
Lung Inflammation in STING-Associated Vasculopathy with Onset in Infancy (SAVI)


Abstract
:1. Introduction
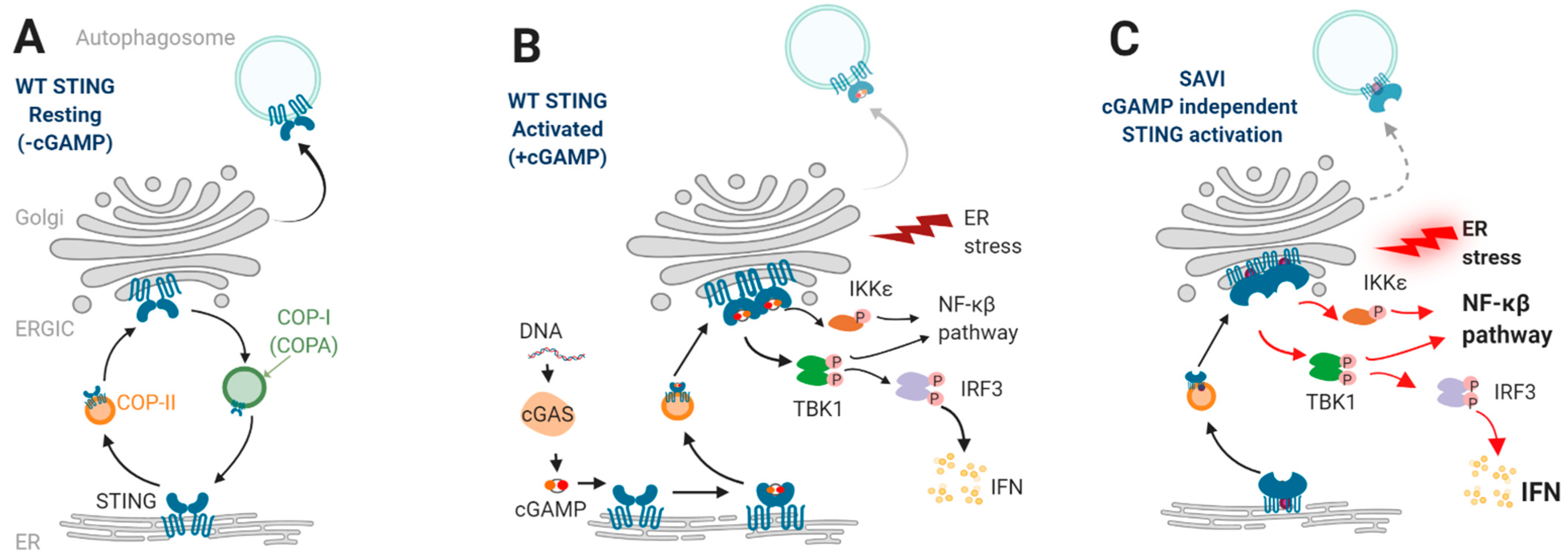
2. STING Signaling
2.1. STING and Type I Interferons
2.2. STING Enhances NF-κβ Pathway
2.3. STING and Autophagy
2.4. STING and Cell Death
3. Sting-Associated Vasculopathy with Onset in Infancy (SAVI)
3.1. Historical Description
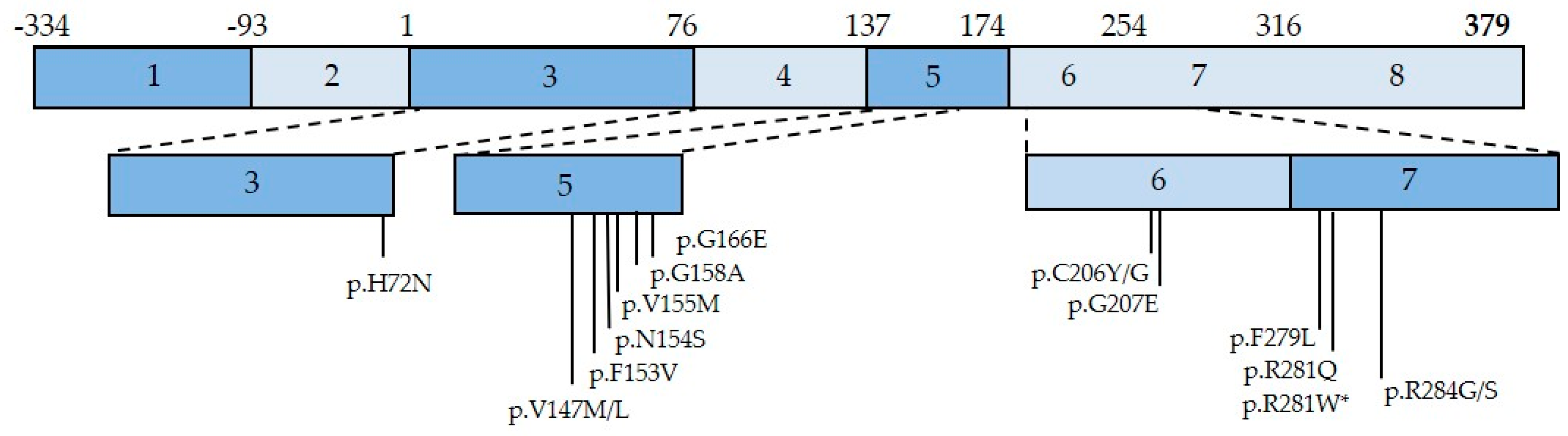
3.2. SAVI Pathogenesis
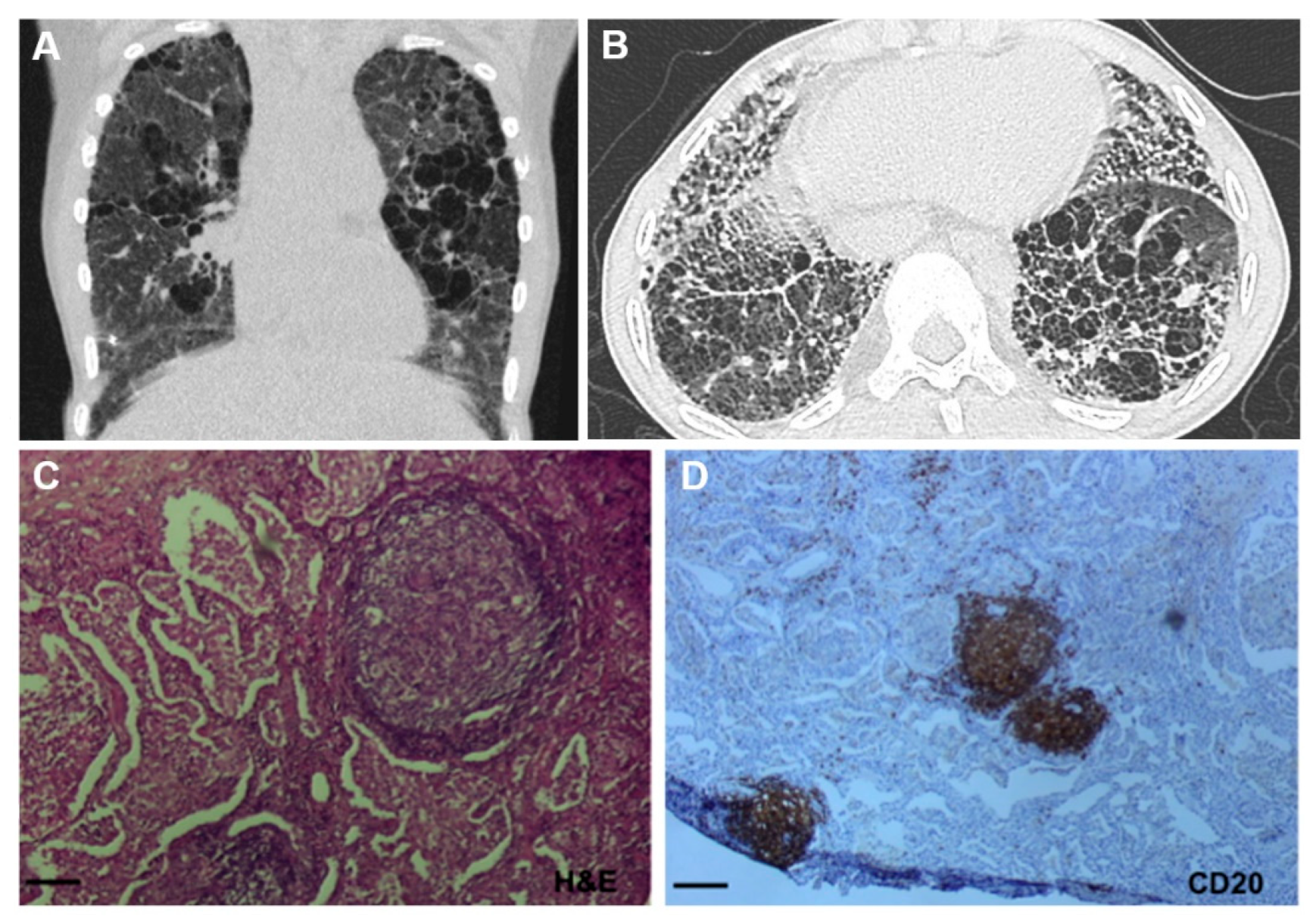
3.3. Lung Involvement in SAVI Patients
3.4. SAVI Lung Disease Pathogenesis
3.5. Futures Perspectives to Decipher SAVI Lung Disease Pathogenesis
4. STING-Mediated Lung Disease beyond SAVI
4.1. COPA Syndrome
4.2. Dermatomyositis and Systemic Lupus Erythematosus (SLE)
4.3. Beyond Type I Interferonopathies
5. Current and Future Therapies
5.1. JAK Inhibitors
5.2. Monoclonal Antibodies to Type I IFN Receptors
5.3. STING Inhibitors
5.4. Antifibrotic Therapy
5.5. Lung Transplantation
5.6. Future Therapeutic Perspectives
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crow, Y.J.; Stetson, D.B. The Type I Interferonopathies: 10 Years On. Nat. Rev. Immunol. 2021, 1–13. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of Type I Interferon Responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Uggenti, C.; Lepelley, A.; Crow, Y.J. Self-Awareness: Nucleic Acid-Driven Inflammation and the Type I Interferonopathies. Annu. Rev. Immunol. 2019, 37, 247–267. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Chase, D.S.; Lowenstein Schmidt, J.; Szynkiewicz, M.; Forte, G.M.A.; Gornall, H.L.; Oojageer, A.; Anderson, B.; Pizzino, A.; Helman, G.; et al. Characterization of Human Disease Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A. 2015, 167A, 296–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokatayev, V.; Hasin, N.; Chon, H.; Cerritelli, S.M.; Sakhuja, K.; Ward, J.M.; Morris, H.D.; Yan, N.; Crouch, R.J. RNase H2 Catalytic Core Aicardi-Goutières Syndrome-Related Mutant Invokes CGAS-STING Innate Immune-Sensing Pathway in Mice. J. Exp. Med. 2016, 213, 329–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crow, Y.J. Type I Interferonopathies: A Novel Set of Inborn Errors of Immunity. Ann. N. Y. Acad. Sci. 2011, 1238, 91–98. [Google Scholar] [CrossRef]
- Sanchez, G.A.M.; Reinhardt, A.; Ramsey, S.; Wittkowski, H.; Hashkes, P.J.; Berkun, Y.; Schalm, S.; Murias, S.; Dare, J.A.; Brown, D.; et al. JAK1/2 Inhibition with Baricitinib in the Treatment of Autoinflammatory Interferonopathies. J. Clin. Investig. 2018, 128, 3041–3052. [Google Scholar] [CrossRef] [Green Version]
- Crow, Y.J.; Manel, N. Aicardi-Goutières Syndrome and the Type I Interferonopathies. Nat. Rev. Immunol. 2015, 15, 429–440. [Google Scholar] [CrossRef]
- Liu, Y.; Jesus, A.A.; Marrero, B.; Yang, D.; Ramsey, S.E.; Sanchez, G.A.M.; Tenbrock, K.; Wittkowski, H.; Jones, O.Y.; Kuehn, H.S.; et al. Activated STING in a Vascular and Pulmonary Syndrome. N. Engl. J. Med. 2014, 371, 507–518. [Google Scholar] [CrossRef] [Green Version]
- Jeremiah, N.; Neven, B.; Gentili, M.; Callebaut, I.; Maschalidi, S.; Stolzenberg, M.-C.; Goudin, N.; Frémond, M.-L.; Nitschke, P.; Molina, T.J.; et al. Inherited STING-Activating Mutation Underlies a Familial Inflammatory Syndrome with Lupus-like Manifestations. J. Clin. Investig. 2014, 124, 5516–5520. [Google Scholar] [CrossRef]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING Regulates Intracellular DNA-Mediated, Type I Interferon-Dependent Innate Immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollock, A.J.; Zaver, S.A.; Woodward, J.J. A STING-Based Biosensor Affords Broad Cyclic Dinucleotide Detection within Single Living Eukaryotic Cells. Nat. Commun. 2020, 11, 3533. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Schmid-Burgk, J.L.; Hemmerling, I.; Horvath, G.L.; Schmidt, T.; Latz, E.; Hornung, V. Cell Intrinsic Immunity Spreads to Bystander Cells via the Intercellular Transfer of CGAMP. Nature 2013, 503, 530–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, G.; Zhang, C.; Chen, Z.J.; Bai, X.-C.; Zhang, X. Cryo-EM Structures of STING Reveal Its Mechanism of Activation by Cyclic GMP-AMP. Nature 2019, 567, 389–393. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Hayashi, T.; Takahara, K.; Satoh, T.; Lee, H.; Matsunaga, K.; Kageyama, S.; Omori, H.; Noda, T.; et al. Atg9a Controls DsDNA-Driven Dynamic Translocation of STING and the Innate Immune Response. Proc. Natl. Acad. Sci. USA 2009, 106, 20842–20846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.-W.; Li, S.; Li, C.; Lian, H.; Yang, Q.; Zhong, B.; Shu, H.-B. IRhom2 Is Essential for Innate Immunity to DNA Viruses by Mediating Trafficking and Stability of the Adaptor STING. Nat. Immunol. 2016, 17, 1057–1066. [Google Scholar] [CrossRef]
- Ran, Y.; Xiong, M.-G.; Xu, Z.-S.; Luo, W.-W.; Wang, S.-Y.; Wang, Y.-Y. YIPF5 Is Essential for Innate Immunity to DNA Virus and Facilitates COPII-Dependent STING Trafficking. J. Immunol. 2019, 203, 1560–1570. [Google Scholar] [CrossRef]
- Ablasser, A.; Chen, Z.J. CGAS in Action: Expanding Roles in Immunity and Inflammation. Science 2019, 363, eaat8657. [Google Scholar] [CrossRef]
- Mukai, K.; Konno, H.; Akiba, T.; Uemura, T.; Waguri, S.; Kobayashi, T.; Barber, G.N.; Arai, H.; Taguchi, T. Activation of STING Requires Palmitoylation at the Golgi. Nat. Commun. 2016, 7, 11932. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of Innate Immune Adaptor Proteins MAVS, STING, and TRIF Induces IRF3 Activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The CGAS-STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef] [PubMed]
- Gonugunta, V.K.; Sakai, T.; Pokatayev, V.; Yang, K.; Wu, J.; Dobbs, N.; Yan, N. Trafficking-Mediated STING Degradation Requires Sorting to Acidified Endolysosomes and Can Be Targeted to Enhance Anti-Tumor Response. Cell Rep. 2017, 21, 3234–3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, T.; Mukai, K.; Takaya, E.; Shindo, R. STING Operation at the ER/Golgi Interface. Front. Immunol. 2021, 12, 646304. [Google Scholar] [CrossRef] [PubMed]
- Lepelley, A.; Martin-Niclós, M.J.; Le Bihan, M.; Marsh, J.A.; Uggenti, C.; Rice, G.I.; Bondet, V.; Duffy, D.; Hertzog, J.; Rehwinkel, J.; et al. Mutations in COPA Lead to Abnormal Trafficking of STING to the Golgi and Interferon Signaling. J. Exp. Med. 2020, 217, e20200600. [Google Scholar] [CrossRef]
- Watkin, L.B.; Jessen, B.; Wiszniewski, W.; Vece, T.J.; Jan, M.; Sha, Y.; Thamsen, M.; Santos-Cortez, R.L.P.; Lee, K.; Gambin, T.; et al. COPA Mutations Impair ER-Golgi Transport and Cause Hereditary Autoimmune-Mediated Lung Disease and Arthritis. Nat. Genet. 2015, 47, 654–660. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira Mann, C.C.; Orzalli, M.H.; King, D.S.; Kagan, J.C.; Lee, A.S.Y.; Kranzusch, P.J. Modular Architecture of the STING C-Terminal Tail Allows Interferon and NF-ΚB Signaling Adaptation. Cell Rep. 2019, 27, 1165–1175.e5. [Google Scholar] [CrossRef] [Green Version]
- Su, J.; Rui, Y.; Lou, M.; Yin, L.; Xiong, H.; Zhou, Z.; Shen, S.; Chen, T.; Zhang, Z.; Zhao, N.; et al. HIV-2/SIV Vpx Targets a Novel Functional Domain of STING to Selectively Inhibit CGAS–STING-Mediated NF-ΚB Signalling. Nat. Microbiol. 2019, 4, 2552–2564. [Google Scholar] [CrossRef]
- Wu, J.; Yan, N. No Longer A One-Trick Pony: STING Signaling Activity Beyond Interferon. J. Mol. Biol. 2021, 167257. [Google Scholar] [CrossRef]
- Wu, X.; Wu, F.-H.; Wang, X.; Wang, L.; Siedow, J.N.; Zhang, W.; Pei, Z.-M. Molecular Evolutionary and Structural Analysis of the Cytosolic DNA Sensor CGAS and STING. Nucleic Acids Res. 2014, 42, 8243–8257. [Google Scholar] [CrossRef] [Green Version]
- Morehouse, B.R.; Govande, A.A.; Millman, A.; Keszei, A.F.A.; Lowey, B.; Ofir, G.; Shao, S.; Sorek, R.; Kranzusch, P.J. STING Cyclic Dinucleotide Sensing Originated in Bacteria. Nature 2020, 586, 429–433. [Google Scholar] [CrossRef]
- Abe, T.; Barber, G.N. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF-ΚB Activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [Green Version]
- Balka, K.R.; Louis, C.; Saunders, T.L.; Smith, A.M.; Calleja, D.J.; D’Silva, D.B.; Moghaddas, F.; Tailler, M.; Lawlor, K.E.; Zhan, Y.; et al. TBK1 and IKKε Act Redundantly to Mediate STING-Induced NF-ΚB Responses in Myeloid Cells. Cell Rep. 2020, 31, 107492. [Google Scholar] [CrossRef]
- Nigrovic, P.A.; Lee, P.Y.; Hoffman, H.M. Monogenic Autoinflammatory Disorders: Conceptual Overview, Phenotype, and Clinical Approach. J. Allergy Clin. Immunol. 2020, 146, 925–937. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of Mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Manzanillo, P.S.; Shiloh, M.U.; Portnoy, D.A.; Cox, J.S. Mycobacterium Tuberculosis Activates the DNA-Dependent Cytosolic Surveillance Pathway within Macrophages. Cell Host Microbe 2012, 11, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Watson, R.O.; Manzanillo, P.S.; Cox, J.S. Extracellular, M. Tuberculosis DNA Targets Bacteria for Autophagy by Activating the Host DNA-Sensing Pathway. Cell 2012, 150, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Wu, H.; Wang, C.; Li, Y.; Tian, H.; Siraj, S.; Sehgal, S.A.; Wang, X.; Wang, J.; Shang, Y.; et al. STING Directly Activates Autophagy to Tune the Innate Immune Response. Cell Death Differ. 2019, 26, 1735–1749. [Google Scholar] [CrossRef]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy Induction via STING Trafficking Is a Primordial Function of the CGAS Pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Racanelli, A.C.; Choi, A.M.K.; Choi, M.E. Autophagy in Chronic Lung Disease. Prog. Mol. Biol. Transl. Sci. 2020, 172, 135–156. [Google Scholar] [CrossRef]
- Ning, X.; Wang, Y.; Jing, M.; Sha, M.; Lv, M.; Gao, P.; Zhang, R.; Huang, X.; Feng, J.-M.; Jiang, Z. Apoptotic Caspases Suppress Type I Interferon Production via the Cleavage of CGAS, MAVS, and IRF3. Mol. Cell 2019, 74, 19–31.e7. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Y.-J.; Dobbs, N.; Sakai, T.; Liou, J.; Miner, J.J.; Yan, N. STING-Mediated Disruption of Calcium Homeostasis Chronically Activates ER Stress and Primes T Cell Death. J. Exp. Med. 2019, 216, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Iracheta-Vellve, A.; Csak, T.; Satishchandran, A.; Kodys, K.; Kurt-Jones, E.A.; Fitzgerald, K.A.; Szabo, G. STING-IRF3 Pathway Links Endoplasmic Reticulum Stress with Hepatocyte Apoptosis in Early Alcoholic Liver Disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16544–16549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Ramshorn, K.; Pinci, F.; Zuber, S.; O’Duill, F.; Schmid-Burgk, J.L.; Hoss, F.; Buhmann, R.; et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 2017, 171, 1110–1124.e18. [Google Scholar] [CrossRef]
- Cerboni, S.; Jeremiah, N.; Gentili, M.; Gehrmann, U.; Conrad, C.; Stolzenberg, M.-C.; Picard, C.; Neven, B.; Fischer, A.; Amigorena, S.; et al. Intrinsic Antiproliferative Activity of the Innate Sensor STING in T Lymphocytes. J. Exp. Med. 2017, 214, 1769–1785. [Google Scholar] [CrossRef] [Green Version]
- Siedel, H.; Roers, A.; Rösen-Wolff, A.; Luksch, H. Type I Interferon-Independent T Cell Impairment in a Tmem173 N153S/WT Mouse Model of STING Associated Vasculopathy with Onset in Infancy (SAVI). Clin. Immunol. 2020, 216, 108466. [Google Scholar] [CrossRef]
- Keskitalo, S.; Haapaniemi, E.; Einarsdottir, E.; Rajamäki, K.; Heikkilä, H.; Ilander, M.; Pöyhönen, M.; Morgunova, E.; Hokynar, K.; Lagström, S.; et al. Novel TMEM173 Mutation and the Role of Disease Modifying Alleles. Front. Immunol. 2019, 10, 2770. [Google Scholar] [CrossRef] [Green Version]
- Staels, F.; Betrains, A.; Doubel, P.; Willemsen, M.; Cleemput, V.; Vanderschueren, S.; Corveleyn, A.; Meyts, I.; Sprangers, B.; Crow, Y.J.; et al. Adult-Onset ANCA-Associated Vasculitis in SAVI: Extension of the Phenotypic Spectrum, Case Report and Review of the Literature. Front. Immunol. 2020, 11, 575219. [Google Scholar] [CrossRef]
- Tang, X.; Xu, H.; Zhou, C.; Peng, Y.; Liu, H.; Liu, J.; Li, H.; Yang, H.; Zhao, S. STING-Associated Vasculopathy with Onset in Infancy in Three Children with New Clinical Aspect and Unsatisfactory Therapeutic Responses to Tofacitinib. J. Clin. Immunol. 2020, 40, 114–122. [Google Scholar] [CrossRef]
- Lin, B.; Berard, R.; Al Rasheed, A.; Aladba, B.; Kranzusch, P.J.; Henderlight, M.; Grom, A.; Kahle, D.; Torreggiani, S.; Aue, A.G.; et al. A Novel STING1 Variant Causes a Recessive Form of STING-Associated Vasculopathy with Onset in Infancy (SAVI). J. Allergy Clin. Immunol. 2020, 146, 1204–1208.e6. [Google Scholar] [CrossRef]
- Sui, H.; Zhou, M.; Imamichi, H.; Jiao, X.; Sherman, B.T.; Lane, H.C.; Imamichi, T. STING Is an Essential Mediator of the Ku70-Mediated Production of IFN-Λ1 in Response to Exogenous DNA. Sci. Signal. 2017, 10, eaah5054. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.; Torreggiani, S.; Kahle, D.; Rumsey, D.G.; Wright, B.L.; Montes-Cano, M.A.; Silveira, L.F.; Alehashemi, S.; Mitchell, J.; Aue, A.G.; et al. Case Report: Novel SAVI-Causing Variants in STING1 Expand the Clinical Disease Spectrum and Suggest a Refined Model of STING Activation. Front. Immunol. 2021, 12, 636225. [Google Scholar] [CrossRef]
- Frémond, M.-L.; Crow, Y.J. STING-Mediated Lung Inflammation and Beyond. J. Clin. Immunol. 2021, 41, 501–514. [Google Scholar] [CrossRef]
- Dobbs, N.; Burnaevskiy, N.; Chen, D.; Gonugunta, V.K.; Alto, N.M.; Yan, N. STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe 2015, 18, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Frémond, M.-L.; Rodero, M.P.; Jeremiah, N.; Belot, A.; Jeziorski, E.; Duffy, D.; Bessis, D.; Cros, G.; Rice, G.I.; Charbit, B.; et al. Efficacy of the Janus Kinase 1/2 Inhibitor Ruxolitinib in the Treatment of Vasculopathy Associated with TMEM173-Activating Mutations in 3 Children. J. Allergy Clin. Immunol. 2016, 138, 1752–1755. [Google Scholar] [CrossRef] [Green Version]
- Broggi, A.; Ghosh, S.; Sposito, B.; Spreafico, R.; Balzarini, F.; Lo Cascio, A.; Clementi, N.; De Santis, M.; Mancini, N.; Granucci, F.; et al. Type III Interferons Disrupt the Lung Epithelial Barrier upon Viral Recognition. Science 2020, 369, 706–712. [Google Scholar] [CrossRef]
- Warner, J.D.; Irizarry-Caro, R.A.; Bennion, B.G.; Ai, T.L.; Smith, A.M.; Miner, C.A.; Sakai, T.; Gonugunta, V.K.; Wu, J.; Platt, D.J.; et al. STING-Associated Vasculopathy Develops Independently of IRF3 in Mice. J. Exp. Med. 2017, 214, 3279–3292. [Google Scholar] [CrossRef] [Green Version]
- Saldanha, R.G.; Balka, K.R.; Davidson, S.; Wainstein, B.K.; Wong, M.; Macintosh, R.; Loo, C.K.C.; Weber, M.A.; Kamath, V.; CIRCA; et al. A Mutation Outside the Dimerization Domain Causing Atypical STING-Associated Vasculopathy With Onset in Infancy. Front. Immunol. 2018, 9, 1535. [Google Scholar] [CrossRef] [Green Version]
- Frémond, M.-L.; Hadchouel, A.; Berteloot, L.; Melki, I.; Bresson, V.; Barnabei, L.; Jeremiah, N.; Belot, A.; Bondet, V.; Brocq, O.; et al. Overview of STING-Associated Vasculopathy with Onset in Infancy (SAVI) Among 21 Patients. J. Allergy Clin. Immunol. Pract. 2020, 9, 803–818. [Google Scholar] [CrossRef]
- Raffaele, C.G.L.; Messia, V.; Moneta, G.; Caiello, I.; Federici, S.; Pardeo, M.; Bracaglia, C.; De Benedetti, F.; Insalaco, A. A Patient with Stimulator of Interferon Genes-Associated Vasculopathy with Onset in Infancy without Skin Vasculopathy. Rheumatol. Oxf. Engl. 2020, 59, 905–907. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Pawaria, S.; Bernier, J.; Moses, S.; Henry, K.; Fang, T.; Burkly, L.; Marshak-Rothstein, A.; Fitzgerald, K.A. Hierarchy of Clinical Manifestations in SAVI N153S and V154M Mouse Models. Proc. Natl. Acad. Sci. USA 2019, 116, 7941–7950. [Google Scholar] [CrossRef] [Green Version]
- Luksch, H.; Stinson, W.A.; Platt, D.J.; Qian, W.; Kalugotla, G.; Miner, C.A.; Bennion, B.G.; Gerbaulet, A.; Rösen-Wolff, A.; Miner, J.J. STING-Associated Lung Disease in Mice Relies on T Cells but Not Type I Interferon. J. Allergy Clin. Immunol. 2019, 144, 254–266.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennion, B.G.; Croft, C.A.; Ai, T.L.; Qian, W.; Menos, A.M.; Miner, C.A.; Frémond, M.-L.; Doisne, J.-M.; Andhey, P.S.; Platt, D.J.; et al. STING Gain-of-Function Disrupts Lymph Node Organogenesis and Innate Lymphoid Cell Development in Mice. Cell Rep. 2020, 31, 107771. [Google Scholar] [CrossRef] [PubMed]
- Bouis, D.; Kirstetter, P.; Arbogast, F.; Lamon, D.; Delgado, V.; Jung, S.; Ebel, C.; Jacobs, H.; Knapp, A.-M.; Jeremiah, N.; et al. Severe Combined Immunodeficiency in Stimulator of Interferon Genes (STING) V154M/Wild-Type Mice. J. Allergy Clin. Immunol. 2019, 143, 712–725.e5. [Google Scholar] [CrossRef] [Green Version]
- Martin, G.R.; Henare, K.; Salazar, C.; Scheidl-Yee, T.; Eggen, L.J.; Tailor, P.P.; Kim, J.H.; Podstawka, J.; Fritzler, M.J.; Kelly, M.M.; et al. Expression of a Constitutively Active Human STING Mutant in Hematopoietic Cells Produces an Ifnar1-Dependent Vasculopathy in Mice. Life Sci. Alliance 2019, 2, e201800215. [Google Scholar] [CrossRef] [Green Version]
- Gruber, C.; Bogunovic, D. Incomplete Penetrance in Primary Immunodeficiency: A Skeleton in the Closet. Hum. Genet. 2020, 139, 745–757. [Google Scholar] [CrossRef]
- Boers, J.E.; Ambergen, A.W.; Thunnissen, F.B. Number and Proliferation of Clara Cells in Normal Human Airway Epithelium. Am. J. Respir. Crit. Care Med. 1999, 159, 1585–1591. [Google Scholar] [CrossRef]
- Rock, J.R.; Randell, S.H.; Hogan, B.L.M. Airway Basal Stem Cells: A Perspective on Their Roles in Epithelial Homeostasis and Remodeling. Dis. Model. Mech. 2010, 3, 545–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobe, A.H. Animal Models, Learning Lessons to Prevent and Treat Neonatal Chronic Lung Disease. Front. Med. 2015, 2, 49. [Google Scholar] [CrossRef] [Green Version]
- Anastasiou, M.; Newton, G.A.; Kaur, K.; Carrillo-Salinas, F.J.; Smolgovsky, S.A.; Bayer, A.L.; Ilyukha, V.; Sharma, S.; Poltorak, A.; Luscinskas, F.W.; et al. Endothelial STING Controls T Cell Transmigration in an IFNI-Dependent Manner. JCI Insight 2021, 6, 149346. [Google Scholar] [CrossRef]
- Shih, A.Y.; Damm-Ganamet, K.L.; Mirzadegan, T. Dynamic Structural Differences between Human and Mouse STING Lead to Differing Sensitivity to DMXAA. Biophys. J. 2018, 114, 32–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Campbell, A.M.; Chan, J.; Schattgen, S.A.; Orlowski, G.M.; Nayar, R.; Huyler, A.H.; Nündel, K.; Mohan, C.; Berg, L.J.; et al. Suppression of Systemic Autoimmunity by the Innate Immune Adaptor STING. Proc. Natl. Acad. Sci. USA 2015, 112, E710–E717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akazawa, S.; Mackin, L.; Jhala, G.; Fynch, S.; Catterall, T.; Selck, C.; Graham, K.L.; Krishnamurthy, B.; Pappas, E.G.; Kwong, C.-T.J.; et al. Deficiency of the Innate Immune Adaptor STING Promotes Autoreactive T Cell Expansion in NOD Mice. Diabetologia 2021, 64, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Barnabei, L.; Castela, M.; Banal, C.; Lefort, N.; Rieux-Laucat, F. Generation of an IPSC Line (IMAGINi011-A) from a Patient Carrying a STING Mutation. Stem Cell Res. 2020, 50, 102107. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Vedaie, M.; Roberts, D.A.; Thomas, D.C.; Villacorta-Martin, C.; Alysandratos, K.-D.; Hawkins, F.; Kotton, D.N. Derivation of Self-Renewing Lung Alveolar Epithelial Type II Cells from Human Pluripotent Stem Cells. Nat. Protoc. 2019, 14, 3303–3332. [Google Scholar] [CrossRef]
- Jacob, A.; Morley, M.; Hawkins, F.; McCauley, K.B.; Jean, J.C.; Heins, H.; Na, C.-L.; Weaver, T.E.; Vedaie, M.; Hurley, K.; et al. Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell 2017, 21, 472–488.e10. [Google Scholar] [CrossRef]
- Yin, X.; Riva, L.; Pu, Y.; Martin-Sancho, L.; Kanamune, J.; Yamamoto, Y.; Sakai, K.; Gotoh, S.; Miorin, L.; De Jesus, P.D.; et al. MDA5 Governs the Innate Immune Response to SARS-CoV-2 in Lung Epithelial Cells. Cell Rep. 2021, 34, 108628. [Google Scholar] [CrossRef]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.-I.; Taylor, C.J.; Jetter, C.; et al. Single-Cell RNA Sequencing Reveals Profibrotic Roles of Distinct Epithelial and Mesenchymal Lineages in Pulmonary Fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef] [PubMed]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.-I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.-I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-Derived Alveolar Macrophages Drive Lung Fibrosis and Persist in the Lung over the Life Span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strikoudis, A.; Cieślak, A.; Loffredo, L.; Chen, Y.-W.; Patel, N.; Saqi, A.; Lederer, D.J.; Snoeck, H.-W. Modeling of Fibrotic Lung Disease Using 3D Organoids Derived from Human Pluripotent Stem Cells. Cell Rep. 2019, 27, 3709–3723.e5. [Google Scholar] [CrossRef] [Green Version]
- De Carvalho, A.L.R.T.; Liu, H.-Y.; Chen, Y.-W.; Porotto, M.; Moscona, A.; Snoeck, H.-W. The in Vitro Multilineage Differentiation and Maturation of Lung and Airway Cells from Human Pluripotent Stem Cell-Derived Lung Progenitors in 3D. Nat. Protoc. 2021, 16, 1802–1829. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-W.; Huang, S.X.; de Carvalho, A.L.R.T.; Ho, S.-H.; Islam, M.N.; Volpi, S.; Notarangelo, L.D.; Ciancanelli, M.; Casanova, J.-L.; Bhattacharya, J.; et al. A Three-Dimensional Model of Human Lung Development and Disease from Pluripotent Stem Cells. Nat. Cell Biol. 2017, 19, 542–549. [Google Scholar] [CrossRef]
- Volpi, S.; Tsui, J.; Mariani, M.; Pastorino, C.; Caorsi, R.; Sacco, O.; Ravelli, A.; Shum, A.K.; Gattorno, M.; Picco, P. Type I Interferon Pathway Activation in COPA Syndrome. Clin. Immunol. 2018, 187, 33–36. [Google Scholar] [CrossRef]
- Crow, M.K. Type I Interferon in the Pathogenesis of Lupus. J. Immunol. 2014, 192, 5459–5468. [Google Scholar] [CrossRef]
- Huard, C.; Gullà, S.V.; Bennett, D.V.; Coyle, A.J.; Vleugels, R.A.; Greenberg, S.A. Correlation of Cutaneous Disease Activity with Type 1 Interferon Gene Signature and Interferon β in Dermatomyositis. Br. J. Dermatol. 2017, 176, 1224–1230. [Google Scholar] [CrossRef]
- Wu, W.; Guo, L.; Fu, Y.; Wang, K.; Zhang, D.; Xu, W.; Chen, Z.; Ye, S. Interstitial Lung Disease in Anti-MDA5 Positive Dermatomyositis. Clin. Rev. Allergy Immunol. 2021, 60, 293–304. [Google Scholar] [CrossRef]
- Ladislau, L.; Suárez-Calvet, X.; Toquet, S.; Landon-Cardinal, O.; Amelin, D.; Depp, M.; Rodero, M.P.; Hathazi, D.; Duffy, D.; Bondet, V.; et al. JAK Inhibitor Improves Type I Interferon Induced Damage: Proof of Concept in Dermatomyositis. Brain J. Neurol. 2018, 141, 1609–1621. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, X.; Ye, S. Tofacitinib in Amyopathic Dermatomyositis-Associated Interstitial Lung Disease. N. Engl. J. Med. 2019, 381, 291–293. [Google Scholar] [CrossRef]
- Li, Y.; Bax, C.; Patel, J.; Vazquez, T.; Ravishankar, A.; Bashir, M.M.; Grinnell, M.; Diaz, D.; Werth, V.P. Plasma-Derived DNA Containing-Extracellular Vesicles Induce STING-Mediated Proinflammatory Responses in Dermatomyositis. Theranostics 2021, 11, 7144–7158. [Google Scholar] [CrossRef]
- Ma, R.; Ortiz Serrano, T.P.; Davis, J.; Prigge, A.D.; Ridge, K.M. The CGAS-STING Pathway: The Role of Self-DNA Sensing in Inflammatory Lung Disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 13156–13170. [Google Scholar] [CrossRef] [PubMed]
- Benmerzoug, S.; Rose, S.; Bounab, B.; Gosset, D.; Duneau, L.; Chenuet, P.; Mollet, L.; Le Bert, M.; Lambers, C.; Geleff, S.; et al. STING-Dependent Sensing of Self-DNA Drives Silica-Induced Lung Inflammation. Nat. Commun. 2018, 9, 5226. [Google Scholar] [CrossRef]
- Nascimento, M.; Gombault, A.; Lacerda-Queiroz, N.; Panek, C.; Savigny, F.; Sbeity, M.; Bourinet, M.; Le Bert, M.; Riteau, N.; Ryffel, B.; et al. Self-DNA Release and STING-Dependent Sensing Drives Inflammation to Cigarette Smoke in Mice. Sci. Rep. 2019, 9, 14848. [Google Scholar] [CrossRef]
- Savigny, F.; Schricke, C.; Lacerda-Queiroz, N.; Meda, M.; Nascimento, M.; Huot-Marchand, S.; Da Gama Monteiro, F.; Ryffel, B.; Gombault, A.; Le Bert, M.; et al. Protective Role of the Nucleic Acid Sensor STING in Pulmonary Fibrosis. Front. Immunol. 2020, 11, 588799. [Google Scholar] [CrossRef] [PubMed]
- Chia, J.; Eroglu, F.K.; Özen, S.; Orhan, D.; Montealegre-Sanchez, G.; de Jesus, A.A.; Goldbach-Mansky, R.; Cowen, E.W. Failure to Thrive, Interstitial Lung Disease, and Progressive Digital Necrosis with Onset in Infancy. J. Am. Acad. Dermatol. 2016, 74, 186–189. [Google Scholar] [CrossRef] [Green Version]
- Balci, S.; Ekinci, R.M.K.; de Jesus, A.A.; Goldbach-Mansky, R.; Yilmaz, M. Baricitinib Experience on STING-Associated Vasculopathy with Onset in Infancy: A Representative Case from Turkey. Clin. Immunol. 2020, 212, 108273. [Google Scholar] [CrossRef]
- Alghamdi, M.A.; Mulla, J.; Saheb Sharif-Askari, N.; Guzmán-Vega, F.J.; Arold, S.T.; Abd-Alwahed, M.; Alharbi, N.; Kashour, T.; Halwani, R. A Novel Biallelic STING1 Gene Variant Causing SAVI in Two Siblings. Front. Immunol. 2020, 11, 599564. [Google Scholar] [CrossRef]
- Kim, H.; Brooks, K.M.; Tang, C.C.; Wakim, P.; Blake, M.; Brooks, S.R.; Montealegre Sanchez, G.A.; de Jesus, A.A.; Huang, Y.; Tsai, W.L.; et al. Pharmacokinetics, Pharmacodynamics, and Proposed Dosing of the Oral JAK1 and JAK2 Inhibitor Baricitinib in Pediatric and Young Adult CANDLE and SAVI Patients. Clin. Pharmacol. Ther. 2018, 104, 364–373. [Google Scholar] [CrossRef]
- Neven, B.; Al Adba, B.; Hully, M.; Desguerre, I.; Pressiat, C.; Boddaert, N.; Duffy, D.; Rice, G.I.; Seabra, L.; Frémond, M.-L.; et al. JAK Inhibition in the Aicardi-Goutières Syndrome. N. Engl. J. Med. 2020, 383, 2190–2191. [Google Scholar] [CrossRef]
- Hadjadj, J.; Frémond, M.-L.; Neven, B. Emerging Place of JAK Inhibitors in the Treatment of Inborn Errors of Immunity. Front. Immunol. 2021, 12, 717388. [Google Scholar] [CrossRef]
- Gadina, M.; Chisolm, D.A.; Philips, R.L.; McInness, I.B.; Changelian, P.S.; O’Shea, J.J. Translating JAKs to Jakinibs. J. Immunol. 2020, 204, 2011–2020. [Google Scholar] [CrossRef]
- Funakoshi-Tago, M.; Tago, K.; Sato, Y.; Tominaga, S.-I.; Kasahara, T. JAK2 Is an Important Signal Transducer in IL-33-Induced NF-ΚB Activation. Cell. Signal. 2011, 23, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Soriano, A.; Soriano, M.; Espinosa, G.; Manna, R.; Emmi, G.; Cantarini, L.; Hernández-Rodríguez, J. Current Therapeutic Options for the Main Monogenic Autoinflammatory Diseases and PFAPA Syndrome: Evidence-Based Approach and Proposal of a Practical Guide. Front. Immunol. 2020, 11, 865. [Google Scholar] [CrossRef] [PubMed]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.-C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N. Engl. J. Med. 2019, 382, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.A.; Morand, E.F.; Bruce, I.N.; Manzi, S.; Kalunian, K.C.; Vital, E.M.; Ford, T.L.; Gupta, R.; Hiepe, F.; Santiago, M.; et al. Type I Interferon Inhibitor Anifrolumab in Active Systemic Lupus Erythematosus (TULIP-1): A Randomised, Controlled, Phase 3 Trial. Lancet Rheumatol. 2019, 1, e208–e219. [Google Scholar] [CrossRef]
- Burki, T.K. FDA Approval for Anifrolumab in Patients with Lupus. Lancet Rheumatol. 2021, 3, e689. [Google Scholar] [CrossRef]
- Li, S.; Hong, Z.; Wang, Z.; Li, F.; Mei, J.; Huang, L.; Lou, X.; Zhao, S.; Song, L.; Chen, W.; et al. The Cyclopeptide Astin C Specifically Inhibits the Innate Immune CDN Sensor STING. Cell Rep. 2018, 25, 3405–3421.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; van der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with Covalent Small-Molecule Inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef]
- Hansen, A.L.; Buchan, G.J.; Rühl, M.; Mukai, K.; Salvatore, S.R.; Ogawa, E.; Andersen, S.D.; Iversen, M.B.; Thielke, A.L.; Gunderstofte, C.; et al. Nitro-Fatty Acids Are Formed in Response to Virus Infection and Are Potent Inhibitors of STING Palmitoylation and Signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E7768–E7775. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.; Chong, Z.; Law, C.S.; Mukai, K.; Ho, F.O.; Martinu, T.; Backes, B.J.; Eckalbar, W.L.; Taguchi, T.; Shum, A.K. A Defect in COPI-Mediated Transport of STING Causes Immune Dysregulation in COPA Syndrome. J. Exp. Med. 2020, 217, e20201045. [Google Scholar] [CrossRef]
- Prabakaran, T.; Troldborg, A.; Kumpunya, S.; Alee, I.; Marinković, E.; Windross, S.J.; Nandakumar, R.; Narita, R.; Zhang, B.-C.; Carstensen, M.; et al. A STING Antagonist Modulating the Interaction with STIM1 Blocks ER-to-Golgi Trafficking and Inhibits Lupus Pathology. EBioMedicine 2021, 66, 103314. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deterding, R.; Griese, M.; Deutsch, G.; Warburton, D.; DeBoer, E.M.; Cunningham, S.; Clement, A.; Schwerk, N.; Flaherty, K.R.; Brown, K.K.; et al. Study Design of a Randomised, Placebo-Controlled Trial of Nintedanib in Children and Adolescents with Fibrosing Interstitial Lung Disease. ERJ Open Res. 2021, 7, 00805–02020. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.L.N.; Robertson, L.; Rice, G.I.; Seabra, L.; Hilliard, T.N.; Crow, Y.J.; Ramanan, A.V. Type 1 Interferonopathy Presenting as Juvenile Idiopathic Arthritis with Interstitial Lung Disease: Report of a New Phenotype. Pediatr. Rheumatol. Online J. 2020, 18, 37. [Google Scholar] [CrossRef]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of Genome Editing Technology in the Targeted Therapy of Human Diseases: Mechanisms, Advances and Prospects. Signal Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef]
- Ewart, D.T.; Peterson, E.J.; Steer, C.J. Gene Editing for Inflammatory Disorders. Ann. Rheum. Dis. 2019, 78, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Gardner, C.L.; Pavel-Dinu, M.; Dobbs, K.; Bosticardo, M.; Reardon, P.K.; Lack, J.; DeRavin, S.S.; Le, K.; Bello, E.; Pala, F.; et al. Gene Editing Rescues In Vitro T Cell Development of RAG2-Deficient Induced Pluripotent Stem Cells in an Artificial Thymic Organoid System. J. Clin. Immunol. 2021, 41, 852–862. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Clinical Symptoms |
|---|
| Cough |
| Tachypnea |
| Exertional dyspnea |
| Hemoptysis |
| Nail clubbing |
| Radiological Findings |
| Ground glass opacities |
| Septal thickening |
| Honeycombing |
| Crazy paving |
| Cysts |
| Lung Function Tests |
| Restrictive ventilatory impairment |
| Obstructive ventilatory impairment |
| Hyperinflation |
| Mixed pattern |
| Broncho-Alveolar Lavage |
| Alveolitis (lymphocytic, neutrophilic, or mixed) |
| Intra alveolar hemorrhage |
| Normal |
| Histopathological Findings |
| Fibrosis |
| Lymphoid follicles with germinal left organization |
| Inflammatory infiltrate |
| Alveolar hemorrhage |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
David, C.; Frémond, M.-L. Lung Inflammation in STING-Associated Vasculopathy with Onset in Infancy (SAVI). Cells 2022, 11, 318. https://doi.org/10.3390/cells11030318
David C, Frémond M-L. Lung Inflammation in STING-Associated Vasculopathy with Onset in Infancy (SAVI). Cells. 2022; 11(3):318. https://doi.org/10.3390/cells11030318
Chicago/Turabian StyleDavid, Clémence, and Marie-Louise Frémond. 2022. "Lung Inflammation in STING-Associated Vasculopathy with Onset in Infancy (SAVI)" Cells 11, no. 3: 318. https://doi.org/10.3390/cells11030318
APA StyleDavid, C., & Frémond, M.-L. (2022). Lung Inflammation in STING-Associated Vasculopathy with Onset in Infancy (SAVI). Cells, 11(3), 318. https://doi.org/10.3390/cells11030318