
Low-Dose-Rate Irradiation Suppresses the Expression of Cell Cycle-Related Genes, Resulting in Modification of Sensitivity to Anti-Cancer Drugs

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. Irradiation Procedure
2.4. Cell Proliferation and Drug Sensitivity Analyses
2.5. RNA-Seq Analysis
2.6. Quantitative Reverse Transcription–Polymerase Chain Reaction (RT-PCR) Analysis
2.7. Immunoblotting Analysis
2.8. Knock-Down Analysis
2.9. Plasmid Constructs and Luciferase Reporter Analysis
2.10. Cell Cycle Analysis
2.11. Immunofluorescence Analysis
2.12. Statistical Analysis
3. Results
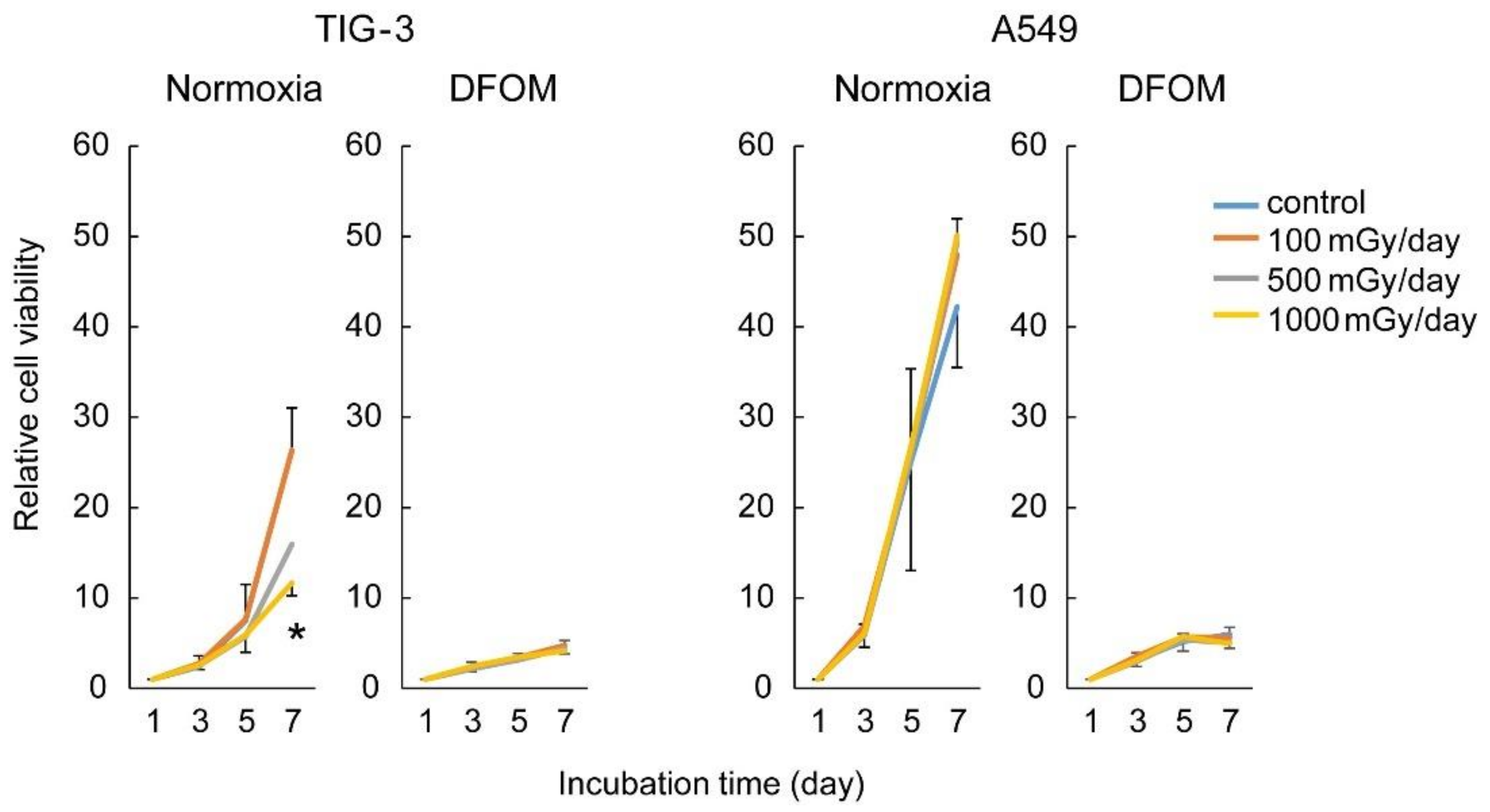
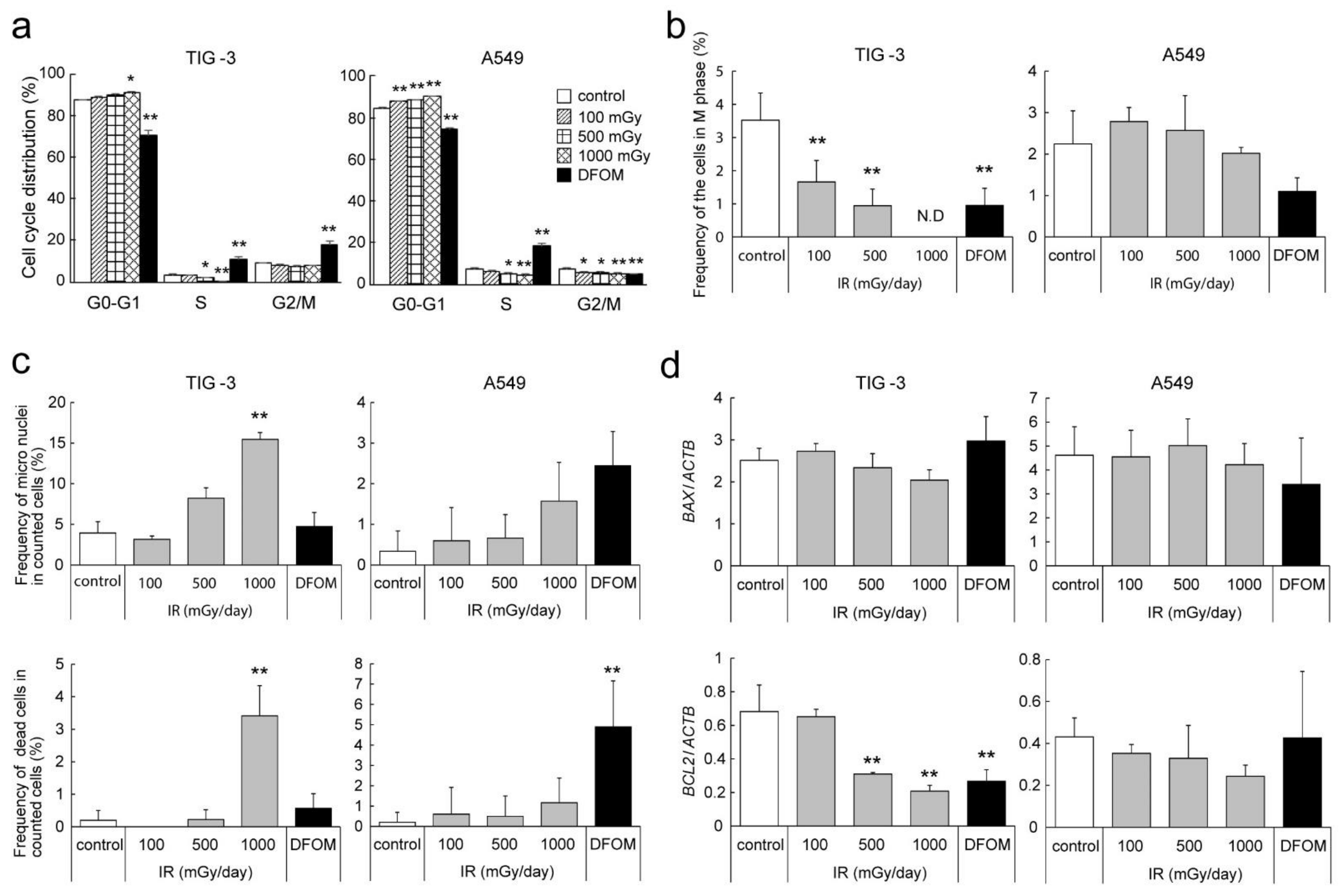
3.1. Effects of LDR-IR on Cell Proliferation
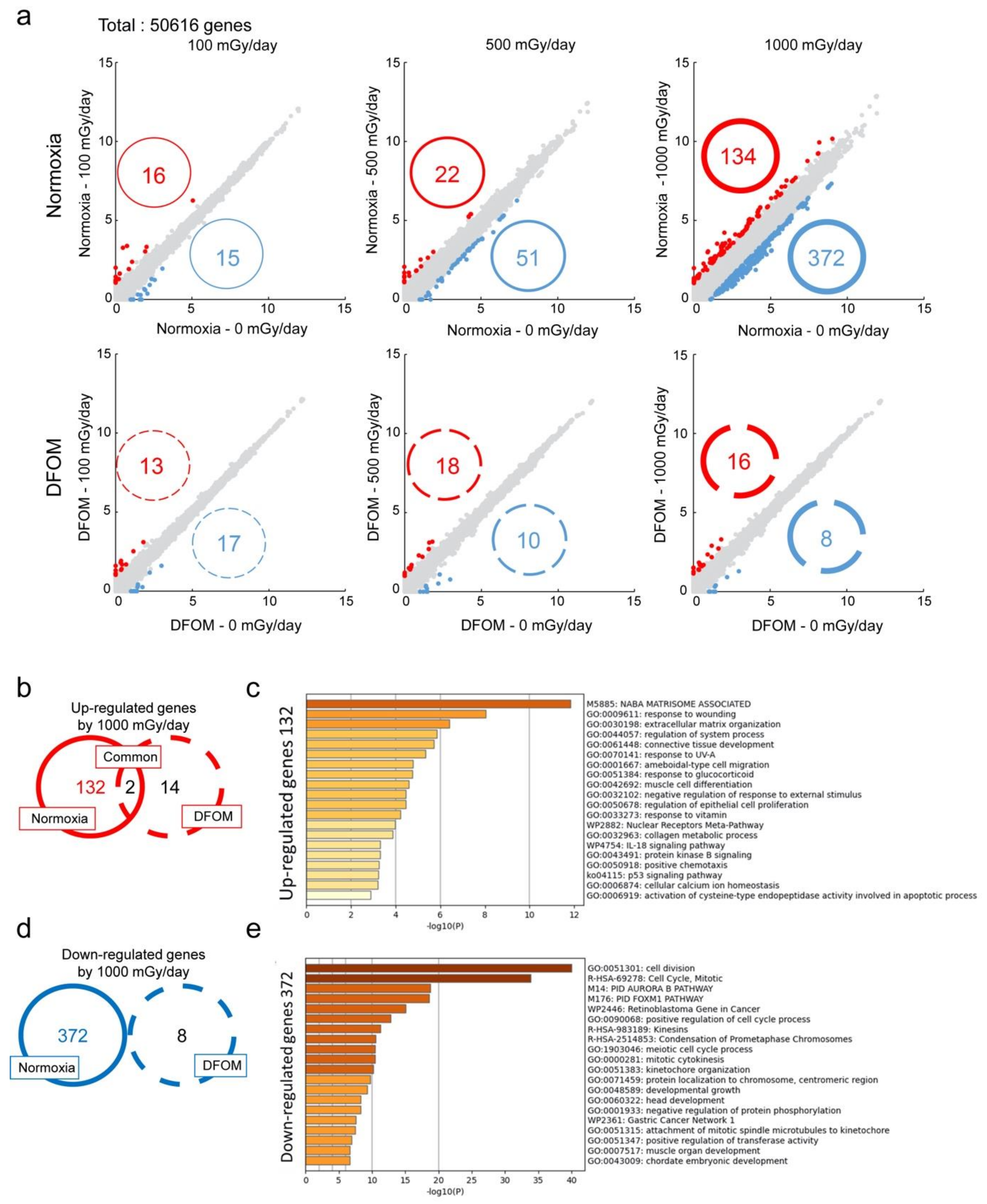
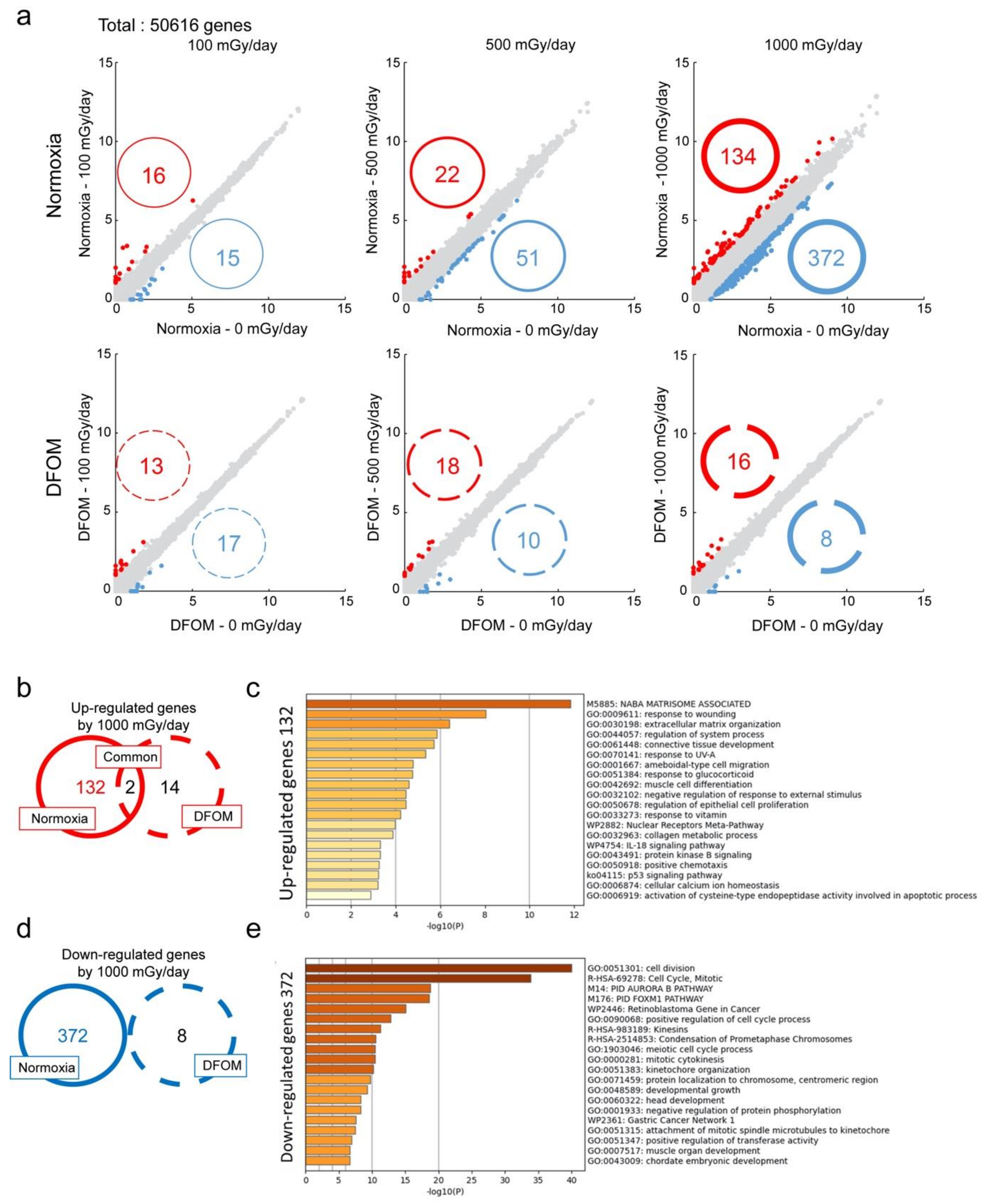
3.2. Effects of LDR-IR on Global Gene Expression
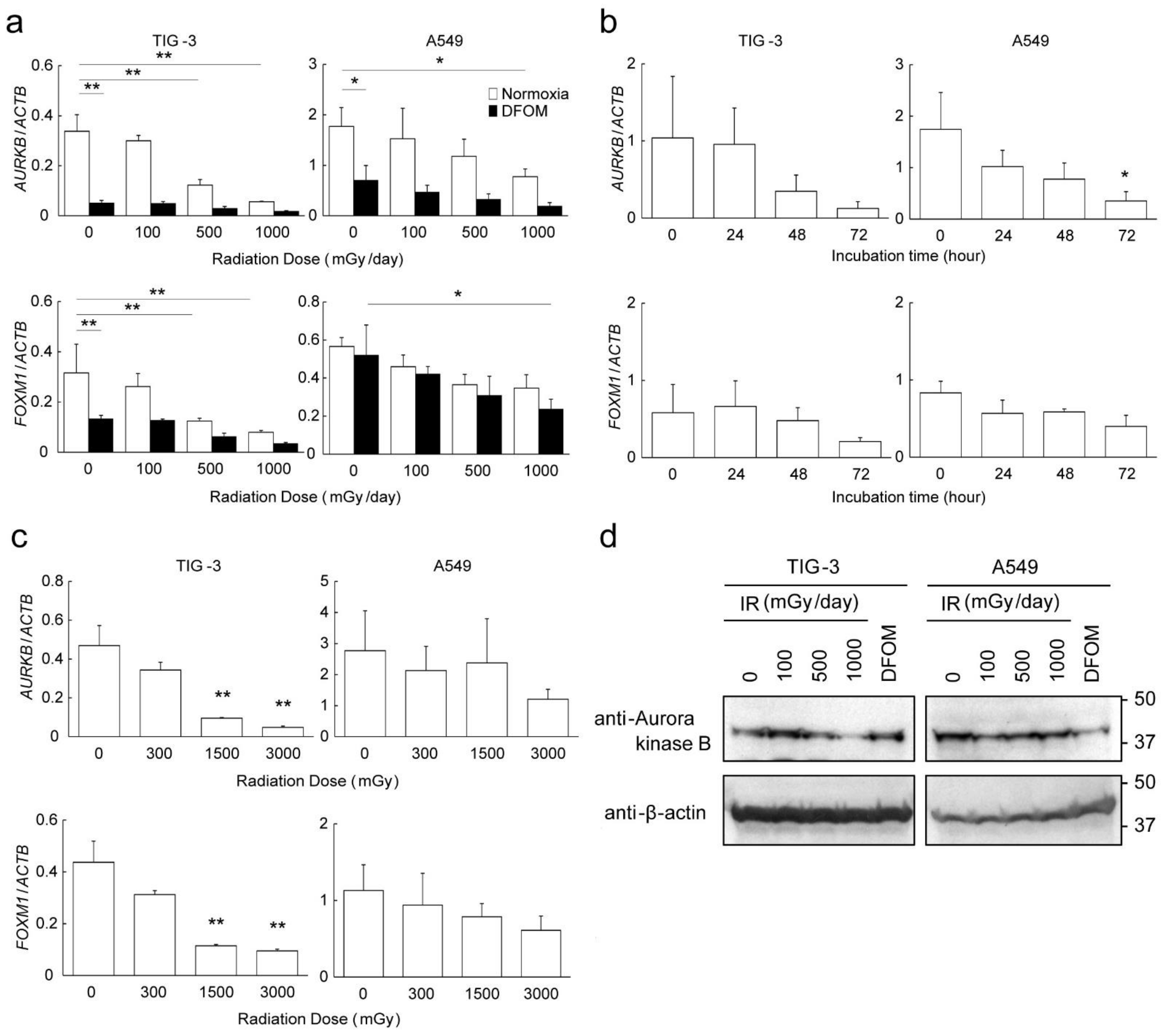
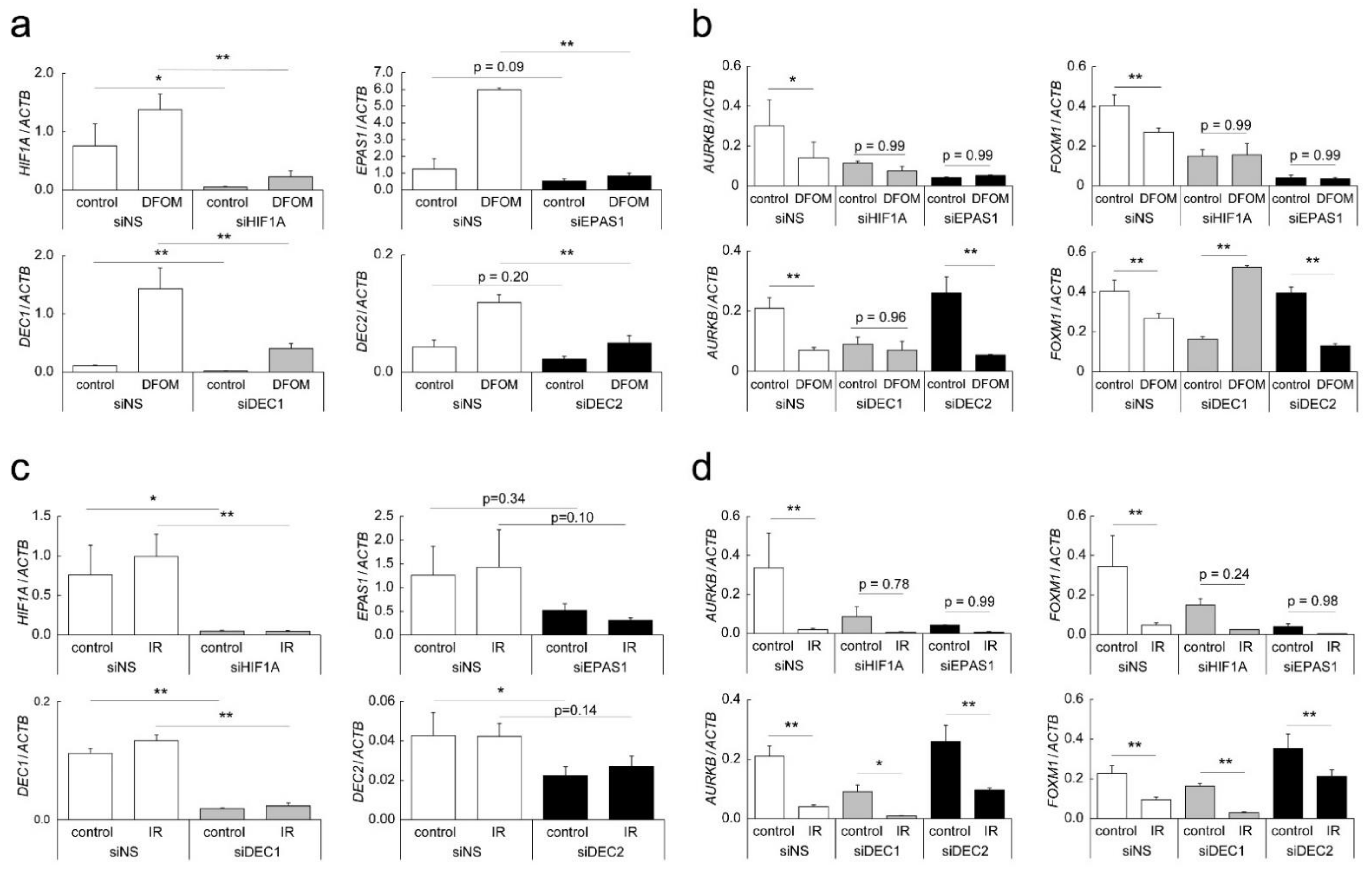
3.3. Effects of LDR-IR on Expression of AURKB and FOXM1
3.4. Mechanisms of Altered AURKB and FOXM1 Expression
3.5. Effects of LDR-IR on the Cell Cycle and Nuclear Morphology
3.6. Effect of LDR-IR on Sensitivity to Cell Cycle-Targeting Anti-Cancer Drugs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de González, A.B.; Darby, S. Risk of cancer from diagnostic X-Rays: Estimates for the UK and 14 other countries. Lancet 2004, 363, 345–351. [Google Scholar] [CrossRef]
- Samet, J.M.; Niwa, O. At the 75th Anniversary of the bombings of Hiroshima and Nagasaki, the radiation effects research foundation continues studies of the atomic bomb survivors and their children. Carcinogenesis 2020, 41, 1471–1472. [Google Scholar] [CrossRef] [PubMed]
- Yeager, M.; Machiela, M.J.; Kothiyal, P.; Dean, M.; Bodelon, C.; Suman, S.; Wang, M.; Mirabello, L.; Nelson, C.W.; Zhou, W.; et al. Lack of transgenerational effects of ionizing radiation exposure from the Chernobyl accident. Science 2021, 372, 725–729. [Google Scholar] [CrossRef]
- Morton, L.M.; Karyadi, D.M.; Stewart, C.; Bogdanova, T.I.; Dawson, E.T.; Steinberg, M.K.; Dai, J.; Hartley, S.W.; Schonfeld, S.J.; Sampson, J.N.; et al. Radiation-related genomic profile of papillary thyroid carcinoma after the Chernobyl accident. Science 2021, 372, eabg2538. [Google Scholar] [CrossRef] [PubMed]
- Imano, N.; Nishibuchi, I.; Kawabata, E.; Kinugasa, Y.; Shi, L.; Sakai, C.; Ishida, M.; Sakane, H.; Akita, T.; Ishida, T.; et al. Evaluating individual radiosensitivity for the prediction of acute toxicities of chemoradiotherapy in esophageal cancer patients. Radiat. Res. 2020, 195, 244–252. [Google Scholar] [CrossRef]
- Le Bourg, É. Hormesis, aging and longevity. Biochim. Biophys. Acta (BBA) Gen. Subj. 2009, 1790, 1030–1039. [Google Scholar] [CrossRef]
- Rühm, W.; Azizova, T.V.; Bouffler, S.D.; Little, M.P.; Shore, R.E.; Walsh, L.; Woloschak, G.E. Dose-rate effects in radiation biology and radiation protection. Ann. ICRP 2016, 45, 262–279. [Google Scholar] [CrossRef] [Green Version]
- Barsoumian, H.B.; Ramapriyan, R.; Younes, A.I.; Caetano, M.S.; Menon, H.; Comeaux, N.I.; Cushman, T.R.; Schoenhals, J.E.; Cadena, A.P.; Reilly, T.P.; et al. Low-dose radiation treatment enhances systemic antitumor immune responses by overcoming the inhibitory stroma. J. Immunother. Cancer 2020, 8, e000537. [Google Scholar] [CrossRef]
- Semenza, G.L. Intratumoral hypoxia, radiation resistance, and HIF-1. Cancer Cell 2004, 5, 405–406. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Bono, H.; Hiyama, K.; Kawamoto, T.; Kato, Y.; Nakanishi, T.; Nishiyama, M.; Hiyama, E.; Hirohashi, N.; Sueoka, E.; et al. Differentiated embryo chondrocyte plays a crucial role in DNA damage response via transcriptional regulation under hypoxic conditions. PLoS ONE 2018, 13, e0192136. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible Factor 1 Is a Basic-Helix-Loop-Helix-PAS heterodimer regulated by cellular 02 tension. Proc. Natl. Acad. Sci. USA 1995, 5, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [Green Version]
- Shimamoto, K.; Tanimoto, K.; Fukazawa, T.; Nakamura, H.; Kanai, A.; Bono, H.; Ono, H.; Eguchi, H.; Hirohashi, N. GLIS1, a novel hypoxia-inducible transcription factor, promotes breast cancer cell motility via activation of WNT5A. Carcinogenesis 2020, 41, 1184–1194. [Google Scholar] [CrossRef]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Cairns, R.; Papandreou, I.; Denko, N. Overcoming physiologic barriers to cancer treatment by molecularly targeting the tumor microenvironment. Mol. Cancer Res. 2006, 4, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horsman, M.R.; Overgaard, J. The impact of hypoxia and its modification of the outcome of radiotherapy. J. Radiat. Res. 2016, 57, i90–i98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalesi, E.; Nakamura, H.; Lee, K.L.; Putra, A.C.; Fukazawa, T.; Kawahara, Y.; Makino, Y.; Poellinger, L.; Yuge, L.; Tanimoto, K. The Krüppel-like Zinc finger transcription factor, GLI-Similar 1, is regulated by hypoxia-inducible factors via non-canonical mechanisms. Biochem. Biophys. Res. Commun. 2013, 441, 499–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, H.; Tanimoto, K.; Hiyama, K.; Yunokawa, M.; Kawamoto, T.; Kato, Y.; Yoshiga, K.; Poellinger, L.; Hiyama, E.; Nishiyama, M. Human mismatch repair gene, MLH1, is transcriptionally repressed by the hypoxia-inducible transcription factors, DEC1 and DEC2. Oncogene 2008, 27, 4200–4209. [Google Scholar] [CrossRef] [Green Version]
- Kanda, Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transpl. 2013, 48, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, B.A. How Taxol/Paclitaxel kills cancer cells. MBoC 2014, 25, 2677–2681. [Google Scholar] [CrossRef] [PubMed]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus Brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Kawai, H.; Sasatani, M.; Iizuka, D.; Masuda, Y.; Inaba, T.; Suzuki, K.; Ootsuyama, A.; Umata, T.; Kamiya, K.; et al. A novel ATM/TP53/P21-mediated checkpoint only activated by chronic γ-irradiation. PLoS ONE 2014, 9, e104279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart-Ornstein, J.; Iwamoto, Y.; Miller, M.A.; Prytyskach, M.A.; Ferretti, S.; Holzer, P.; Kallen, J.; Furet, P.; Jambhekar, A.; Forrester, W.C.; et al. P53 dynamics vary between tissues and are linked with radiation sensitivity. Nat. Commun. 2021, 12, 898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, F.; Wang, Z.; Yang, X.; Yu, H.; Si, S.; Lu, J.; Zhou, Z.; Lu, Q.; Wang, Z.; et al. ALKBH5 promotes the proliferation of renal cell carcinoma by regulating AURKB expression in an M6A-dependent manner. Ann. Transl. Med. 2020, 8, 646. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Shang, J.; Ji, W. ALKBH5-M6A-FOXM1 signaling axis promotes proliferation and invasion of lung adenocarcinoma cells under intermittent hypoxia. Biochem. Biophys. Res. Commun. 2020, 521, 499–506. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bögler, O.; et al. M 6 a demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell 2017, 31, 591–606.e6. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.-M.; Huang, W.-J.; Wang, B.; Liu, M.; Zhang, Q.; Yan, W.; Zhu, Q.; Luo, M.; Zhou, Z.-Z.; Tian, D.-A. Transcriptional up-regulation of FoxM1 in response to hypoxia is mediated by HIF-1. J. Cell. Biochem. 2009, 106, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Valerie, K.; Povirk, L.F. Regulation and mechanisms of mammalian double-strand break repair. Oncogene 2003, 22, 5792–5812. [Google Scholar] [CrossRef] [Green Version]
- Haber, J.E. Partners and pathways. Trends Genet. 2000, 16, 259–264. [Google Scholar] [CrossRef]
- Morrison, C. The controlling role of ATM in homologous recombinational Repair of DNA damage. EMBO J. 2000, 19, 463–471. [Google Scholar] [CrossRef]
- Lee, S.E.; Mitchell, R.A.; Cheng, A.; Hendrickson, E.A. Evidence for DNA-PK-dependent and -independent DNA double-strand break repair pathways in mammalian cells as a function of the cell cycle. Mol. Cell. Biol. 1997, 17, 1425–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, M.; Sasaki, M.S.; Sonoda, E.; Morrison, C.; Hashimoto, M.; Utsumi, H.; Yamaguchi-Iwai, Y.; Shinohara, A.; Takeda, S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998, 17, 5497–5508. [Google Scholar] [CrossRef] [PubMed]
- Al-Khafaji, A.S.; Davies, M.P.; Risk, J.M.; Marcus, M.W.; Koffa, M.; Gosney, J.R.; Shaw, R.J.; Field, J.K.; Liloglou, T. Aurora B Expression modulates paclitaxel response in non-small cell lung cancer. Br. J. Cancer 2017, 116, 592–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoellein, A.; Pickhard, A.; von Keitz, F.; Schoeffmann, S.; Piontek, G.; Rudelius, M.; Baumgart, A.; Wagenpfeil, S.; Peschel, C.; Dechow, T.; et al. Aurora kinase inhibition overcomes cetuximab resistance in squamous cell cancer of the head and neck. Oncotarget 2011, 2, 599–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phadke, M.S.; Sini, P.; Smalley, K.S.M. The novel ATP-competitive MEK/Aurora Kinase inhibitor BI-847325 overcomes acquired BRAF inhibitor resistance through suppression of Mcl-1 and MEK expression. Mol. Cancer Ther. 2015, 14, 1354–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, J.R.; Park, H.J.; Wang, Z.; Kiefer, M.M.; Raychaudhuri, P. FoxM1 Mediates resistance to Herceptin and paclitaxel. Cancer Res. 2010, 70, 5054–5063. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimabukuro, K.; Fukazawa, T.; Kanai, A.; Kawai, H.; Mekata, K.; Hirohashi, N.; Kakimoto, N.; Tanimoto, K. Low-Dose-Rate Irradiation Suppresses the Expression of Cell Cycle-Related Genes, Resulting in Modification of Sensitivity to Anti-Cancer Drugs. Cells 2022, 11, 501. https://doi.org/10.3390/cells11030501
Shimabukuro K, Fukazawa T, Kanai A, Kawai H, Mekata K, Hirohashi N, Kakimoto N, Tanimoto K. Low-Dose-Rate Irradiation Suppresses the Expression of Cell Cycle-Related Genes, Resulting in Modification of Sensitivity to Anti-Cancer Drugs. Cells. 2022; 11(3):501. https://doi.org/10.3390/cells11030501
Chicago/Turabian StyleShimabukuro, Kiichi, Takahiro Fukazawa, Akinori Kanai, Hidehiko Kawai, Kengo Mekata, Nobuyuki Hirohashi, Naoya Kakimoto, and Keiji Tanimoto. 2022. "Low-Dose-Rate Irradiation Suppresses the Expression of Cell Cycle-Related Genes, Resulting in Modification of Sensitivity to Anti-Cancer Drugs" Cells 11, no. 3: 501. https://doi.org/10.3390/cells11030501
APA StyleShimabukuro, K., Fukazawa, T., Kanai, A., Kawai, H., Mekata, K., Hirohashi, N., Kakimoto, N., & Tanimoto, K. (2022). Low-Dose-Rate Irradiation Suppresses the Expression of Cell Cycle-Related Genes, Resulting in Modification of Sensitivity to Anti-Cancer Drugs. Cells, 11(3), 501. https://doi.org/10.3390/cells11030501