
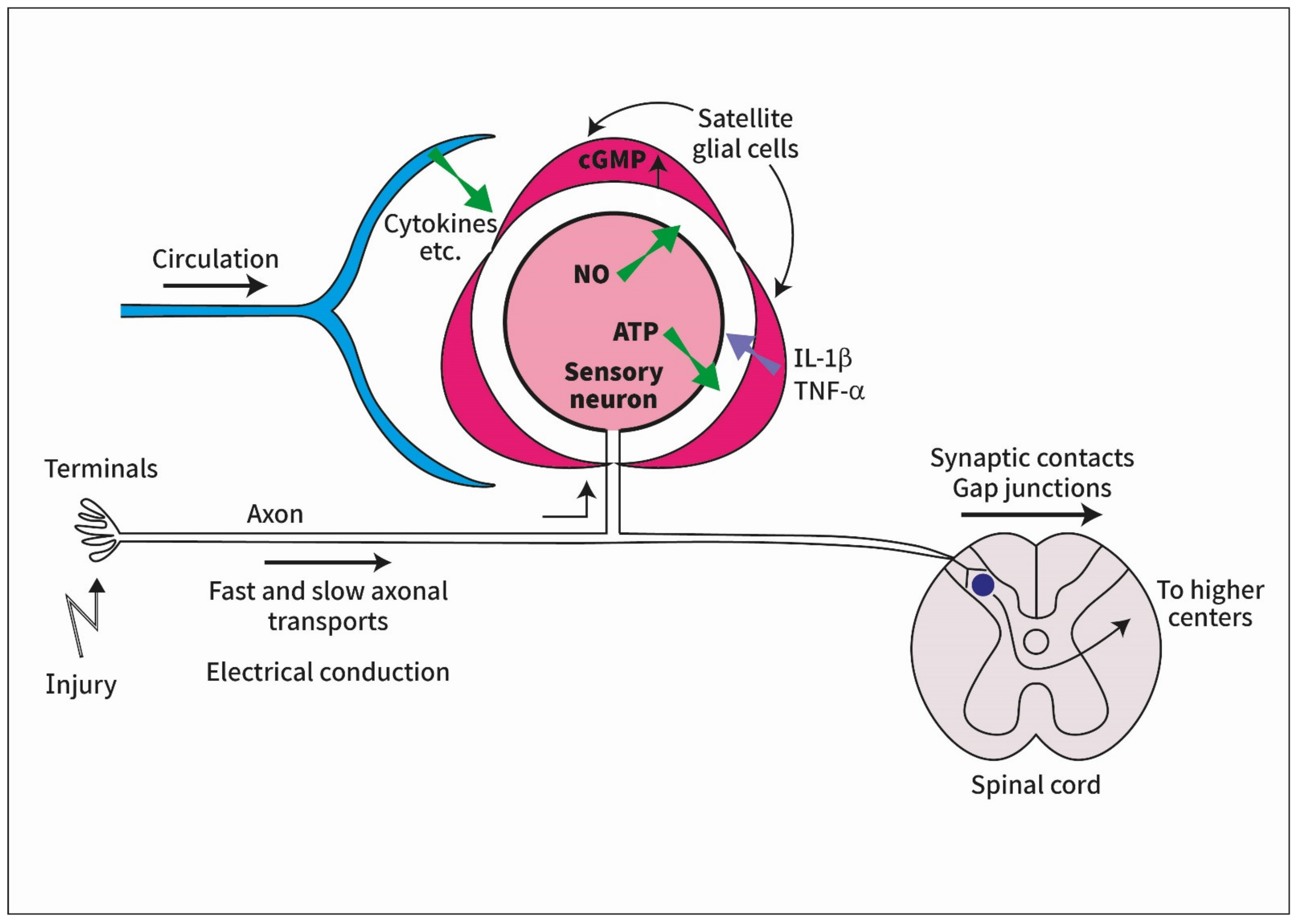
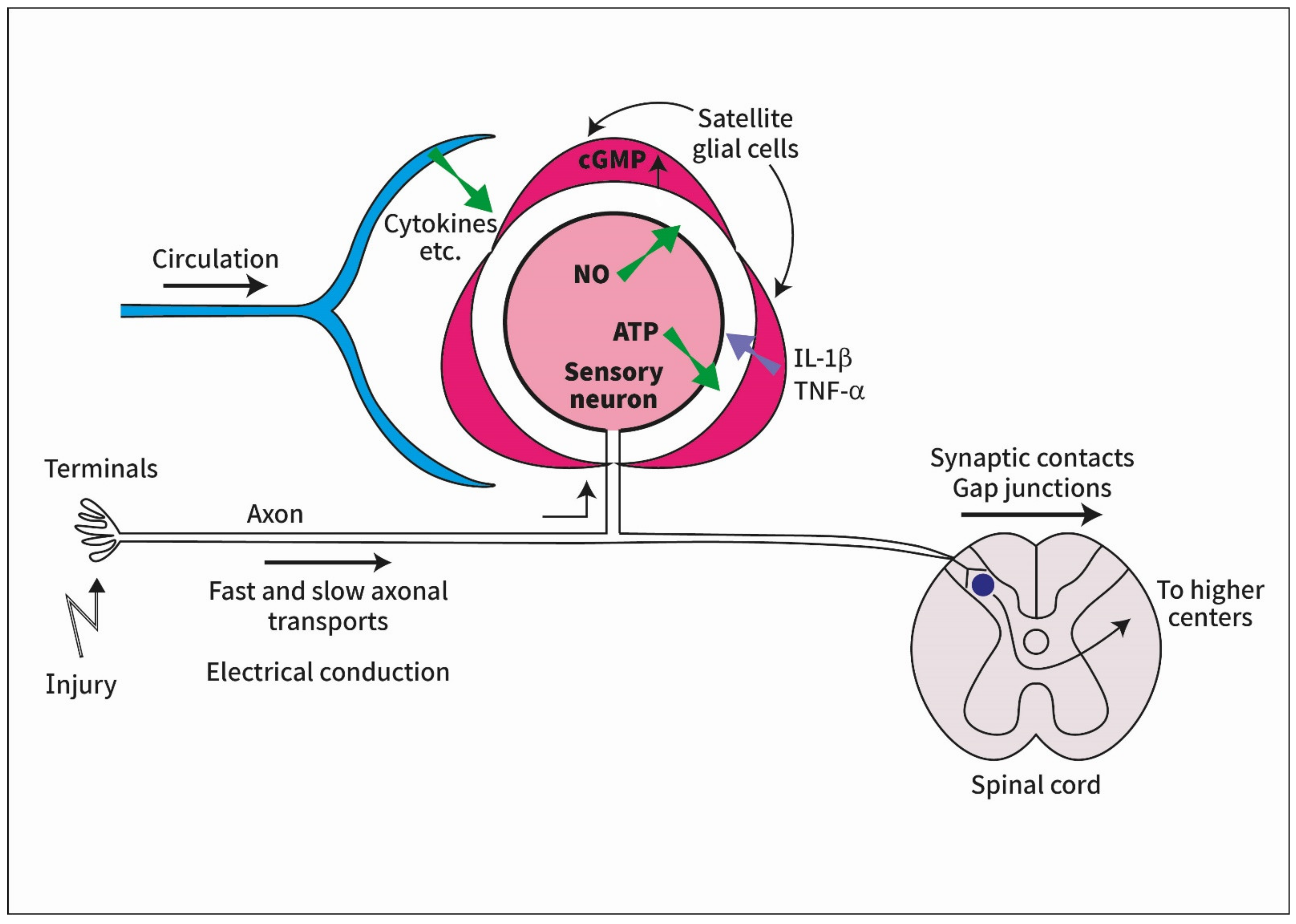
How Is Peripheral Injury Signaled to Satellite Glial Cells in Sensory Ganglia?

{kind=link}
Abstract
:1. Introduction
2. Assays for Changes in Neurons and SGCs
3. Pathways from the Periphery to Sensory Ganglia
3.1. Electrical Conduction
3.2. Signaling across the Spinal Cord
3.3. Axonal Transport
3.4. Humoral Signaling
4. How Do Neurons Signal to SGC?
5. Clinical Implications
Funding
Acknowledgments
Conflicts of Interest
References
- Rotshenker, S. Wallerian degeneration: The innate-immune response to traumatic nerve injury. J. Neuroinflamm. 2011, 8, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldskogius, H.; Kozlova, E.N. Central neuron-glial and glial-glial interactions following axon injury. Prog. Neurobiol. 1998, 55, 1–26. [Google Scholar] [CrossRef]
- Aldskogius, H.; Kozlova, E.N. Dorsal Root Injury-A Model for Exploring Pathophysiology and Therapeutic Strategies in Spinal Cord Injury. Cells 2021, 10, 2185. [Google Scholar] [CrossRef] [PubMed]
- Devor, M. Neuropathic pain pathophysiological response of nerves to injury. In Wall and Melzack’s Textbook of Pain, 6th ed.; McMahon, S.B., Koltzenburg, M., Tracey, I., Turk, D., Eds.; Elsevier Churchill Livingstone: Philadelphia, PA, USA, 2013; pp. 867–888. [Google Scholar]
- Hanani, M.; Spray, D.C. Emerging importance of satellite glia in nervous system function and dysfunction. Nat. Rev. Neurosci. 2020, 21, 485–498. [Google Scholar] [CrossRef]
- Hol, E.M.; Pekny, M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr. Opin. Cell Biol. 2015, 32, 121–130. [Google Scholar] [CrossRef]
- Hatashita, S.; Sekiguchi, M.; Kobayashi, H.; Konno, S.I.; Kikuchi, S.I. Contralateral neuropathic pain and neuropathology in dorsal root ganglion and spinal cord following hemilateral nerve injury in rats. Spine 2008, 33, 1344–1351. [Google Scholar] [CrossRef]
- Xie, W.; Strong, J.A.; Zhang, J.M. Early blockade of injured primary sensory afferents reduces glial cell activation in two rat neuropathic pain models. Neuroscience 2009, 160, 847–857. [Google Scholar] [CrossRef] [Green Version]
- Romero, A.; Romero-Alejo, E.; Vasconcelos, N.; Puig, M.M. Glial cell activation in the spinal cord and dorsal root ganglia induced by surgery in mice. Eur. J. Pharmacol. 2013, 702, 126–134. [Google Scholar] [CrossRef]
- Zhuang, Z.Y.; Gerner, P.; Woolf, C.J.; Ji, R.R. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain 2005, 114, 149–159. [Google Scholar] [CrossRef]
- Dai, Y.; Iwata, K.; Fukuoka, T.; Kondo, E.; Tokunaga, A.; Yamanaka, H.; Tachibana, T.; Liu, Y.; Noguchi, K. Phosphorylation of extracellular signal-regulated kinase in primary afferent neurons by noxious stimuli and its involvement in peripheral sensitization. J. Neurosci. 2002, 22, 7737–7745. [Google Scholar] [CrossRef]
- Yamakita, S.; Horii, Y.; Takemura, H.; Matsuoka, Y.; Yamashita, A.; Yamaguchi, Y.; Matsuda, M.; Sawa, T.; Amaya, F. Synergistic activation of ERK1/2 between A-fiber neurons and glial cells in the DRG contributes to pain hypersensitivity after tissue injury. Mol. Pain 2018, 14, 1744806918767508. [Google Scholar] [CrossRef] [Green Version]
- Harper, A.A.; Lawson, S.N. Electrical properties of rat dorsal root ganglion neurones with different peripheral nerve conduction velocities. J. Physiol. 1985, 359, 47–63. [Google Scholar] [CrossRef] [Green Version]
- Katz, J.; Clarke, H.; Seltzer, Z. Review article: Preventive analgesia: Quo vadimus? Anesth. Analg. 2011, 113, 1242–1253. [Google Scholar] [CrossRef]
- Wall, P.D.; Waxman, S.; Basbaum, A.I. Ongoing activity in peripheral nerve: Injury discharge. Exp. Neurol. 1974, 45, 576–589. [Google Scholar] [CrossRef]
- Xie, W.; Strong, J.A.; Meij, J.T.A.; Zhang, J.M.; Yu, L. Neuropathic pain: Early spontaneous afferent activity is the trigger. Pain 2005, 116, 243–256. [Google Scholar] [CrossRef]
- Sun, Q.; Tu, H.; Xing, G.G.; Han, J.S.; Wan, Y. Ectopic discharges from injured nerve fibers are highly correlated with tactile allodynia only in early, but not late, stage in rats with spinal nerve ligation. Exp. Neurol. 2005, 191, 128–136. [Google Scholar] [CrossRef]
- Tal, M.; Wall, P.D.; Devor, M. Myelinated afferent fiber types that become spontaneously active and mechanosensitive following nerve transection in the rat. Brain Res. 1999, 824, 218–223. [Google Scholar] [CrossRef]
- Mohr, K.M.; Pallesen, L.T.; Richner, M.; Vaegter, C.B. Discrepancy in the Usage of GFAP as a Marker of Satellite Glial Cell Reactivity. Biomedicines 2021, 9, 1022. [Google Scholar] [CrossRef]
- Blum, E.; Procacci, P.; Conte, V.; Hanani, M. Systemic inflammation alters satellite glial cell function and structure. A possible contribution to pain. Neuroscience 2014, 274, 209–217. [Google Scholar] [CrossRef]
- Dublin, P.; Hanani, M. Satellite glial cells in sensory ganglia: Their possible contribution to inflammatory pain. Brain Behav. Immun. 2007, 21, 592–598. [Google Scholar] [CrossRef]
- Eliav, E.; Herzberg, U.; Ruda, M.A.; Bennett, G.J. Neuropathic pain from an experimental neuritis of the rat sciatic nerve. Pain 1999, 83, 169–182. [Google Scholar] [CrossRef]
- Bourquin, A.F.; Süveges, M.; Pertin, M.; Gilliard, N.; Sardy, S.; Davison, A.C.; Spahn, D.R.; Decosterd, I. Assessment and analysis of mechanical allodynia-like behavior induced by spared nerve injury (SNI) in the mouse. Pain 2006, 122, 14.e1–14.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringkamp, M.; Eschenfelder, S.; Grethel, E.J.; Häbler, H.J.; Meyer, R.A.; Jänig, W.; Raja, S.N. Lumbar sympathectomy failed to reverse mechanical allodynia- and hyperalgesia-like behavior in rats with L5 spinal nerve injury. Pain 1999, 79, 143–153. [Google Scholar] [CrossRef]
- Wolf, G.; Gabay, E.; Tal, M.; Yirmiya, R.; Shavit, Y. Genetic impairment of interleukin-1 signaling attenuates neuropathic pain, autotomy, and spontaneous ectopic neuronal activity, following nerve injury in mice. Pain 2006, 120, 315–324. [Google Scholar] [CrossRef]
- Chen, Q.; Heinricher, M.M. Plasticity in the Link between Pain-Transmitting and Pain-Modulating Systems in Acute and Persistent Inflammation. J. Neurosci. 2019, 39, 2065–2079. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Chung, J.M. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 1992, 50, 355–363. [Google Scholar] [CrossRef]
- Ruskin, D.N.; Sturdevant, I.C.; Wyss, L.S.; Masino, S.A. Ketogenic diet effects on inflammatory allodynia and ongoing pain in rodents. Sci. Rep. 2021, 11, 725. [Google Scholar] [CrossRef]
- Woodham, P.; Anderson, P.N.; Nadim, W.; Turmaine, M. Satellite cells surrounding axotomised rat dorsal root ganglion cells increase expression of a GFAP-like protein. Neurosci. Lett. 1989, 98, 8–12. [Google Scholar] [CrossRef]
- Chacur, M.; Milligan, E.D.; Gazda, L.S.; Armstrong, C.; Wang, H.; Tracey, K.J.; Maier, S.F.; Watkins, L.R. A new model of sciatic inflammatory neuritis (SIN): Induction of unilateral and bilateral mechanical allodynia following acute unilateral peri-sciatic immune activation in rats. Pain 2001, 94, 231–244. [Google Scholar] [CrossRef]
- Dubový, P.; Brázda, V.; Klusáková, I.; Hradilová-Svíženská, I. Bilateral elevation of interleukin-6 protein and mRNA in both lumbar and cervical dorsal root ganglia following unilateral chronic compression injury of the sciatic nerve. J. Neuroinflamm. 2013, 10, 55. [Google Scholar] [CrossRef] [Green Version]
- Dubový, P.; Jancálek, R.; Klusáková, I.; Svízenská, I.; Pejchalová, K. Intra- and extraneuronal changes of immunofluorescence staining for TNF-alpha and TNFR1 in the dorsal root ganglia of rat peripheral neu ropathic pain models. Cell Mol. Neurobiol. 2006, 26, 1205–1217. [Google Scholar] [CrossRef]
- Koltzenburg, M.; Wall, P.D.; McMahon, S.B. Does the right side know what the left is doing? Trends Neurosci. 1999, 22, 122–127. [Google Scholar] [CrossRef]
- Pitcher, G.M.; Ritchie, J.; Henry, J.L. Nerve constriction in the rat: Model of neuropathic, surgical and central pain. Pain 1999, 83, 37–46. [Google Scholar] [CrossRef]
- Sluka, K.A.; Kalra, A.; Moore, S.A. Unilateral intramuscular injections of acidic saline produce a bilateral, long-lasting hyperalgesia. Muscle Nerve 2001, 24, 37–46. [Google Scholar] [CrossRef]
- Spataro, L.E.; Sloane, E.M.; Milligan, E.D.; Wieseler-Frank, J.; Schoeniger, D.; Jekich, B.M.; Barrientos, R.M.; Maier, S.F.; Watkins, L.R. Spinal gap junctions: Potential involvement in pain facilitation. J. Pain 2004, 5, 392–405. [Google Scholar] [CrossRef]
- Choi, H.S.; Roh, D.H.; Yoon, S.Y.; Kwon, S.G.; Choi, S.R.; Kang, S.Y.; Moon, J.Y.; Han, H.J.; Kim, H.W.; Beitz, A.J.; et al. The role of spinal interleukin-1beta and astrocyte connexin 43 in the development of mirror-image pain in an inflammatory pain model. Exp. Neurol. 2017, 287, 1–13. [Google Scholar] [CrossRef]
- Jancálek, R.; Dubový, P.; Svízenská, I.; Klusáková, I.J. Bilateral changes of TNF-alpha and IL-10 protein in the lumbar and cervical dorsal root ganglia following a unilateral chronic constriction injury of the sciatic nerve. Neuroinflammation 2010, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Dahlstrom, A.B. Fast intra-axonal transport: Beginning, development and post-genome advances. Prog. Neurobiol. 2010, 90, 119–145. [Google Scholar] [CrossRef]
- Miller, K.E.; Heidemann, S.R. What is slow axonal transport? Exp. Cell Res. 2008, 314, 1981–1990. [Google Scholar] [CrossRef]
- Kristensson, K. Retrograde transport of macromolecules in axons. Annu. Rev. Pharmacol. Toxicol. 1978, 18, 97–110. [Google Scholar] [CrossRef]
- Whiteley, S.J.; Townsend, J.; Tomlinson, D.R.; Brown, A.M. Fast orthograde axonal transport in sciatic motoneurones and nerve temperature in streptozotocin-diabetic rats. Diabetologia 1985, 28, 847–851. [Google Scholar] [CrossRef] [Green Version]
- DiStefano, P.S.; Curtis, R. Receptor mediated retrograde axonal transport of neurotrophic factors is increased after peripheral nerve injury. Prog. Brain Res. 1994, 103, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Dubový, P.; Hradilová-Svíženská, I.; Brázda, V.; Joukal, M. Toll-Like Receptor 9-Mediated Neuronal Innate Immune Reaction Is Associated with Initiating a Pro-Regenerative State in Neurons of the Dorsal Root Ganglia Non-Associated with Sciatic Nerve Lesion. Int. J. Mol. Sci. 2021, 22, 7446. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.; Devor, M. Chemically mediated cross-excitation in rat dorsal root ganglia. J. Neurosci. 1996, 16, 4733–4741. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, G.B.; Mulpuri, Y.; Damasio, A.; Spigelman, I. A Role for The P2Y1 Receptor in Nonsynaptic Cross-depolarization in the Rat Dorsal Root Ganglia. Neuroscience 2019, 423, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Rozanski, G.M.; Kim, H.; Li, Q.; Wong, F.K.; Stanley, E.F. Slow chemical transmission between dorsal root ganglion neuron somata. Eur. J. Neurosci. 2012, 36, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Anderson, M.; Park, K.; Zheng, Q.; Agarwal, A.; Gong, C.; Saijilafu; Young, L.; He, S.; LaVinka, P.C.; et al. Coupled Activation of Primary Sensory Neurons Contributes to Chronic Pain. Neuron 2016, 91, 1085–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephenson, J.L.; Byers, M.R. GFAP immunoreactivity in trigeminal ganglion satellite cells after tooth injury in rats. Exp. Neurol. 1995, 131, 11–22. [Google Scholar] [CrossRef]
- Thalakoti, S.; Patil, V.V.; Damodaram, S.; Vause, C.V.; Langford, L.E.; Freeman, S.E.; Durham, P.L. Neuron-glia signaling in trigeminal ganglion: Implications for migraine pathology. Headache 2007, 47, 1008–1023, discussion 24–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.Y.; Belzer, V.; Hanani, M. Gap junctions in dorsal root ganglia: Possible contribution to visceral pain. Eur. J. Pain 2010, 14, 49.e1–49.e11. [Google Scholar] [CrossRef]
- Suadicani, S.O.; Cherkas, P.S.; Zuckerman, J.; Smith, D.N.; Spray, D.C.; Hanani, M. Bidirectional calcium signaling between satellite glial cells and neurons in cultured mouse trigeminal ganglia. Neuron Glia Biol. 2010, 6, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Thippeswamy, T.; Morris, R. Evidence that nitric oxide-induced synthesis of cGMP occurs in a paracrine but not an autocrine fashion and that the site of its release can be regulated: Studies in dorsal root ganglia in vivo and in vitro. Nitric Oxide 2001, 5, 105–115. [Google Scholar] [CrossRef]
- Belzer, V.; Hanani, M. Nitric oxide as a messenger between neurons and satellite glial cells in dorsal root ganglia. Glia 2019, 67, 1296–1307. [Google Scholar] [CrossRef]
- Feldman-Goriachnik, R.; Hanani, M. How do neurons in sensory ganglia communicate with satellite glial cells? Brain Res. 2021, 1760, 147384. [Google Scholar] [CrossRef]
- Magni, G.; Riccio, D.; Ceruti, S. Tackling Chronic Pain and Inflammation through the Purinergic System. Curr. Med. Chem. 2018, 25, 3830–3865. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.Y.; Gu, Y.; Chen, Y. Communication between neuronal somata and satellite glial cells in sensory ganglia. Glia 2013, 61, 1571–1581. [Google Scholar] [CrossRef] [Green Version]
- Matsuka, Y.; Afroz, S.; Dalanon, J.C.; Iwasa, T.; Waskitho, A.; Oshima, M. The role of chemical transmitters in neuron-glia interaction and pain in sensory ganglion. Neurosci. Biobehav. Rev. 2020, 108, 393–399. [Google Scholar] [CrossRef]
- Binshtok, A.M.; Wang, H.; Zimmermann, K.; Amaya, F.; Vardeh, D.; Shi, L.; Brenner, G.J.; Ji, R.R.; Bean, B.P.; Woolf, C.J.; et al. Nociceptors are interleukin-1 β sensors. J. Neurosci. 2008, 28, 14062–14073. [Google Scholar] [CrossRef] [Green Version]
- Takeda, M.; Takahashi, M.; Matsumoto, S. Contribution of the activation of satellite glia in sensory ganglia to pathological pain. Neurosci. Biobehav. Rev. 2009, 33, 784–792. [Google Scholar] [CrossRef]
- Haroutiunian, S.; Nikolajsen, L.; Finnerup, N.B.; Jensen, T.S. The neuropathic component in persistent postsurgical pain: A systematic literature review. Pain 2013, 154, 95–102. [Google Scholar] [CrossRef]
- Wylde, V.; Dennis, J.; Beswick, A.D.; Bruce, J.; Eccleston, C.; Howells, N.; Peters, T.J.; Gooberman-Hill, R. Systematic review of management of chronic pain after surgery. Brit. J. Surg. 2017, 104, 1293–1306. [Google Scholar] [CrossRef]
- Grape, S.; Tramèr, M.R. Do we need preemptive analgesia for the treatment of postoperative pain? Best Pract. Res. Clin. Anaesthesiol. 2007, 21, 51–63. [Google Scholar] [CrossRef]
- Sotgiu, M.L.; Biella, G.; Firmi, L.; Pasqualucci, V. Topical axonal transport blocker vincristine prevents nerve injury-induced spinal neuron sensitization in rats. J. Neurotrauma 1998, 15, 1077–1082. [Google Scholar] [CrossRef]
- Devor, M.; Govrin-Lippmann, R. Axoplasmic transport block reduces ectopic impulse generation in injured peripheral nerves. Pain 1983, 16, 73–85. [Google Scholar] [CrossRef]
- Stein, C.; Lang, L.J. Peripheral mechanisms of opioid analgesia. Curr. Opin. Pharmacol. 2009, 9, 3–8. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanani, M. How Is Peripheral Injury Signaled to Satellite Glial Cells in Sensory Ganglia? Cells 2022, 11, 512. https://doi.org/10.3390/cells11030512
Hanani M. How Is Peripheral Injury Signaled to Satellite Glial Cells in Sensory Ganglia? Cells. 2022; 11(3):512. https://doi.org/10.3390/cells11030512
Chicago/Turabian StyleHanani, Menachem. 2022. "How Is Peripheral Injury Signaled to Satellite Glial Cells in Sensory Ganglia?" Cells 11, no. 3: 512. https://doi.org/10.3390/cells11030512
APA StyleHanani, M. (2022). How Is Peripheral Injury Signaled to Satellite Glial Cells in Sensory Ganglia? Cells, 11(3), 512. https://doi.org/10.3390/cells11030512