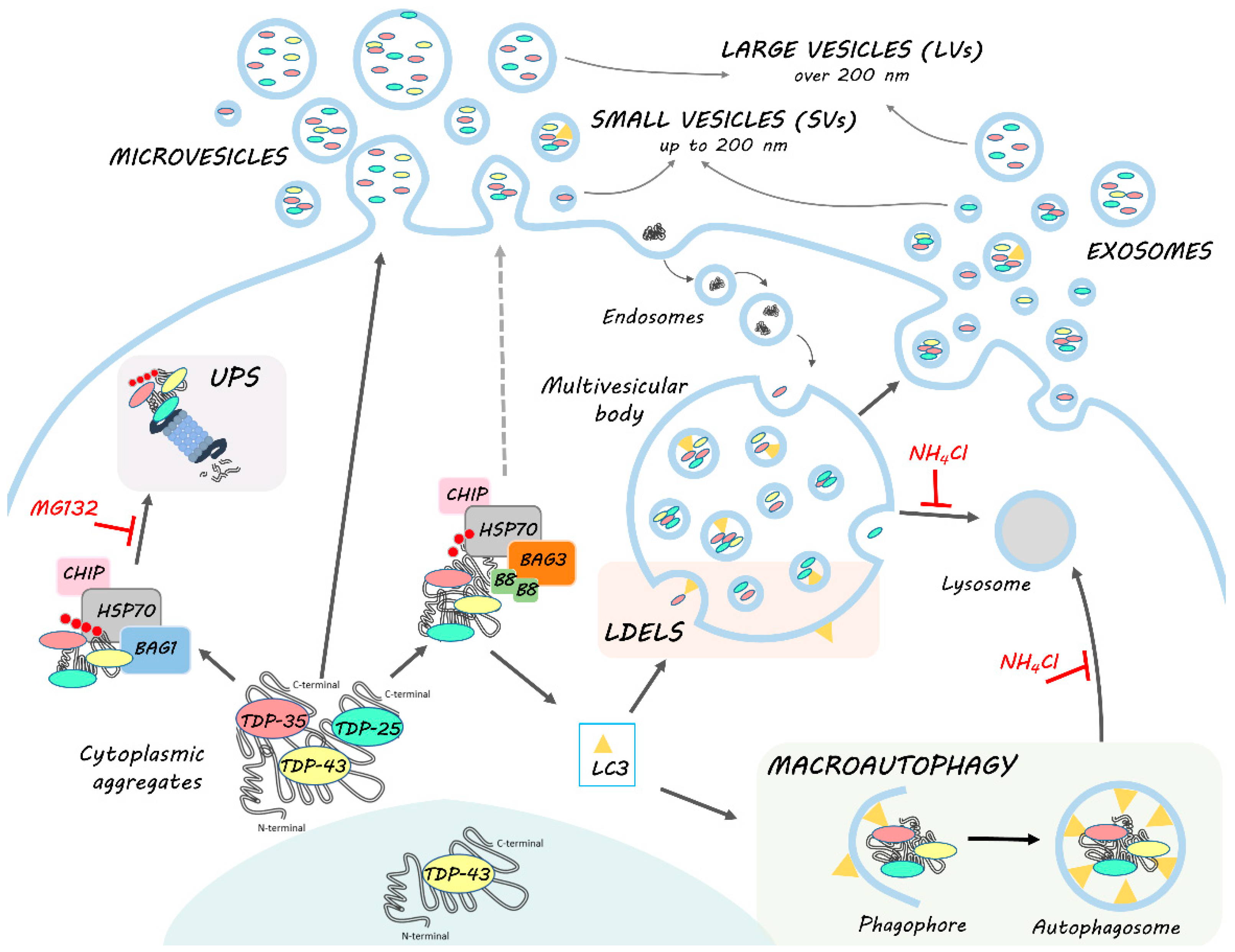
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Cultures
2.3. Subjects
2.4. EVs Isolation from NSC-34 Culture Medium
2.5. EVs Isolation from Plasma
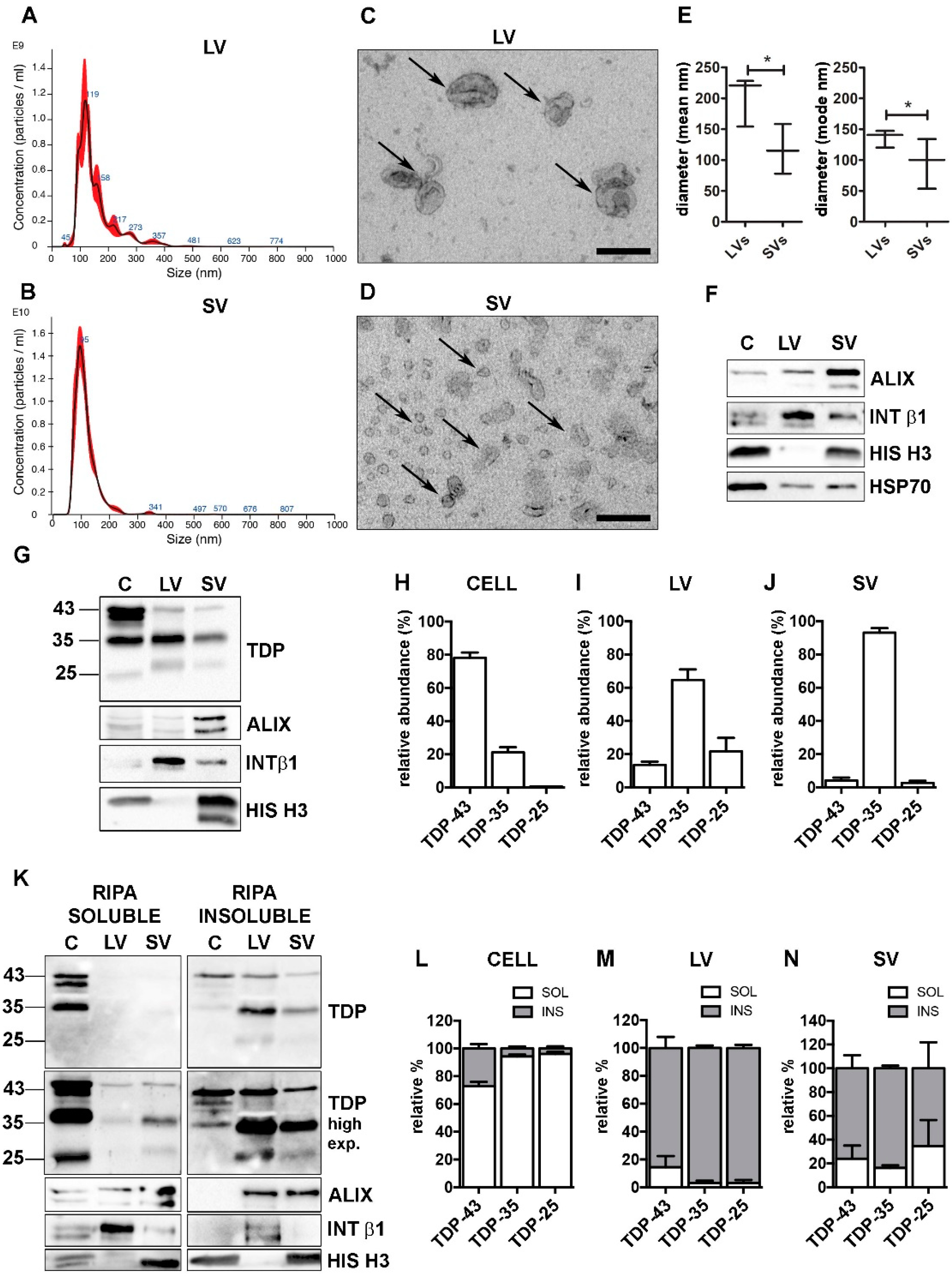
2.6. Nanoparticle-Tracking Analysis
2.7. Transmission Electron Microscopy Analysis
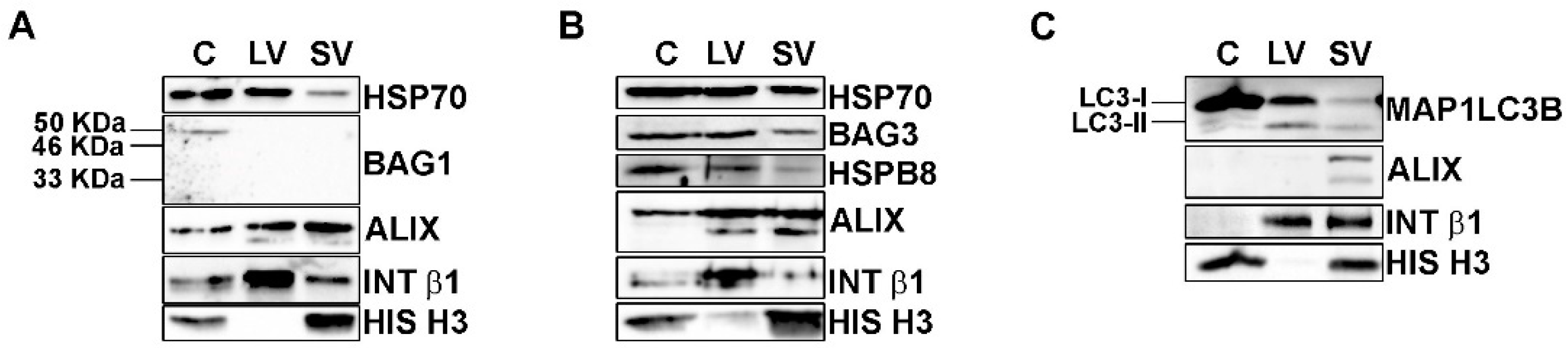
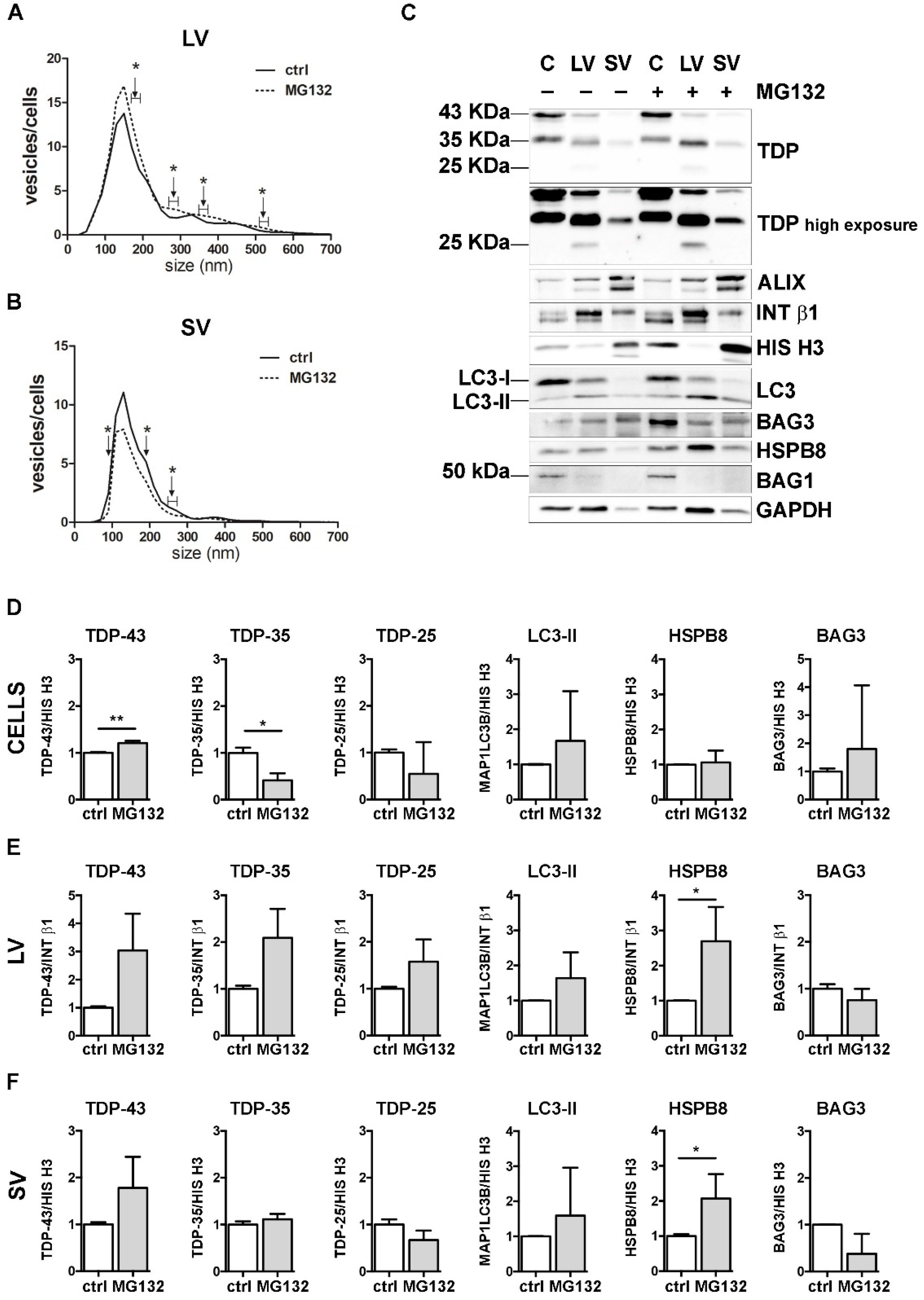
2.8. Western Blot
2.9. Statistical Analysis
3. Results and Discussions
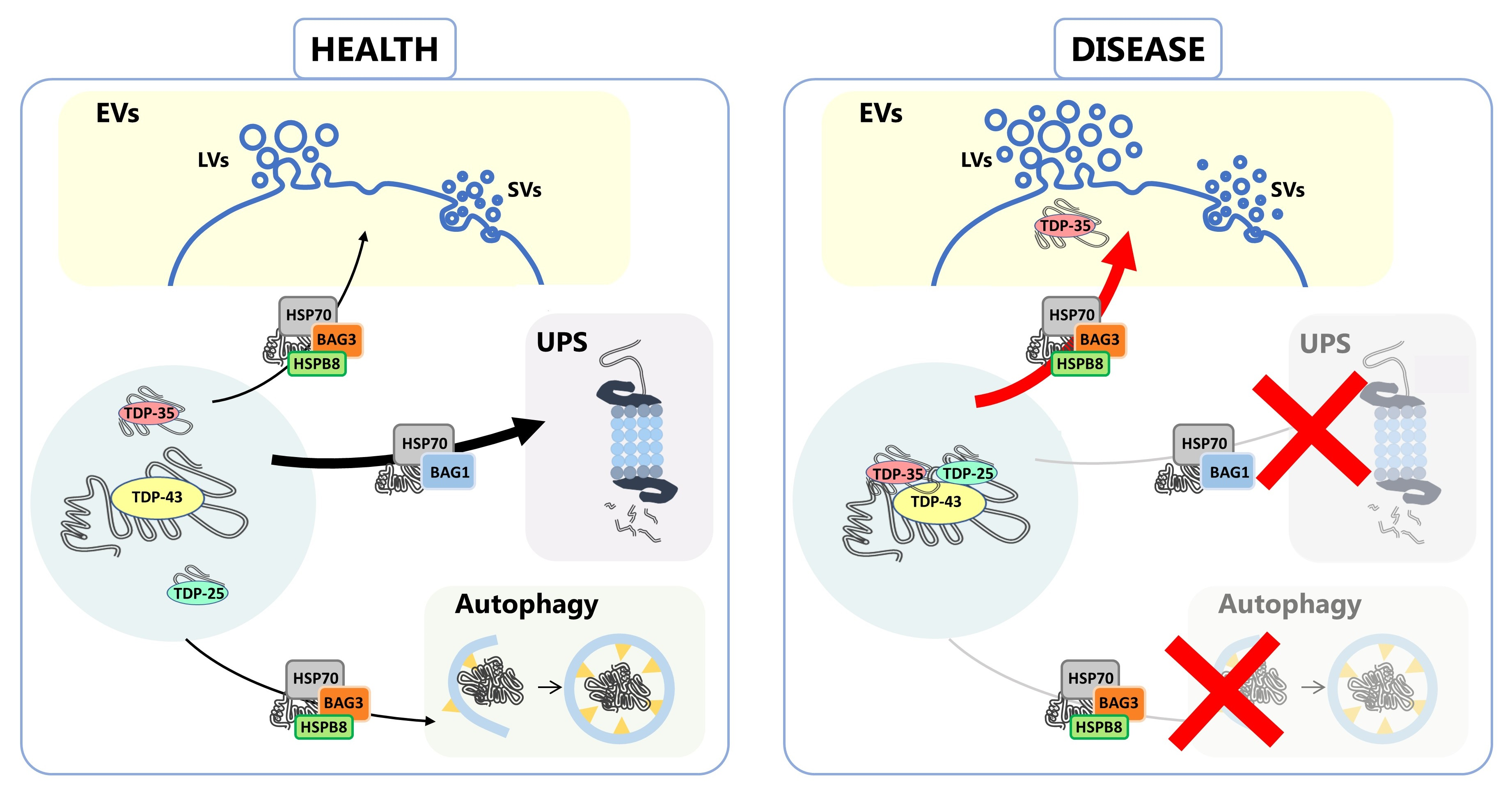
3.1. NSC-34 Cells-Secreted LVs and SVs Were Enriched in Insoluble TDP-43 Species That Were Almost Absent in Cells
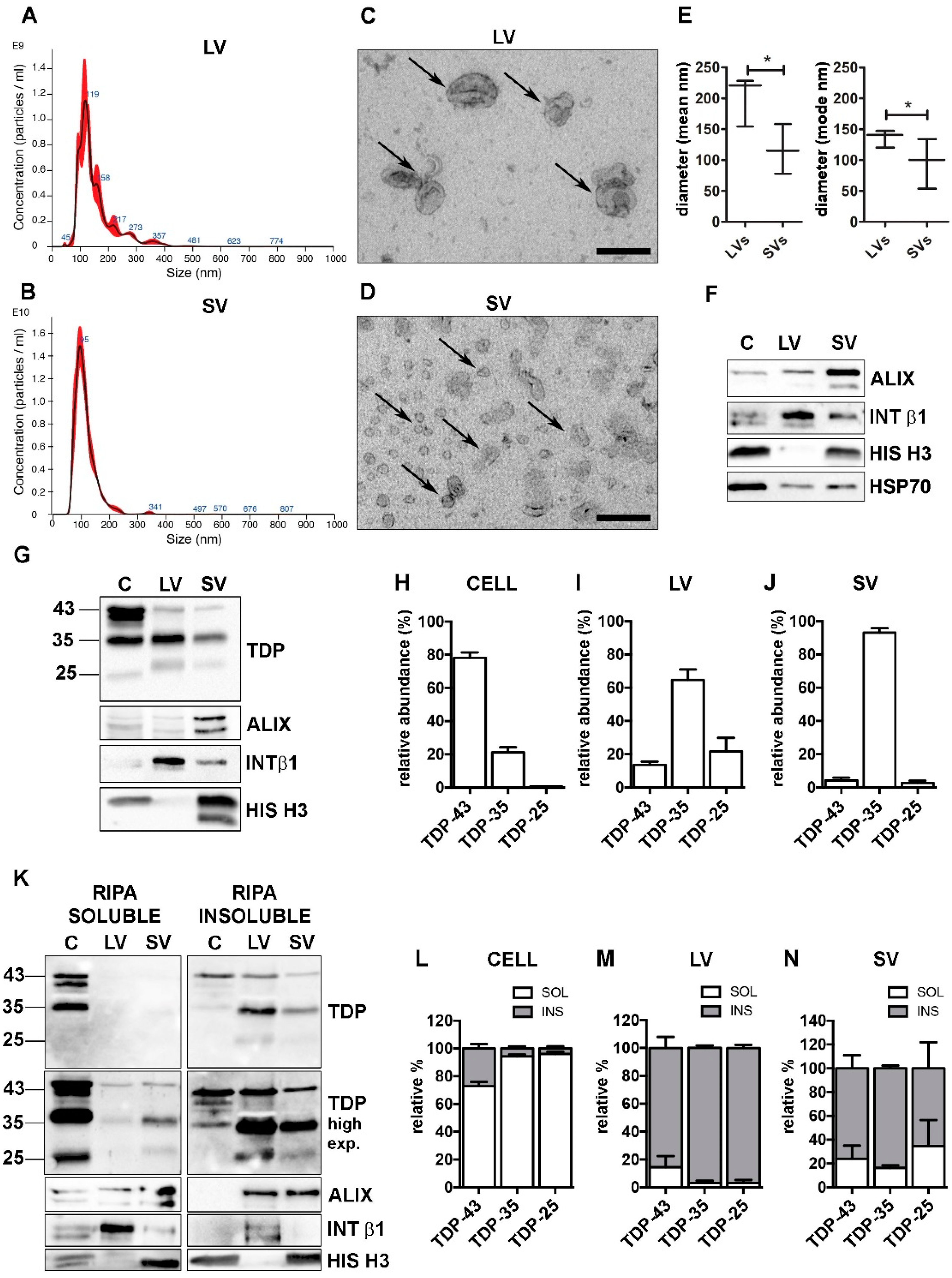
3.2. EVs Contained PQC System Components Involved in the Intracellular Clearance of Insoluble TDP-43 Species
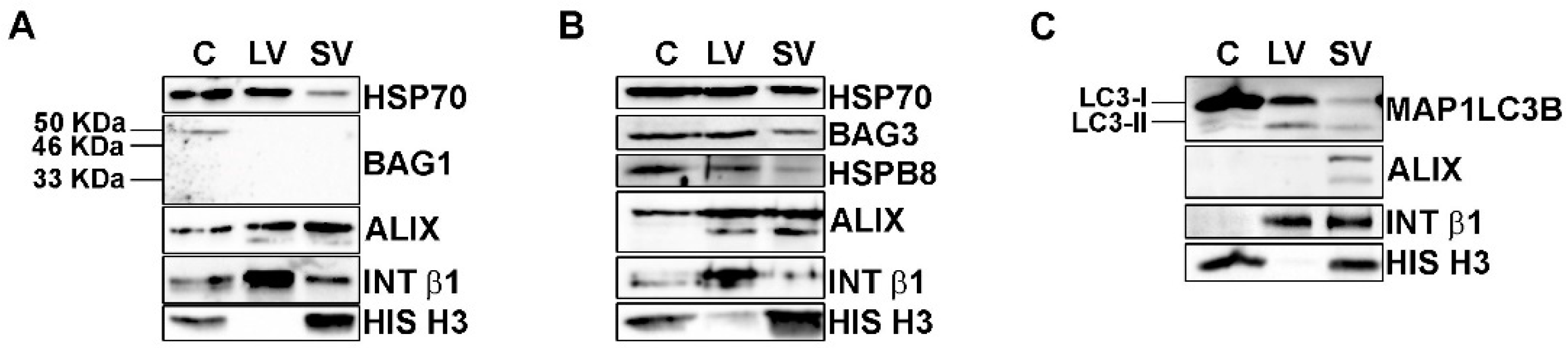
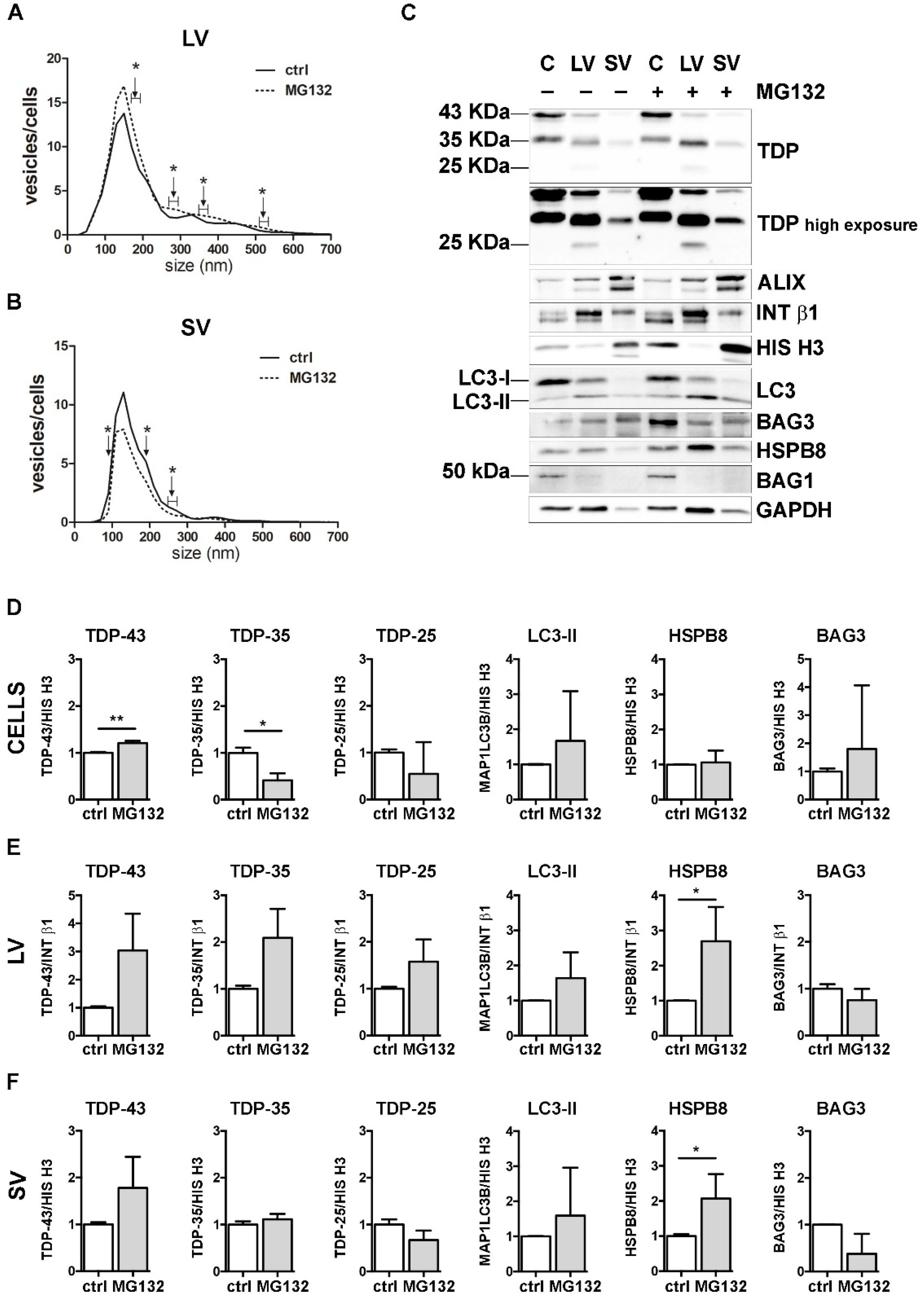
3.3. UPS Inhibition Increased The LVs Secretion of TDP-43 Species
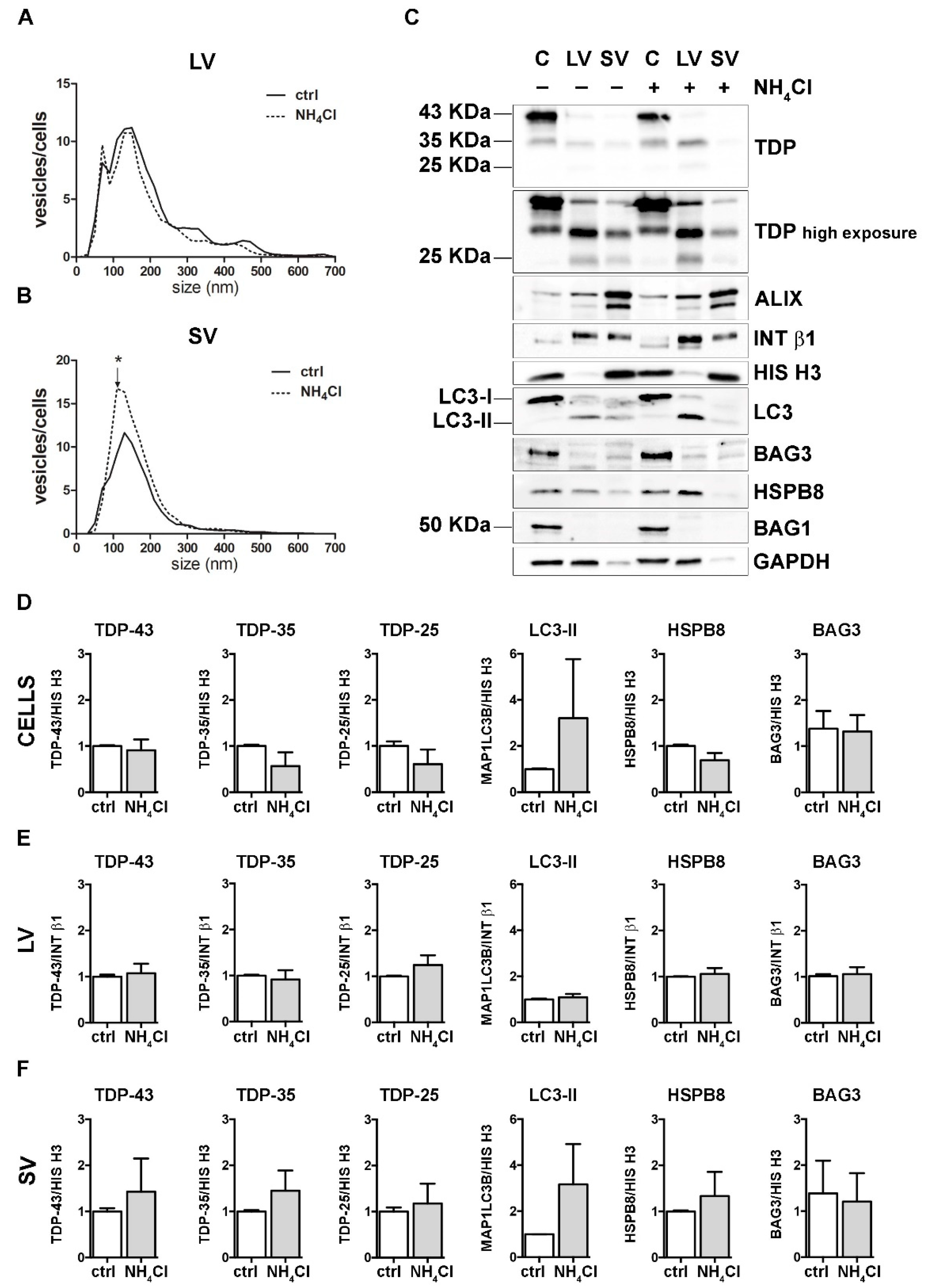
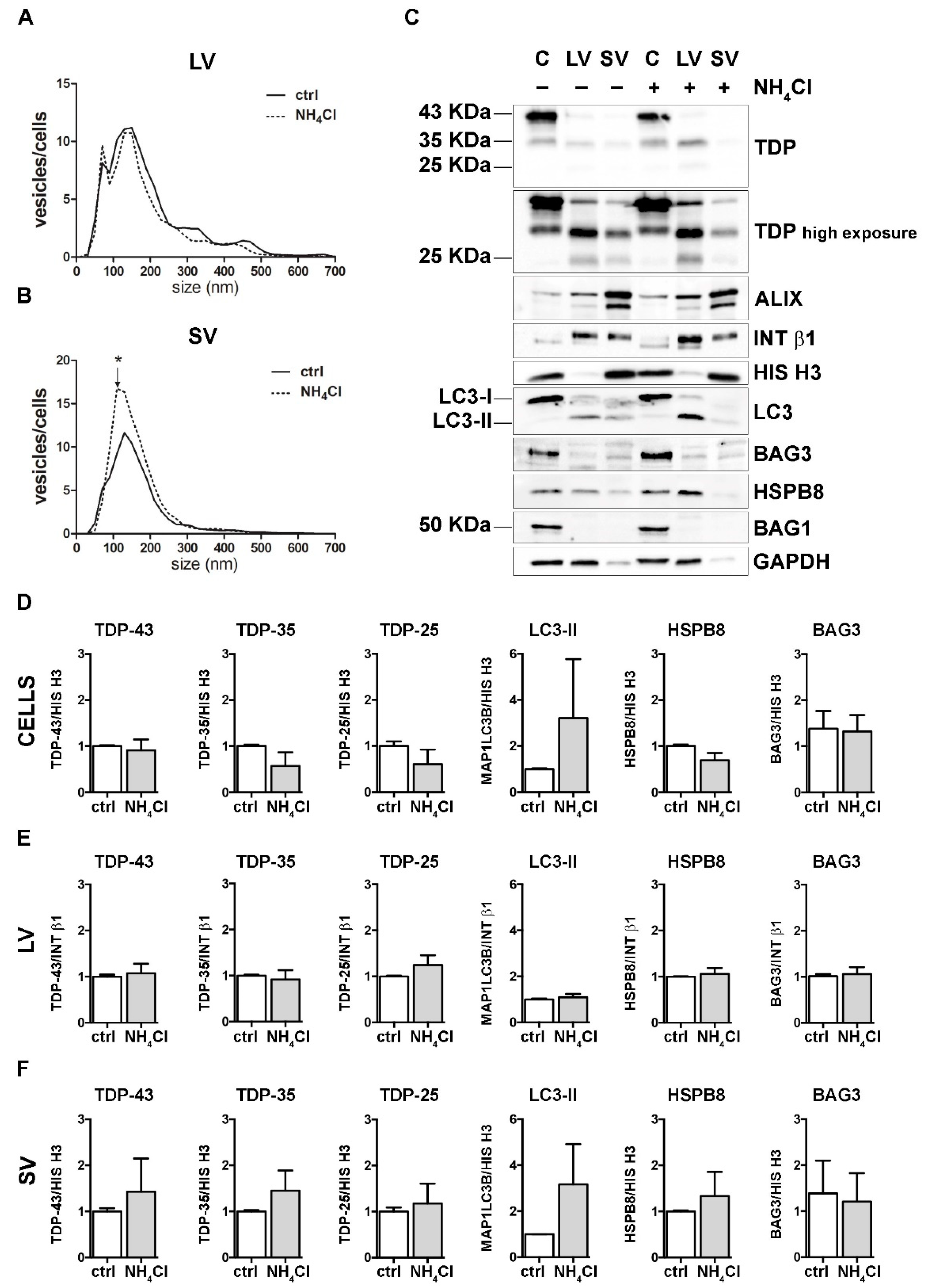
3.4. Autophagy Blockage Did Not Affect Extracellular TDP-43 Species Release
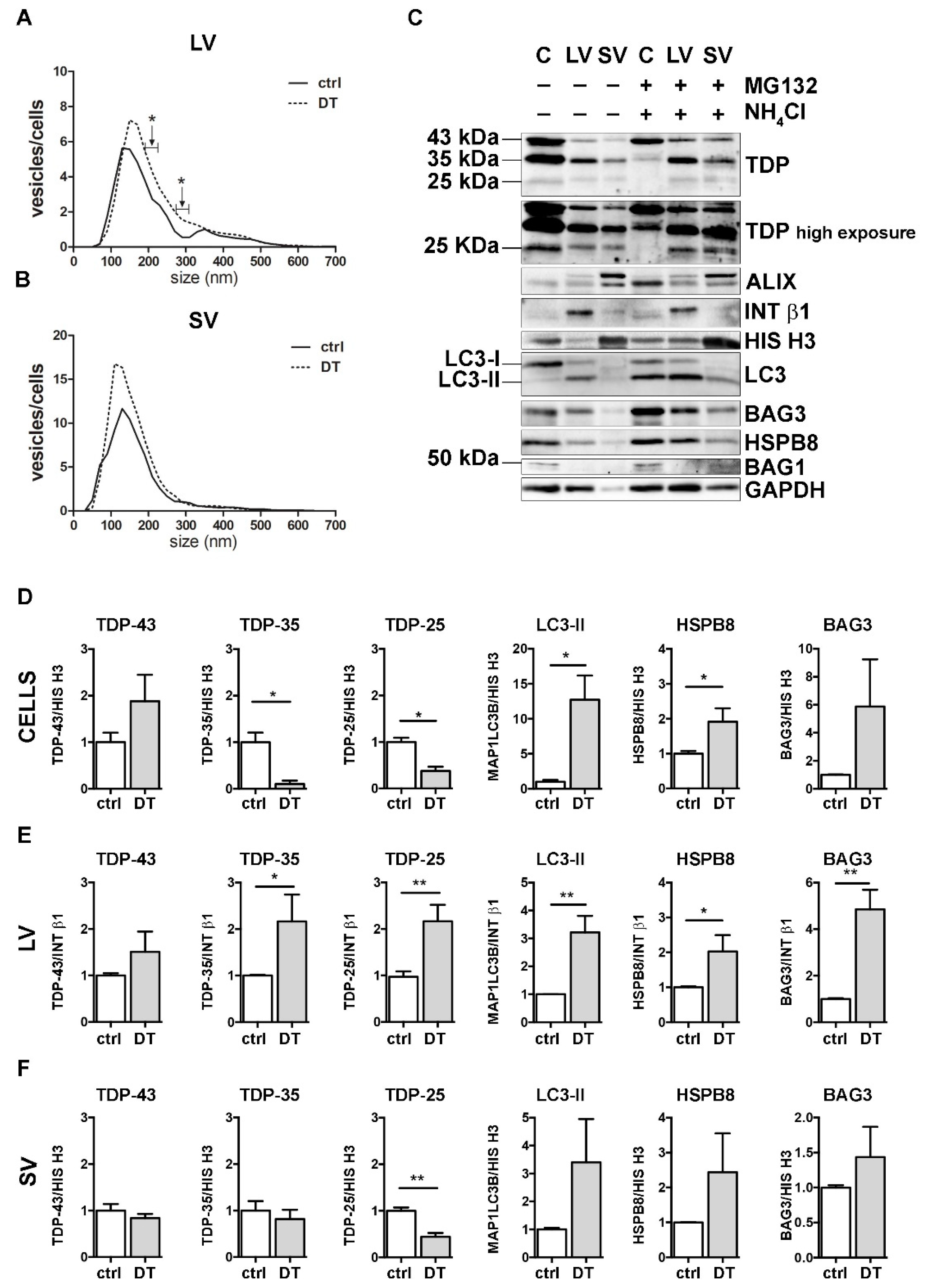
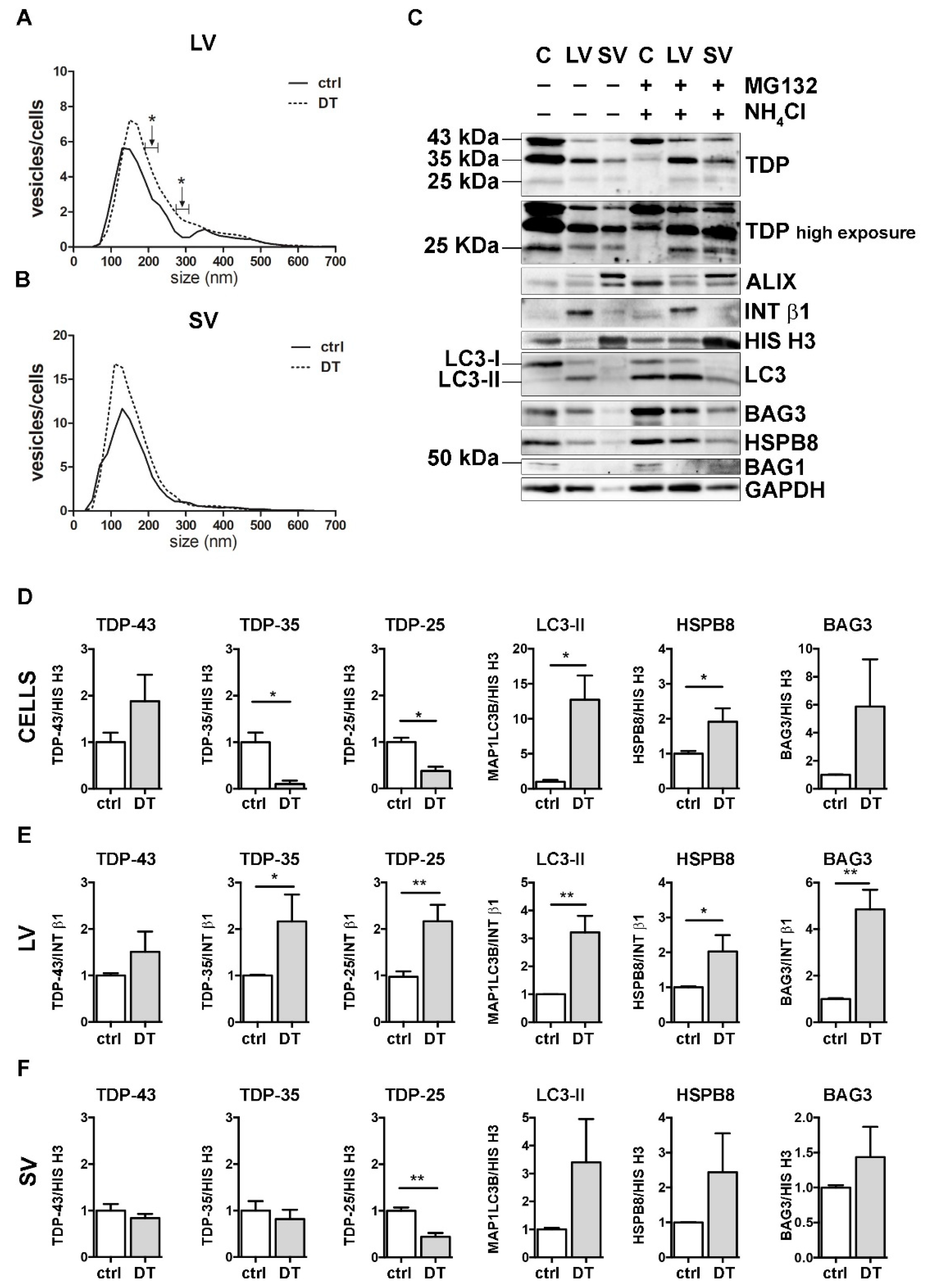
3.5. The Simultaneous Blockage of UPS and Autophagy Increased the Extracellular LV-Mediated Release of TDP-35, TDP-25 and PQC System Components
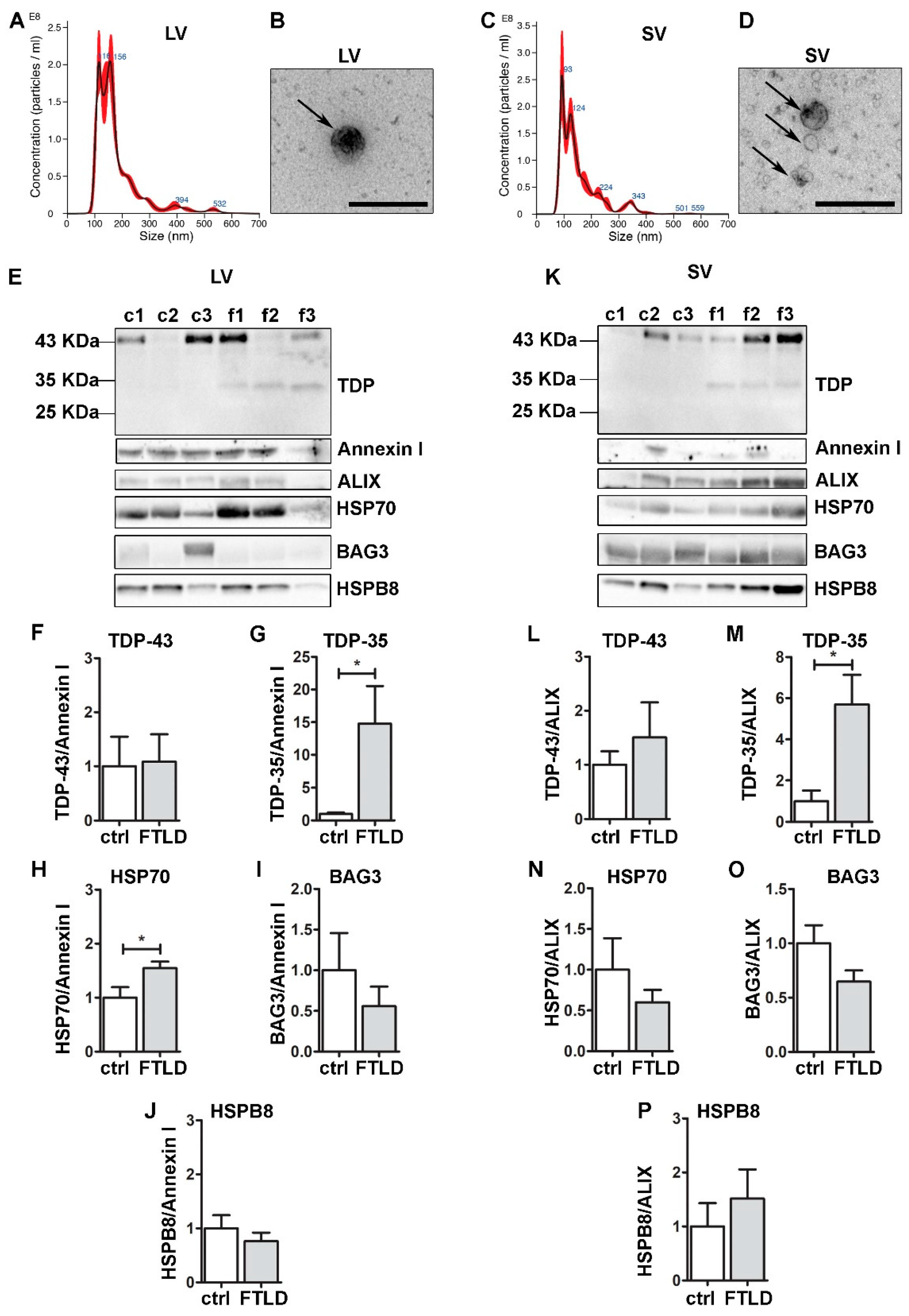
3.6. FTLD Patients-Derived EVs Were Enriched in TDP-35 Fragments and Contained CASA Complex Components
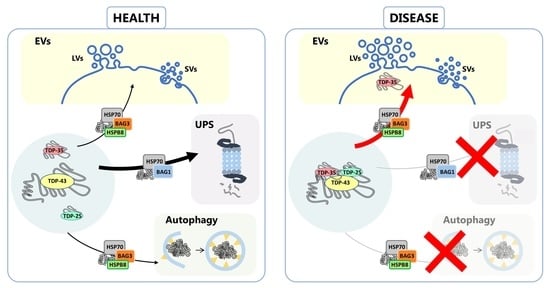
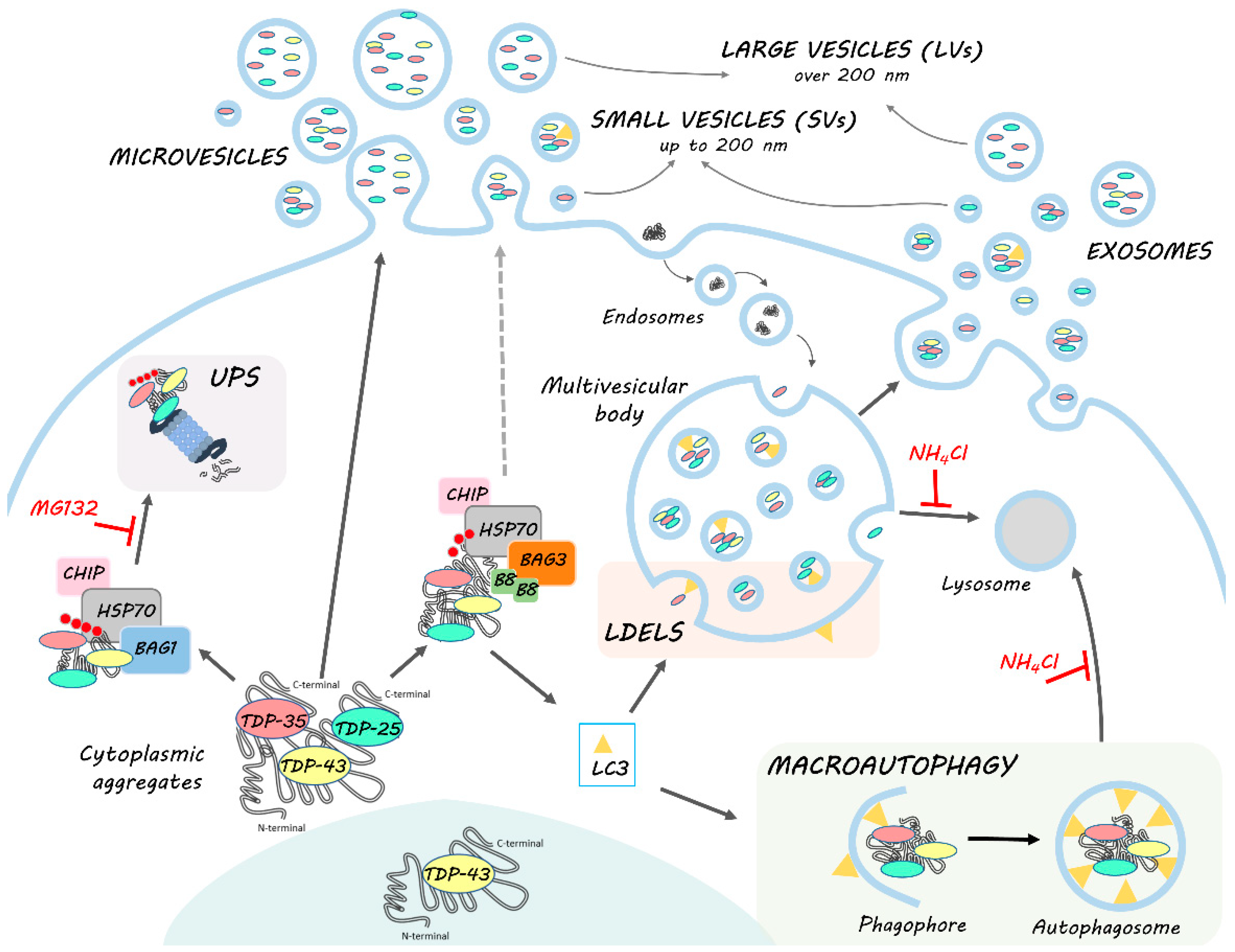
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Spires-Jones, T.L.; Attems, J.; Thal, D.R. Interactions of Pathological Proteins in Neurodegenerative Diseases. Acta Neuropathol. 2017, 134, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M.Y. Protein Transmission in Neurodegenerative Disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Lee, S.; Jeon, Y.-M.; Kim, S.; Kwon, Y.; Kim, H.-J. The Role of TDP-43 Propagation in Neurodegenerative Diseases: Integrating Insights from Clinical and Experimental Studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Luh, L.M.; Bertolotti, A. Potential Benefit of Manipulating Protein Quality Control Systems in Neurodegenerative Diseases. Curr. Opin. Neurobiol. 2020, 61, 125–132. [Google Scholar] [CrossRef]
- Jin, T.; Gu, J.; Li, Z.; Xu, Z.; Gui, Y. Recent Advances on Extracellular Vesicles in Central Nervous System Diseases. Clin. Interv. Aging 2021, 16, 257–274. [Google Scholar] [CrossRef]
- Cicardi, M.E.; Marrone, L.; Azzouz, M.; Trotti, D. Proteostatic Imbalance and Protein Spreading in Amyotrophic Lateral Sclerosis. EMBO J. 2021, 40, e106389. [Google Scholar] [CrossRef]
- Pinnell, J.R.; Cui, M.; Tieu, K. Exosomes in Parkinson Disease. J. Neurochem. 2021, 157, 413–428. [Google Scholar] [CrossRef]
- Uemura, N.; Uemura, M.T.; Luk, K.C.; Lee, V.M.Y.; Trojanowski, J.Q. Cell-to-Cell Transmission of Tau and α-Synuclein. Trends Mol. Med. 2020, 26, 936–952. [Google Scholar] [CrossRef]
- Vassileff, N.; Cheng, L.; Hill, A.F. Extracellular Vesicles—Propagators of Neuropathology and Sources of Potential Biomarkers and Therapeutics for Neurodegenerative Diseases. J. Cell Sci. 2020, 133, jcs243139. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of Secretion and Uptake of Exosomes and Other Extracellular Vesicles for Cell-to-Cell Communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Cocozza, F.; Grisard, E.; Martin-Jaular, L.; Mathieu, M.; Théry, C. SnapShot: Extracellular Vesicles. Cell 2020, 182. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedrioli, G.; Paganetti, P. Hijacking Endocytosis and Autophagy in Extracellular Vesicle Communication: Where the Inside Meets the Outside. Front. Cell Dev. Biol. 2021, 8, 595515. [Google Scholar] [CrossRef]
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. Int. J. Mol. Sci. 2020, 21, 2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delorme-Axford, E.; Klionsky, D.J. The LC3-Conjugation Machinery Specifies Cargo Loading and Secretion of Extracellular Vesicles. Autophagy 2020, 16, 1169–1171. [Google Scholar] [CrossRef] [PubMed]
- Leidal, A.M.; Debnath, J. LC3-Dependent Extracellular Vesicle Loading and Secretion (LDELS). Autophagy 2020, 16, 1162–1163. [Google Scholar] [CrossRef]
- Leidal, A.M.; Huang, H.H.; Marsh, T.; Solvik, T.; Zhang, D.; Ye, J.; Kai, F.B.; Goldsmith, J.; Liu, J.Y.; Huang, Y.H.; et al. The LC3-Conjugation Machinery Specifies the Loading of RNA-Binding Proteins into Extracellular Vesicles. Nat. Cell Biol. 2020, 22, 187–199. [Google Scholar] [CrossRef]
- Iguchi, Y.; Eid, L.; Parent, M.; Soucy, G.; Bareil, C.; Riku, Y.; Kawai, K.; Takagi, S.; Yoshida, M.; Katsuno, M.; et al. Exosome Secretion Is a Key Pathway for Clearance of Pathological TDP-43. Brain 2016, 139, 3187–3201. [Google Scholar] [CrossRef] [Green Version]
- Sproviero, D.; La Salvia, S.; Giannini, M.; Crippa, V.; Gagliardi, S.; Bernuzzi, S.; Diamanti, L.; Ceroni, M.; Pansarasa, O.; Poletti, A.; et al. Pathological Proteins Are Transported by Extracellular Vesicles of Sporadic Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2018, 12, 487. [Google Scholar] [CrossRef]
- Feneberg, E.; Gray, E.; Ansorge, O.; Talbot, K.; Turner, M.R. Towards a TDP-43-Based Biomarker for ALS and FTLD. Mol. Neurobiol. 2018, 55, 7789–7801. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Ma, M.; Teng, J.; Teng, R.K.F.; Zhou, S.; Yin, J.; Fonkem, E.; Huang, J.H.; Wu, E.; Wang, X. Exposure to ALS-FTD-CSF Generates TDP-43 Aggregates in Glioblastoma Cells through Exosomes and TNTs-like Structure. Oncotarget 2015, 6, 24178–24191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.R.; Walther, P.; Ludolph, A.C.; et al. TDP-43 Is Intercellularly Transmitted across Axon Terminals. J. Cell Biol. 2015, 211, 897–911. [Google Scholar] [CrossRef]
- Sproviero, D.; Gagliardi, S.; Zucca, S.; Arigoni, M.; Giannini, M.; Garofalo, M.; Olivero, M.; Dell’orco, M.; Pansarasa, O.; Bernuzzi, S.; et al. Different MiRNA Profiles in Plasma Derived Small and Large Extracellular Vesicles from Patients with Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 2737. [Google Scholar] [CrossRef] [PubMed]
- Sproviero, D.; La Salvia, S.; Colombo, F.; Zucca, S.; Pansarasa, O.; Diamanti, L.; Costa, A.; Lova, L.; Giannini, M.; Gagliardi, S.; et al. Leukocyte Derived Microvesicles as Disease Progression Biomarkers in Slow Progressing Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2019, 13, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 Is a Component of Ubiquitin-Positive Tau-Negative Inclusions in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Klim, J.R.; Pintacuda, G.; Nash, L.A.; Guerra San Juan, I.; Eggan, K. Connecting TDP-43 Pathology with Neuropathy. Trends Neurosci. 2021, 44, 424–440. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Ratti, A.; Buratti, E. Physiological Functions and Pathobiology of TDP-43 and FUS/TLS Proteins. J. Neurochem. 2016, 138, 95–111. [Google Scholar] [CrossRef]
- Afroz, T.; Pérez-Berlanga, M.; Polymenidou, M. Structural Transition, Function and Dysfunction of TDP-43 in Neurodegenerative Diseases. Chimia 2019, 73, 380–390. [Google Scholar] [CrossRef]
- François-Moutal, L.; Perez-Miller, S.; Scott, D.D.; Miranda, V.G.; Mollasalehi, N.; Khanna, M. Structural Insights Into TDP-43 and Effects of Post-Translational Modifications. Front. Mol. Neurosci. 2019, 12, 301. [Google Scholar] [CrossRef] [PubMed]
- Berning, B.A.; Walker, A.K. The Pathobiology of TDP-43 C-Terminal Fragments in ALS and FTLD. Front. Neurosci. 2019, 13, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laferrière, F.; Maniecka, Z.; Pérez-Berlanga, M.; Hruska-Plochan, M.; Gilhespy, L.; Hock, E.M.; Wagner, U.; Afroz, T.; Boersema, P.J.; Barmettler, G.; et al. TDP-43 Extracted from Frontotemporal Lobar Degeneration Subject Brains Displays Distinct Aggregate Assemblies and Neurotoxic Effects Reflecting Disease Progression Rates. Nat. Neurosci. 2019, 22, 65–77. [Google Scholar] [CrossRef]
- Wood, A.; Gurfinkel, Y.; Polain, N.; Lamont, W.; Lyn Rea, S. Molecular Mechanisms Underlying TDP-43 Pathology in Cellular and Animal Models of ALS and FTLD. Int. J. Mol. Sci. 2021, 22, 4705. [Google Scholar] [CrossRef]
- Scotter, E.L.; Vance, C.; Nishimura, A.L.; Lee, Y.B.; Chen, H.J.; Urwin, H.; Sardone, V.; Mitchell, J.C.; Rogelj, B.; Rubinsztein, D.C.; et al. Differential Roles of the Ubiquitin Proteasome System and Autophagy in the Clearance of Soluble and Aggregated TDP-43 Species. J. Cell Sci. 2014, 127, 1263–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crippa, V.; Cicardi, M.E.; Ramesh, N.; Seguin, S.J.; Ganassi, M.; Bigi, I.; Diacci, C.; Zelotti, E.; Baratashvili, M.; Gregory, J.M.; et al. The Chaperone HSPB8 Reduces the Accumulation of Truncated TDP-43 Species in Cells and Protects against TDP-43-Mediated Toxicity. Hum. Mol. Genet. 2016, 25, 3908–3924. [Google Scholar] [CrossRef] [PubMed]
- Cicardi, M.E.; Cristofani, R.; Rusmini, P.; Meroni, M.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Piccolella, M.; Messi, E.; Galbiati, M.; et al. Tdp-25 Routing to Autophagy and Proteasome Ameliorates Its Aggregation in Amyotrophic Lateral Sclerosis Target Cells. Sci. Rep. 2018, 8, 12390. [Google Scholar] [CrossRef]
- Ormeño, F.; Hormazabal, J.; Moreno, J.; Riquelme, F.; Rios, J.; Criollo, A.; Albornoz, A.; Alfaro, I.E.; Budini, M. Chaperone Mediated Autophagy Degrades TDP-43 Protein and Is Affected by TDP-43 Aggregation. Front. Mol. Neurosci. 2020, 13, 19. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-C.; Bose, J.K.; Majumder, P.; Lee, K.-H.; Huang, J.-T.J.; Huang, J.K.; Shen, C.-K.J. Metabolism and Mis-Metabolism of the Neuropathological Signature Protein TDP-43. J. Cell Sci. 2014, 127, 3024–3038. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Hidalgo, L.; Altuntas, S.; Rossin, F.; D’Eletto, M.; Marsella, C.; Farrace, M.G.; Falasca, L.; Antonioli, M.; Fimia, G.M.; Piacentini, M. Transglutaminase Type 2-Dependent Selective Recruitment of Proteins into Exosomes under Stressful Cellular Conditions. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2084–2092. [Google Scholar] [CrossRef]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose Induces Autophagy via Lysosomal-Mediated TFEB Activation in Models of Motoneuron Degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef] [PubMed]
- Cristofani, R.; Crippa, V.; Vezzoli, G.; Rusmini, P.; Galbiati, M.; Cicardi, M.E.; Meroni, M.; Ferrari, V.; Tedesco, B.; Piccolella, M.; et al. The Small Heat Shock Protein B8 (HSPB8) Efficiently Removes Aggregating Species of Dipeptides Produced in C9ORF72-Related Neurodegenerative Diseases. Cell Stress Chaperones 2018, 23, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chare, L.; Hodges, J.R.; Leyton, C.E.; McGinley, C.; Tan, R.H.; Kril, J.J.; Halliday, G.M. New Criteria for Frontotemporal Dementia Syndromes: Clinical and Pathological Diagnostic Implications. J. Neurol. Neurosurg. Psychiatry 2014, 85, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, S.; Gagliani, M.C.; Bellese, G.; Marconi, S.; Lechiara, A.; Dameri, M.; Aiello, C.; Tagliatti, E.; Castagnola, P.; Cortese, K. Imaging of Endocytic Trafficking and Extracellular Vesicles Released Under Neratinib Treatment in ERBB2+ Breast Cancer Cells. J. Histochem. Cytochem. 2021, 69, 461–473. [Google Scholar] [CrossRef]
- Cristofani, R.; Crippa, V.; Rusmini, P.; Cicardi, M.E.; Meroni, M.; Licata, N.V.; Sala, G.; Giorgetti, E.; Grunseich, C.; Galbiati, M.; et al. Inhibition of Retrograde Transport Modulates Misfolded Protein Accumulation and Clearance in Motoneuron Diseases. Autophagy 2017, 13, 1280–1303. [Google Scholar] [CrossRef] [PubMed]
- Grad, L.I.; Yerbury, J.J.; Turner, B.J.; Guest, W.C.; Pokrishevsky, E.; O’Neill, M.A.; Yanai, A.; Silverman, J.M.; Zeineddine, R.; Corcoran, L.; et al. Intercellular Propagated Misfolding of Wild-Type Cu/Zn Superoxide Dismutase Occurs via Exosome-Dependent and -Independent Mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 3620–3625. [Google Scholar] [CrossRef] [Green Version]
- Sarko, D.K.; Staats, K.A.; Pinto, S.; Cunha, C.; Barbosa, M.; Vaz, A.R.; Brites, D. Exosomes from NSC-34 Cells Transfected with HSOD1-G93A Are Enriched in MiR-124 and Drive Alterations in Microglia Phenotype. Front. Neurosci. 2017, 1, 273. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, Z.; Shi, X.; Tong, H.; Su, L. Prognostic Value of Plasma Exosomal Levels of Histone H3 Protein in Patients with Heat Stroke. Exp. Ther. Med. 2021, 22, 922. [Google Scholar] [CrossRef]
- Cascella, R.; Fani, G.; Bigi, A.; Chiti, F.; Cecchi, C. Partial Failure of Proteostasis Systems Counteracting TDP-43 Aggregates in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3685. [Google Scholar] [CrossRef] [Green Version]
- Mariotto, E.; Viola, G.; Zanon, C.; Aveic, S. A BAG’s Life: Every Connection Matters in Cancer. Pharmacol. Ther. 2020, 209, 107498. [Google Scholar] [CrossRef]
- Arndt, V.; Dick, N.; Tawo, R.; Dreiseidler, M.; Wenzel, D.; Hesse, M.; Fürst, D.O.; Saftig, P.; Saint, R.; Fleischmann, B.K.; et al. Chaperone-Assisted Selective Autophagy Is Essential for Muscle Maintenance. Curr. Biol. 2010, 20, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Crippa, V.; Carra, S.; Rusmini, P.; Sau, D.; Bolzoni, E.; Bendotti, C.; De Biasi, S.; Poletti, A. A Role of Small Heat Shock Protein B8 (HspB8) in the Autophagic Removal of Misfolded Proteins Responsible for Neurodegenerative Diseases. Autophagy 2010, 7, 958–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristofani, R.; Crippa, V.; Cicardi, M.E.; Tedesco, B.; Ferrari, V.; Chierichetti, M.; Casarotto, E.; Piccolella, M.; Messi, E.; Galbiati, M.; et al. A Crucial Role for the Protein Quality Control System in Motor Neuron Diseases. Front. Aging Neurosci. 2020, 12, 191. [Google Scholar] [CrossRef] [PubMed]
- Chowdary, T.K.; Raman, B.; Ramakrishna, T.; Rao, C.M. Interaction of Mammalian Hsp22 with Lipid Membranes. Biochem. J. 2007, 401, 437–445. [Google Scholar] [CrossRef]
- Crippa, V.; D’Agostino, V.G.; Cristofani, R.; Rusmini, P.; Cicardi, M.E.; Messi, E.; Loffredo, R.; Pancher, M.; Piccolella, M.; Galbiati, M.; et al. Transcriptional Induction of the Heat Shock Protein B8 Mediates the Clearance of Misfolded Proteins Responsible for Motor Neuron Diseases. Sci. Rep. 2016, 6, 22827. [Google Scholar] [CrossRef]
- Crippa, V.; Sau, D.; Rusmini, P.; Boncoraglio, A.; Onesto, E.; Bolzoni, E.; Galbiati, M.; Fontana, E.; Marino, M.; Carra, S.; et al. The Small Heat Shock Protein B8 (HspB8) Promotes Autophagic Removal of Misfolded Proteins Involved in Amyotrophic Lateral Sclerosis (ALS). Hum. Mol. Genet. 2010, 19, 3440–3456. [Google Scholar] [CrossRef] [Green Version]
- Minoia, M.; Boncoraglio, A.; Vinet, J.; Morelli, F.F.; Brunsting, J.F.; Poletti, A.; Krom, S.; Reits, E.; Kampinga, H.H.; Carra, S. BAG3 Induces the Sequestration of Proteasomal Clients into Cytoplasmic Puncta Implications for a Proteasome-to-Autophagy Switch. Autophagy 2014, 10, 1603–1621. [Google Scholar] [CrossRef] [Green Version]
- Carra, S.; Seguin, S.J.; Landry, J. HspB8 and Bag3: A New Chaperone Complex Targeting Misfolded Proteins to Macroautophagy. Autophagy 2008, 4, 237–239. [Google Scholar] [CrossRef] [Green Version]
- Chanteloup, G.; Cordonnier, M.; Moreno-Ramos, T.; Pytel, V.; Matías-Guiu, J.; Gobbo, J.; Cabrera-Martín, M.N.; Gómez-Pinedo, U.; Garrido, C.; Matías-Guiu, J.A. Exosomal HSP70 for Monitoring of Frontotemporal Dementia and Alzheimer’s Disease: Clinical and FDG-PET Correlation. J. Alzheimer’s Dis. 2019, 71, 1263–1269. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casarotto, E.; Sproviero, D.; Corridori, E.; Gagliani, M.C.; Cozzi, M.; Chierichetti, M.; Cristofani, R.; Ferrari, V.; Galbiati, M.; Mina, F.; et al. Neurodegenerative Disease-Associated TDP-43 Fragments Are Extracellularly Secreted with CASA Complex Proteins. Cells 2022, 11, 516. https://doi.org/10.3390/cells11030516
Casarotto E, Sproviero D, Corridori E, Gagliani MC, Cozzi M, Chierichetti M, Cristofani R, Ferrari V, Galbiati M, Mina F, et al. Neurodegenerative Disease-Associated TDP-43 Fragments Are Extracellularly Secreted with CASA Complex Proteins. Cells. 2022; 11(3):516. https://doi.org/10.3390/cells11030516
Chicago/Turabian StyleCasarotto, Elena, Daisy Sproviero, Eleonora Corridori, Maria Cristina Gagliani, Marta Cozzi, Marta Chierichetti, Riccardo Cristofani, Veronica Ferrari, Mariarita Galbiati, Francesco Mina, and et al. 2022. "Neurodegenerative Disease-Associated TDP-43 Fragments Are Extracellularly Secreted with CASA Complex Proteins" Cells 11, no. 3: 516. https://doi.org/10.3390/cells11030516
APA StyleCasarotto, E., Sproviero, D., Corridori, E., Gagliani, M. C., Cozzi, M., Chierichetti, M., Cristofani, R., Ferrari, V., Galbiati, M., Mina, F., Piccolella, M., Rusmini, P., Tedesco, B., Gagliardi, S., Cortese, K., Cereda, C., Poletti, A., & Crippa, V. (2022). Neurodegenerative Disease-Associated TDP-43 Fragments Are Extracellularly Secreted with CASA Complex Proteins. Cells, 11(3), 516. https://doi.org/10.3390/cells11030516