
Nuclear Receptor PXR in Drug-Induced Hypercholesterolemia



Abstract
:1. Introduction
2. Evidence for Induction of Hypercholesterolemia by PXR Activation in Humans
3. Mechanisms of PXR-Induced Hypercholesterolemia
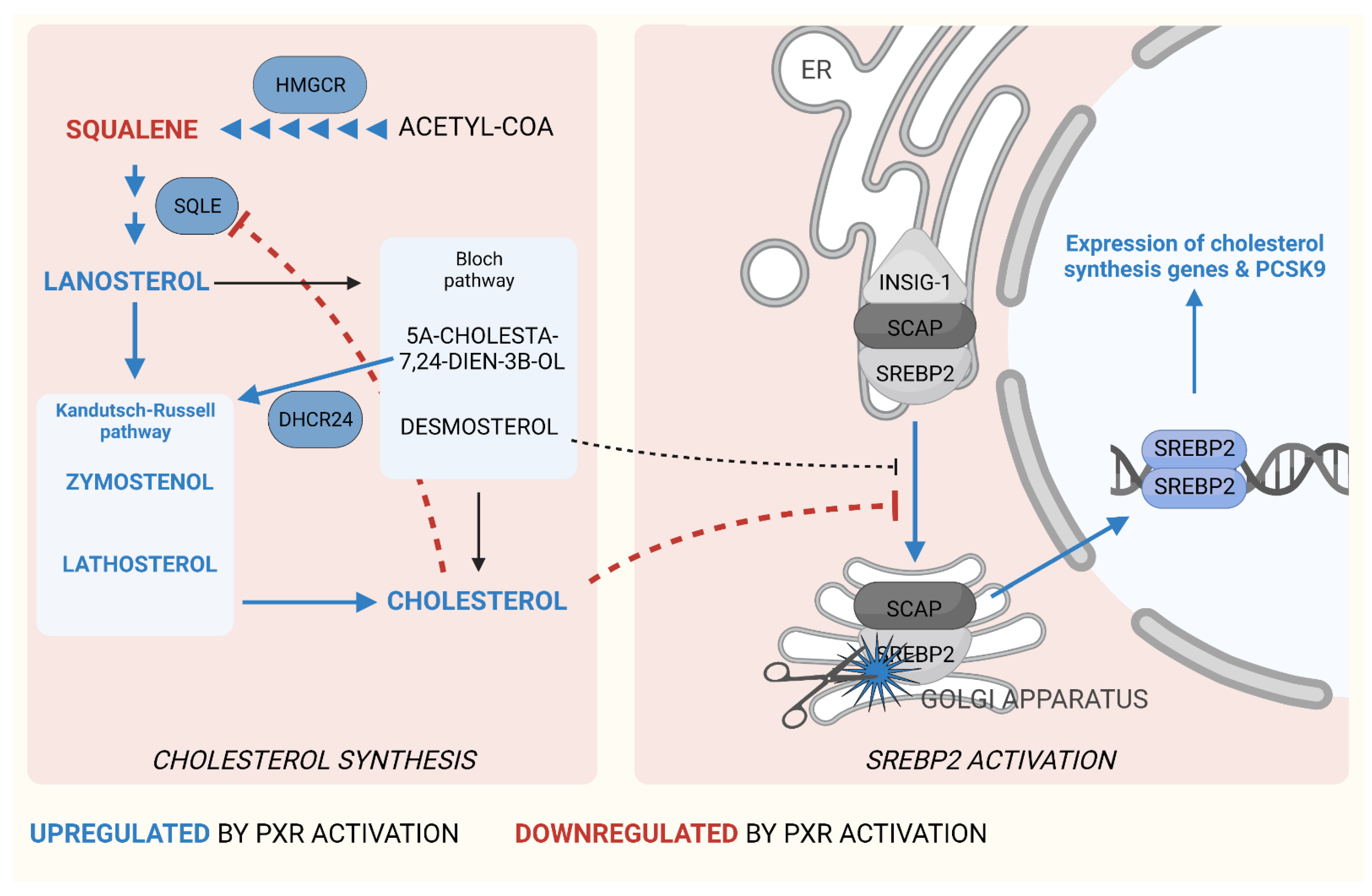
3.1. PXR in Cholesterol Synthesis
3.2. PCSK9 Induction by PXR Activation
3.3. PXR in Intestinal Cholesterol Absorption
3.4. PXR in Bile Acid Synthesis and Cholesterol Metabolism
4. PXR in HDL Homeostasis
5. Drug-Induced Hypercholesterolemia
6. Identification of PXR Ligands among the Drugs Inducing Hypercholesterolemia
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Prim. 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Karr, S. Epidemiology and management of hyperlipidemia. Am. J. Manag. Care 2017, 23, S139–S148. [Google Scholar] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Abbafati, C.; Abbas, K.M.; Abbasi-Kangevari, M.; Abd-Allah, F.; Abdelalim, A.; Abdollahi, M.; Abdollahpour, I.; Abegaz, K.H.; Abolhassani, H.; Aboyans, V.; et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Song, Y.; Liu, J.; Zhao, K.; Gao, L.; Zhao, J. Cholesterol-induced toxicity: An integrated view of the role of cholesterol in multiple diseases. Cell Metab. 2021, 33, 1911–1925. [Google Scholar] [CrossRef] [PubMed]
- Chaggar, P.S.; Shaw, S.M.; Williams, S.G. Effect of antipsychotic medications on glucose and lipid levels. J. Clin. Pharmacol. 2011, 51, 631–638. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Moore, J.T.; Wade, L.; Staudinger, J.L.; Watson, M.A.; Jones, S.A.; McKee, D.D.; Oliver, B.B.; Willson, T.M.; Zetterström, R.H.; et al. An Orphan Nuclear Receptor Activated by Pregnanes Defines a Novel Steroid Signaling Pathway. Cell 1998, 92, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Hakkola, J.; Hukkanen, J.; Turpeinen, M.; Pelkonen, O. Inhibition and induction of CYP enzymes in humans: An update. Arch. Toxicol. 2020, 94, 3671–3722. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Young, G.M.; Xie, W. The xenobiotic receptors PXR and CAR in liver physiology, an update. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166101. [Google Scholar] [CrossRef]
- Ihunnah, C.A.; Jiang, M.; Xie, W. Nuclear receptor PXR, transcriptional circuits and metabolic relevance. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 956–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hukkanen, J.; Hakkola, J. PXR and 4β-Hydroxycholesterol Axis and the Components of Metabolic Syndrome. Cells 2020, 9, 2445. [Google Scholar] [CrossRef] [PubMed]
- Hukkanen, J.; Hakkola, J.; Rysä, J. Pregnane X receptor (PXR)—A contributor to the diabetes epidemic? Drug Metabol. Drug Interact. 2014, 29, 3–15. [Google Scholar] [CrossRef]
- Toporova, L.; Balaguer, P. Nuclear receptors are the major targets of endocrine disrupting chemicals. Mol. Cell. Endocrinol. 2020, 502, 110665. [Google Scholar] [CrossRef] [PubMed]
- Isojärvi, J.I.T.; Myllylä, V.V.; Pakarinen, A.J. Serum lipid levels during carbamazepine medication. A prospective study. Arch. Neurol. 1993, 50, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Eirís, J.; Novo-Rodríguez, M.I.; Del Río, M.; Meseguer, P.; Del Río, M.C.; Castro-Gago, M. The effects on lipid and apolipoprotein serum levels of long-term carbamazepine, valproic acid and phenobarbital therapy in children with epilepsy. Epilepsy Res. 2000, 41, 1–7. [Google Scholar] [CrossRef]
- Luoma, P.V.; Reunanen, M.I.; Sotaniemi, E.A. Changes in serum triglyceride and cholesterol levels during long-term phenytoin treatment for epilepsy. Acta Med. Scand. 1979, 206, 229–231. [Google Scholar] [CrossRef]
- Katsiki, N.; Mikhailidis, D.P.; Nair, D.R. The effects of antiepileptic drugs on vascular risk factors: A narrative review. Seizure 2014, 23, 677–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calandre, E.P.; Rodriguez-Lopez, C.; Blazquez, A.; Cano, D. Serum lipids, lipoproteins and apolipoproteins A and B in epileptic patients treated with valproic acid, carbamazepine or phenobarbital. Acta Neurol. Scand. 1991, 83, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Nikolaos, T.; Stylianos, G.; Chryssoula, N.; Irini, P.; Christos, M.; Dimitrios, T.; Konstantinos, P.; Antonis, T. The effect of long-term antiepileptic treatment on serum cholesterol (TC, HDL, LDL) and triglyceride levels in adult epileptic patients on monotherapy. Med. Sci. Monit. 2004, 10, MT50–MT52. [Google Scholar] [PubMed]
- Coyne, M.J.; Bonorris, G.G.; Goldstein, L.I.; Schoenfield, L.J. Effect of chenodeoxycholic acid and phenobarbital on the rate-limiting enzymes of hepatic cholesterol and bile acid synthesis in patients with gallstones. J. Lab. Clin. Med. 1976, 87, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Sberna, A.L.; Assem, M.; Gautier, T.; Grober, J.; Guiu, B.; Jeannin, A.; Pais De Barros, J.P.; Athias, A.; Lagrost, L.; Masson, D. Constitutive androstane receptor activation stimulates faecal bile acid excretion and reverse cholesterol transport in mice. J. Hepatol. 2011, 55, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Raymond, K. Roles of rifampicin in drug-drug interactions: Underlying molecular mechanisms involving the nuclear pregnane X receptor. Ann. Clin. Microbiol. Antimicrob. 2006, 5, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feely, J.; Clee, M.; Pereira, L.; Guy, E. Enzyme induction with rifampicin; lipoproteins and drug binding to alpha 1-acid glycoprotein. Br. J. Clin. Pharmacol. 1983, 16, 195–197. [Google Scholar] [CrossRef] [Green Version]
- Lütjohann, D.; Hahn, C.; Prange, W.; Sudhop, T.; Axelson, M.; Sauerbruch, T.; von Bergmann, K.; Reichel, C. Influence of rifampin on serum markers of cholesterol and bile acid synthesis in men. Int. J. Clin. Pharmacol. Ther. 2004, 42, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Ohnhaus, E.E.; Kirchhof, B.; Peheim, E. Effect of enzyme induction on plasma lipids using antipyrine, phenobarbital, and rifampicin. Clin. Pharmacol. Ther. 1979, 25, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Bjorkhem, I.; Miettinen, T.; Reihner, E.; Ewerth, S.; Angelin, B.; Einarsson, K. Correlation between serum levels of some cholesterol precursors and activity of HMG-CoA reductase in human liver. J. Lipid Res. 1987, 28, 1137–1143. [Google Scholar] [CrossRef]
- Kasichayanula, S.; Boulton, D.W.; Luo, W.L.; Rodrigues, A.D.; Yang, Z.; Goodenough, A.; Lee, M.; Jemal, M.; LaCreta, F. Validation of 4β-hydroxycholesterol and evaluation of other endogenous biomarkers for the assessment of CYP3A activity in healthy subjects. Br. J. Clin. Pharmacol. 2014, 78, 1122–1134. [Google Scholar] [CrossRef] [Green Version]
- Rysä, J.; Buler, M.; Savolainen, M.J.; Ruskoaho, H.; Hakkola, J.; Hukkanen, J. Pregnane X Receptor Agonists Impair Postprandial Glucose Tolerance. Clin. Pharmacol. Ther. 2013, 93, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Hassani-Nezhad-Gashti, F.; Salonurmi, T.; Hautajärvi, H.; Rysä, J.; Hakkola, J.; Hukkanen, J. Pregnane X receptor activator rifampin increases blood pressure and stimulates plasma renin activity. Clin. Pharmacol. Ther. 2020, 108, 856–865. [Google Scholar] [CrossRef]
- Karpale, M.; Käräjämäki, A.J.; Kummu, O.; Gylling, H.; Hyötyläinen, T.; Orešič, M.; Tolonen, A.; Hautajärvi, H.; Savolainen, M.J.; Ala-Korpela, M.; et al. Activation of nuclear receptor PXR induces atherogenic lipids and PCSK9 through SREBP2-mediated mechanism. Br. J. Pharmacol. 2021, 178, 2461–2481. [Google Scholar] [CrossRef] [PubMed]
- Diczfalusy, U.; Nylén, H.; Elander, P.; Bertilsson, L. 4β-hydroxycholesterol, an endogenous marker of CYP3A4/5 activity in humans. Br. J. Clin. Pharmacol. 2011, 71, 183–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theile, D.; Haefeli, W.E.; Weiss, J. Effects of adrenolytic mitotane on drug elimination pathways assessed in vitro. Endocrine 2015, 49, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Kroiss, M.; Quinkler, M.; Lutz, W.K.; Allolio, B.; Fassnacht, M. Drug interactions with mitotane by induction of CYP3A4 metabolism in the clinical management of adrenocortical carcinoma. Clin. Endocrinol. 2011, 75, 585–591. [Google Scholar] [CrossRef]
- Maher, V.; Trainer, P.J.; Scoppola, A.; Anderson, J.V.; Thompson, G.R.; Besser, G.M. Possible Mechanism and Treatment of o,p’DDD-induced Hypercholesterolaemia. Q. J. Med. 1992, 84, 671–679. [Google Scholar] [PubMed]
- Vikner, M.E.; Krogh, J.; Daugaard, G.; Andreassen, M. Metabolic and hormonal side effects of mitotane treatment for adrenocortical carcinoma: A retrospective study in 50 Danish patients. Clin. Endocrinol. 2021, 94, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Molnar, G.D.; Nunn, S.L.; Tauxe, W.N. The effect of o,p’-DDD therapy on plasma cholesterol in adrenal carcinoma. Proc. Staff Meet. Mayo Clin. 1961, 36, 618–620. [Google Scholar] [PubMed]
- Lu, Y.; Feskens, E.J.M.; Boer, J.M.A.; Müller, M. The potential influence of genetic variants in genes along bile acid and bile metabolic pathway on blood cholesterol levels in the population. Atherosclerosis 2010, 210, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, J.M.; Turley, S.D. Control of cholesterol turnover in the mouse. J. Biol. Chem. 2002, 277, 3801–3804. [Google Scholar] [CrossRef] [Green Version]
- Mitsche, M.A.; McDonald, J.G.; Hobbs, H.H.; Cohen, J.C. Flux analysis of cholesterol biosynthesis in vivo reveals multiple tissue and cell-type specific pathways. Elife 2015, 4, e07999. [Google Scholar] [CrossRef]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2019, 21, 225–245. [Google Scholar] [CrossRef]
- Sever, N.; Yang, T.; Brown, M.S.; Goldstein, J.L.; DeBose-Boyd, R.A. Accelerated Degradation of HMG CoA Reductase Mediated by Binding of Insig-1 to Its Sterol-Sensing Domain. Mol. Cell 2003, 11, 25–33. [Google Scholar] [CrossRef]
- Roth, A.; Looser, R.; Kaufmann, M.; Blättler, S.M.; Rencurel, F.; Huang, W.; Moore, D.D.; Meyer, U.A. Regulatory Cross-Talk between Drug Metabolism and Lipid Homeostasis: Constitutive Androstane Receptor and Pregnane X Receptor Increase Insig-1 Expression. Mol. Pharmacol. 2008, 73, 1282–1289. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhai, Y.; Mu, Y.; Gong, H.; Uppal, H.; Toma, D.; Ren, S.; Evans, R.M.; Xie, W. A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J. Biol. Chem. 2006, 281, 15013–15020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitter, A.; Rümmele, P.; Klein, K.; Kandel, B.A.; Rieger, J.K.; Nüssler, A.K.; Zanger, U.M.; Trauner, M.; Schwab, M.; Burk, O. Pregnane X receptor activation and silencing promote steatosis of human hepatic cells by distinct lipogenic mechanisms. Arch. Toxicol. 2015, 89, 2089–2103. [Google Scholar] [CrossRef]
- Ann Barretto, S.; Lasserre, F.; Fougerat, A.; Smith, L.; Fougeray, T.; Lukowicz, C.; Polizzi, A.; Smati, S.; Régnier, M.; Naylies, C.; et al. Gene expression profiling reveals that PXR activation inhibits hepatic PPARα activity and decreases FGF21 secretion in male C57BL6/J mice. Int. J. Mol. Sci. 2019, 20, 3767. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Pan, X.; Wu, F.; Ye, D.; Zhang, Y.; Wang, Y.; Jin, L.; Lian, Q.; Huang, Y.; Ding, H.; et al. Fibroblast growth factor 21 prevents atherosclerosis by suppression of hepatic sterol regulatory element-binding protein-2 and induction of adiponectin in mice. Circulation 2015, 131, 1861–1871. [Google Scholar] [CrossRef] [Green Version]
- Bloch, K. The biological synthesis of cholesterol. Science 1965, 150, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Kandutsch, A.A.; Russell, A.E. Preputial gland tumor sterols. 2. The identification of 4 alpha-methyl-Delta 8-cholesten-3 beta-ol. J. Biol. Chem. 1960, 235, 2253–2255. [Google Scholar] [CrossRef]
- Gwag, T.; Meng, Z.; Sui, Y.; Helsley, R.N.; Park, S.H.; Wang, S.; Greenberg, R.N.; Zhou, C. Non-nucleoside reverse transcriptase inhibitor efavirenz activates PXR to induce hypercholesterolemia and hepatic steatosis. J. Hepatol. 2019, 70, 930–940. [Google Scholar] [CrossRef]
- Gill, S.; Stevenson, J.; Kristiana, I.; Brown, A.J. Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab. 2011, 13, 260–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, N.K.; Howe, V.; Jatana, N.; Thukral, L.; Brown, A.J. A conserved degron containing an amphipathic helix regulates the cholesterol-mediated turnover of human squalene monooxygenase, a rate-limiting enzyme in cholesterol synthesis. J. Biol. Chem. 2017, 292, 19959–19973. [Google Scholar] [CrossRef] [Green Version]
- Spann, N.J.; Garmire, L.X.; McDonald, J.G.; Myers, D.S.; Milne, S.B.; Shibata, N.; Reichart, D.; Fox, J.N.; Shaked, I.; Heudobler, D.; et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell 2012, 151, 138–152. [Google Scholar] [CrossRef] [Green Version]
- Warden, B.A.; Fazio, S.; Shapiro, M.D. The PCSK9 revolution: Current status, controversies, and future directions. Trends Cardiovasc. Med. 2020, 30, 179–185. [Google Scholar] [CrossRef]
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Seidah, N.G.; Awan, Z.; Chrétien, M.; Mbikay, M. PCSK9: A key modulator of cardiovascular health. Circ. Res. 2014, 114, 1022–1036. [Google Scholar] [CrossRef]
- Dong, B.; Wu, M.; Li, H.; Kraemer, F.B.; Adeli, K.; Seidah, N.G.; Park, S.W.; Liu, J. Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: Mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J. Lipid Res. 2010, 51, 1486–1495. [Google Scholar] [CrossRef] [Green Version]
- Sahebkar, A.; Simental-Mendía, L.E.; Guerrero-Romero, F.; Golledge, J.; Watts, G.F. Effect of statin therapy on plasma proprotein convertase subtilisin kexin 9 (PCSK9) concentrations: A systematic review and meta-analysis of clinical trials. Diabetes Obes. Metab. 2015, 17, 1042–1055. [Google Scholar] [CrossRef]
- Abumiya, M.; Akamine, Y.; Sato, S.; Takahashi, S.; Yoshioka, T.; Kameoka, Y.; Takahashi, N.; Miura, M. Effects of proprotein convertase subtilisin/kexin type 9 and nilotinib plasma concentrations on nilotinib-induced hypercholesterolaemia in patients with chronic myeloid leukaemia. J. Clin. Pharm. Ther. 2021, 46, 382–387. [Google Scholar] [CrossRef]
- Glerup, S.; Schulz, R.; Laufs, U.; Schlüter, K.-D. Physiological and therapeutic regulation of PCSK9 activity in cardiovascular disease. Basic Res. Cardiol. 2017, 112, 32. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Akamine, Y.; Kagaya, H.; Saito, M.; Inoue, T.; Numakura, K.; Habuchi, T.; Satoh, S.; Miura, M. Changes in PCSK9 and LDL cholesterol concentrations by everolimus treatment and their effects on polymorphisms in PCSK9 and mTORC1. Pharmacol. Rep. 2020, 72, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Simha, V.; Qin, S.; Shah, P.; Smith, B.H.; Kremers, W.K.; Kushwaha, S.; Wang, L.; Pereira, N.L. Sirolimus Therapy Is Associated with Elevation in Circulating PCSK9 Levels in Cardiac Transplant Patients. J. Cardiovasc. Transl. Res. 2017, 10, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Phan, B.A.P.; Dayspring, T.D.; Toth, P.P. Ezetimibe therapy: Mechanism of action and clinical update. Vasc. Health Risk Manag. 2012, 8, 415–427. [Google Scholar]
- Miettinen, T.A.; Puska, P.; Gylling, H.; Vanhanen, H.; Vartiainen, E. Reduction of Serum Cholesterol with Sitostanol-Ester Margarine in a Mildly Hypercholesterolemic Population. N. Engl. J. Med. 1995, 333, 1308–1312. [Google Scholar] [CrossRef] [Green Version]
- Sui, Y.; Helsley, R.N.; Park, S.-H.; Song, X.; Liu, Z.; Zhou, C. Intestinal pregnane X receptor links xenobiotic exposure and hypercholesterolemia. Mol. Endocrinol. 2015, 29, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Betters, J.L.; Yu, L. Niemann-Pick C1-Like 1 (NPC1L1) protein in intestinal and hepatic cholesterol transport. Annu. Rev. Physiol. 2011, 73, 239–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Shah, Y.M.; Guo, G.L.; Wang, T.; Krausz, K.W.; Idle, J.R.; Gonzalez, F.J. Rifaximin is a gut-specific human pregnane X receptor activator. J. Pharmacol. Exp. Ther. 2007, 322, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Krausz, K.W.; Tanaka, N.; Gonzalez, F.J. Chronic exposure to rifaximin causes hepatic steatosis in pregnane X receptor-humanized mice. Toxicol. Sci. 2012, 129, 456–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonoda, J.; Chong, L.W.; Downes, M.; Barish, G.D.; Coulter, S.; Liddle, C.; Lee, C.H.; Evans, R.M. Pregnane X receptor prevents hepatorenal toxicity from cholesterol metabolites. Proc. Natl. Acad. Sci. USA 2005, 102, 2198–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Radominska-Pandya, A.; Shi, Y.; Simon, C.M.; Nelson, M.C.; Ong, E.S.; Waxman, D.J.; Evans, R.M. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc. Natl. Acad. Sci. USA 2001, 98, 3375–3380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Nishida, S.; Xu, M.; Makishima, M.; Xie, W. PXR Prevents Cholesterol Gallstone Disease by Regulating Biosynthesis and Transport of Bile Salts. Gastroenterology 2011, 140, 2095–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; LaTour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef] [Green Version]
- Kast, H.R.; Goodwin, B.; Tarr, P.T.; Jones, S.A.; Anisfeld, A.M.; Stoltz, C.M.; Tontonoz, P.; Kliewer, S.; Willson, T.M.; Edwards, P.A. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J. Biol. Chem. 2002, 277, 2908–2915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhalla, S.; Ozalp, C.; Fang, S.; Xiang, L.; Kemper, J.K. Ligand-activated pregnane X receptor interferes with HNF-4 signaling by targeting a common coactivator PGC-1α. Functional implications in hepatic cholesterol and glucose metabolism. J. Biol. Chem. 2004, 279, 45139–45147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Chiang, J.Y.L. Mechanism of rifampicin and pregnane X receptor inhibition of human cholesterol 7α-hydroxylase gene transcription. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassani-Nezhad-Gashti, F.; Kummu, O.; Karpale, M.; Rysä, J.; Hakkola, J. Nutritional status modifies pregnane X receptor regulated transcriptome. Sci. Rep. 2019, 9, 16728. [Google Scholar] [CrossRef]
- Marschall, H.-U.; Wagner, M.; Zollner, G.; Fickert, P.; Diczfalusy, U.; Gumhold, J.; Silbert, D.; Fuchsbichler, A.; Benthin, L.; Grundström, R.; et al. Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology 2005, 129, 476–485. [Google Scholar] [CrossRef]
- Temel, R.E.; Brown, J.M. A new model of reverse cholesterol transport: EnTICEing strategies to stimulate intestinal cholesterol excretion. Trends Pharmacol. Sci. 2015, 36, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Calandre, E.P.; Porta, B.S.; de la Calzada, D.G. The Effect of Chronic Phenytoin Treatment on Serum Lipid Profile in Adult Epileptic Patients. Epilepsia 1992, 33, 154–157. [Google Scholar] [CrossRef]
- Jakubus, T.; Michalska-Jakubus, M.; Łukawski, K.; Janowska, A.; Czuczwar, S.J. Atherosclerotic risk among children taking antiepileptic drugs. Pharmacol. Rep. 2009, 61, 411–423. [Google Scholar] [CrossRef]
- O’Neill, B.; Callaghan, N.; Stapleton, M.; Molloy, W. Serum elevation of high density lipoprotein (HDL) cholesterol in epileptic patients taking carbamazepine or phenytoin. Acta Neurol. Scand. 1982, 65, 104–109. [Google Scholar] [CrossRef]
- Salonurmi, T.; Nabil, H.; Ronkainen, J.; Hyötyläinen, T.; Hautajärvi, H.; Savolainen, M.J.; Tolonen, A.; Orešič, M.; Känsäkoski, P.; Rysä, J.; et al. 4β-Hydroxycholesterol Signals From the Liver to Regulate Peripheral Cholesterol Transporters. Front. Pharmacol. 2020, 11, 361. [Google Scholar] [CrossRef] [PubMed]
- Domecq, J.P.; Prutsky, G.; Leppin, A.; Sonbol, M.B.; Altayar, O.; Undavalli, C.; Wang, Z.; Elraiyah, T.; Brito, J.P.; Mauck, K.F.; et al. Drugs commonly associated with weight change: A systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 2015, 100, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Bertilsson, G.; Heidrich, J.; Svensson, K.; Åsman, M.; Jendeberg, L.; Sydow-Bäckman, M.; Ohlsson, R.; Postlind, H.; Blomquist, P.; Berkenstam, A. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc. Natl. Acad. Sci. USA 1998, 95, 12208–12213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grewal, G.K.; Singh, K.D.; Kanojia, N.; Rawat, C.; Kukal, S.; Jajodia, A.; Singhal, A.; Misra, R.; Nagamani, S.; Muthusamy, K.; et al. Exploring the Carbamazepine Interaction with Human Pregnane X Receptor and Effect on ABCC2 Using in Vitro and in Silico Approach. Pharm. Res. 2017, 34, 1444–1458. [Google Scholar] [CrossRef]
- Moreland, T.; Park, B.; Rylance, G. Microsomal enzyme induction in children: The influence of carbamazepine treatment on antipyrine kinetics, 6 beta-hydroxycortisol excretion and plasma gamma-glutamyltranspeptidase activity. Br. J. Clin. Pharmacol. 1982, 14, 861–865. [Google Scholar] [CrossRef] [Green Version]
- Oscarson, M.; Zanger, U.M.; Rifki, O.F.; Klein, K.; Eichelbaum, M.; Meyer, U.A. Transcriptional profiling of genes induced in the livers of patients treated with carbamazepine. Clin. Pharmacol. Ther. 2006, 80, 440–456. [Google Scholar] [CrossRef] [PubMed]
- Crawford, P.; Chadwick, D.; Martin, C.; Tjia, J.; Back, D.; Orme, M. The interaction of phenytoin and carbamazepine with combined oral contraceptive steroids. Br. J. Clin. Pharmacol. 1990, 30, 892. [Google Scholar] [CrossRef] [Green Version]
- Lynch, C.; Sakamuru, S.; Huang, R.; Niebler, J.; Ferguson, S.S.; Xia, M. Characterization of human pregnane X receptor activators identified from a screening of the Tox21 compound library. Biochem. Pharmacol. 2021, 184. [Google Scholar] [CrossRef]
- Duran, I.; Carles, J.; Bulat, I.; Hellemans, P.; Mitselos, A.; Ward, P.; Jiao, J.; Armas, D.; Chien, C. Pharmacokinetic Drug-Drug Interaction of Apalutamide, Part 1: Clinical Studies in Healthy Men and Patients with Castration-Resistant Prostate Cancer. Clin. Pharmacokinet. 2020, 59, 1135–1148. [Google Scholar] [CrossRef]
- Chortis, V.; Taylor, A.E.; Schneider, P.; Tomlinson, J.W.; Hughes, B.A.; O’Neil, D.M.; Libé, R.; Allolio, B.; Bertagna, X.; Bertherat, J.; et al. Mitotane therapy in adrenocortical cancer induces CYP3A4 and inhibits 5α-reductase, explaining the need for personalized glucocorticoid and androgen replacement. J. Clin. Endocrinol. Metab. 2013, 98, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Van Erp, N.P.; Guchelaar, H.J.; Ploeger, B.A.; Romijn, J.A.; Den Hartigh, J.; Gelderblom, H. Mitotane has a strong and a durable inducing effect on CYP3A4 activity. Eur. J. Endocrinol. 2011, 164, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmsen, S.; Meijerman, I.; Maas-Bakker, R.F.; Beijnen, J.H.; Schellens, J.H.M. PXR-mediated P-glycoprotein induction by small molecule tyrosine kinase inhibitors. Eur. J. Pharm. Sci. 2012, 48, 644–649. [Google Scholar] [CrossRef]
- Danek, P.J.; Basińska-Ziobroń, A.; Wójcikowski, J.; Daniel, W.A. Levomepromazine and clozapine induce the main human cytochrome P450 drug metabolizing enzyme CYP3A4. Pharmacol. Rep. 2021, 73, 303–308. [Google Scholar] [CrossRef]
- Meng, Z.; Gwag, T.; Sui, Y.; Park, S.-H.H.; Zhou, X.; Zhou, C. The atypical antipsychotic quetiapine induces hyperlipidemia by activating intestinal PXR signaling. JCI Insight 2019, 4, e125657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hariparsad, N.; Nallani, S.C.; Sane, R.S.; Buckley, D.J.; Buckley, A.R.; Desai, P.B. Induction of CYP3A4 by efavirenz in primary human hepatocytes: Comparison with rifampin and phenobarbital. J. Clin. Pharmacol. 2004, 44, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Mouly, S.; Lown, K.S.; Kornhauser, D.; Joseph, J.L.; Fiske, W.D.; Benedek, I.H.; Watkins, P.B. Hepatic but not intestinal CYP3A4 displays dose-dependent induction by efavirenz in humans. Clin. Pharmacol. Ther. 2002, 72, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Fellay, J.; Marzolini, C.; Decosterd, L.; Golay, K.P.; Baumann, P.; Buclin, T.; Telenti, A.; Eap, C.B. Variations of CYP3A activity induced by antiretroviral treatment in HIV-1 infected patients. Eur. J. Clin. Pharmacol. 2005, 60, 865–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, D.; Lau, A.J.; Sherman, M.A.; Chang, T.K.H. Agonism of human pregnane X receptor by rilpivirine and etravirine: Comparison with first generation non-nucleoside reverse transcriptase inhibitors. Biochem. Pharmacol. 2013, 85, 1700–1711. [Google Scholar] [CrossRef]
- Schöller-Gyüre, M.; Kakuda, T.N.; Raoof, A.; De Smedt, G.; Hoetelmans, R.M.W. Clinical pharmacokinetics and pharmacodynamics of etravirine. Clin. Pharmacokinet. 2009, 48, 561–574. [Google Scholar] [CrossRef]
- Kakuda, T.N.; Van Solingen-Ristea, R.M.; Onkelinx, J.; Stevens, T.; Aharchi, F.; De Smedt, G.; Peeters, M.; Leopold, L.; Hoetelmans, R.M.W. The effect of single- and multiple-dose etravirine on a drug cocktail of representative cytochrome P450 probes and digoxin in healthy subjects. J. Clin. Pharmacol. 2014, 54, 422–431. [Google Scholar] [CrossRef]
- Tibotec Intelence (etravirine) full prescribing information 2011. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/022187s008lbl.pdf (accessed on 13 December 2021).
- Solas, C.; Poizot-Martin, I.; Drogoul, M.P.; Ravaux, I.; Dhiver, C.; Lafeuillade, A.; Allegre, T.; Mokhtari, M.; Moreau, J.; Lepeu, G.; et al. Therapeutic drug monitoring of lopinavir/ritonavir given alone or with a non-nucleoside reverse transcriptase inhibitor. Br. J. Clin. Pharmacol. 2004, 57, 436–440. [Google Scholar] [CrossRef] [Green Version]
- Mildvan, D.; Yarrish, R.; Marshak, A.; Hutman, H.W.; McDonough, M.; Lamson, M.; Robinson, P. Pharmacokinetic interaction between nevirapine and ethinyl estradiol/norethindrone when administered concurrently to HIV-infected women. J. Acquir. Immune Defic. Syndr. 2002, 29, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Dailly, E.; Tribut, O.; Tattevin, P.; Arvieux, C.; Perré, P.; Raffi, F.; Jolliet, P. Influence of tenofovir, nevirapine and efavirenz on ritonavir-boosted atazanavir pharmacokinetics in HIV-infected patients. Eur. J. Clin. Pharmacol. 2006, 62, 523–526. [Google Scholar] [CrossRef]
- Svärd, J.; Spiers, J.P.; Mulcahy, F.; Hennessy, M. Nuclear receptor-mediated induction of CYP450 by antiretrovirals: Functional consequences of NR1I2 (PXR) polymorphisms and differential prevalence in whites and sub-Saharan Africans. J. Acquir. Immune Defic. Syndr. 2010, 55, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Dussault, I.; Lin, M.; Hollister, K.; Wang, E.H.; Synold, T.W.; Forman, B.M. Peptide Mimetic HIV Protease Inhibitors Are Ligands for the Orphan Receptor SXR. J. Biol. Chem. 2001, 276, 33309–33312. [Google Scholar] [CrossRef] [Green Version]
- Ouellet, D.; Hsu, A.; Qian, J.; Locke, C.S.; Eason, C.J.; Cavanaugh, J.H.; Leonard, J.M.; Granneman, G.R. Effect of ritonavir on the pharmacokinetics of ethinyl oestradiol in healthy female volunteers. Br. J. Clin. Pharmacol. 1998, 46, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Hsu, A.; Granneman, G.R.; Witt, G.; Locke, C.; Denissen, J.; Molla, A.; Valdes, J.; Smith, J.; Erdman, K.; Lyons, N.; et al. Multiple-dose pharmacokinetics of ritonavir in human immunodeficiency virus-infected subjects. Antimicrob. Agents Chemother. 1997, 41, 898. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Welch, M.A.; Li, Z.; Mackowiak, B.; Heyward, S.; Swaan, P.W.; Wang, H. Mechanistic insights of phenobarbital-mediated activation of human but not mouse pregnane X receptor. Mol. Pharmacol. 2019, 96, 345–354. [Google Scholar] [CrossRef]
- Burstein, S.; Klaiber, E.L. Phenobarbital-induced increase in 6-beta-hydroxycortisol excretion: Clue to its significance in human urine. J. Clin. Endocrinol. Metab. 1965, 25, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.; Cowie, D.E.; Konstantinou, D.K.; Hill, S.J.; Tjelle, T.E.; Axon, A.; Koruth, M.; White, S.A.; Carlsen, H.; Mann, D.A.; et al. The PXR is a drug target for chronic inflammatory liver disease. J. Steroid Biochem. Mol. Biol. 2010, 120, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.J.; Sakamuru, S.; Huang, R.; Moeller, T.A.; Shinn, P.; Van Leer, D.; Auld, D.S.; Austin, C.P.; Xia, M. Identification of Clinically Used Drugs That Activate Pregnane X Receptors. Drug Metab. Dispos. 2011, 39, 151. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Li, L.; Kim, G.; Ekins, S.; Wang, H.; Swaan, P.W. Identification and Validation of Novel Human Pregnane X Receptor Activators among Prescribed Drugs via Ligand-Based Virtual Screening. Drug Metab. Dispos. 2011, 39, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascussi, J.M.; Drocourt, L.; Fabre, J.M.; Maurel, P.; Vilarem, M.J. Dexamethasone induces pregnane X receptor and retinoid X receptor-alpha expression in human hepatocytes: Synergistic increase of CYP3A4 induction by pregnane X receptor activators. Mol. Pharmacol. 2000, 58, 361–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichard, L.; Fabre, I.; Daujat, M.; Domergue, J.; Joyeux, H.; Maurel, P. Effect of corticosteroids on the expression of cytochromes P450 and on cyclosporin A oxidase activity in primary cultures of human hepatocytes. Mol. Pharmacol. 1992, 41, 1047–1055. [Google Scholar]
- McCune, J.S.; Hawke, R.L.; LeCluyse, E.L.; Gillenwater, H.H.; Hamilton, G.; Ritchie, J.; Lindley, C. In vivo and in vitro induction of human cytochrome P4503A4 by dexamethasone. Clin. Pharmacol. Ther. 2000, 68, 356–366. [Google Scholar] [CrossRef]
- Roberts, P.J.; Rollins, K.D.; Kashuba, A.D.M.; Paine, M.F.; Nelsen, A.C.; Williams, E.E.; Moran, C.; Lamba, J.K.; Schuetz, E.G.; Hawke, R.L. The Influence of CYP3A5 Genotype on Dexamethasone Induction of CYP3A Activity in African Americans. Drug Metab. Dispos. 2008, 36, 1465. [Google Scholar] [CrossRef] [Green Version]
- Watkins, P.B.; Murray, S.A.; Winkelman, L.G.; Heuman, D.M.; Wrighton, S.A.; Guzelian, P.S. Erythromycin breath test as an assay of glucocorticoid-inducible liver cytochromes P-450. Studies in rats and patients. J. Clin. Investig. 1989, 83, 688–697. [Google Scholar] [CrossRef] [Green Version]
- Van Duijnhoven, E.M.; Boots, J.M.M.; Christiaans, M.H.L.; Stolk, L.M.L.; Undre, N.A.; Van Hooff, J.P. Increase in tacrolimus trough levels after steroid withdrawal. Transpl. Int. 2003, 16, 721–725. [Google Scholar] [CrossRef]
- Press, R.R.; Ploeger, B.A.; Den Hartigh, J.; Van Der Straaten, T.; Van Pelt, H.; Danhof, M.; De Fijter, H.; Guchelaar, H.J. Explaining variability in ciclosporin exposure in adult kidney transplant recipients. Eur. J. Clin. Pharmacol. 2010, 66, 579. [Google Scholar] [CrossRef] [Green Version]
- Anglicheau, D.; Flamant, M.; Schlageter, M.H.; Martinez, F.; Cassinat, B.; Beaune, P.; Legendre, C.; Thervet, E. Pharmacokinetic interaction between corticosteroids and tacrolimus after renal transplantation. Nephrol. Dial. Transplant 2003, 18, 2409–2414. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.P.; Tan, Z.R.; Huang, S.L.; Huang, Z.; Ou-Yang, D.S.; Zhou, H.H. Isozyme-specific induction of low-dose aspirin on cytochrome P450 in healthy subjects. Clin. Pharmacol. Ther. 2003, 73, 264–271. [Google Scholar] [CrossRef]
- Novotna, A.; Dvorak, Z. Omeprazole and lansoprazole enantiomers induce CYP3A4 in human hepatocytes and cell lines via glucocorticoid receptor and pregnane X receptor axis. PLoS ONE 2014, 9, e105580. [Google Scholar] [CrossRef]
- Wang, K.; Chen, S.; Xie, W.; Wan, Y.J.Y. Retinoids induce cytochrome P450 3A4 through RXR/VDR-mediated pathway. Biochem. Pharmacol. 2008, 75, 2204–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padda, S.K.; Chhatwani, L.; Zhou, L.; Jacobs, C.D.; Lopez-Anaya, A.; Wakelee, H.A. Phase I and pharmacokinetic study of bexarotene in combination with gefitinib in the third-line treatment of non-small-cell lung cancer: Brief report. Anticancer. Drugs 2013, 24, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Wakelee, H.A.; Takimoto, C.H.; Lopez-Anaya, A.; Chu, Q.; Middleton, G.; Dunlop, D.; Ramlau, R.; Leighl, N.; Rowinsky, E.K.; Hao, D.; et al. The effect of bexarotene on atorvastatin pharmacokinetics: Results from a phase I trial of bexarotene plus chemotherapy in patients with advanced non-small cell lung cancer. Cancer Chemother. Pharmacol. 2012, 69, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Novotna, A.; Kamenickova, A.; Pecova, M.; Korhonova, M.; Bartonkova, I.; Dvorak, Z. Profiling of enantiopure drugs towards aryl hydrocarbon (AhR), glucocorticoid (GR) and pregnane X (PXR) receptors in human reporter cell lines. Chem. Biol. Interact. 2014, 208, 64–76. [Google Scholar] [CrossRef]
- Rowland, A.; van Dyk, M.; Warncken, D.; Mangoni, A.A.; Sorich, M.J.; Rowland, A. Evaluation of modafinil as a perpetrator of metabolic drug–drug interactions using a model informed cocktail reaction phenotyping trial protocol. Br. J. Clin. Pharmacol. 2018, 84, 501–509. [Google Scholar] [CrossRef] [Green Version]
- Pillinger, T.; McCutcheon, R.A.; Vano, L.; Mizuno, Y.; Arumuham, A.; Hindley, G.; Beck, K.; Natesan, S.; Efthimiou, O.; Cipriani, A.; et al. Comparative effects of 18 antipsychotics on metabolic function in patients with schizophrenia, predictors of metabolic dysregulation, and association with psychopathology: A systematic review and network meta-analysis. Lancet Psychiatry 2020, 7, 64–77. [Google Scholar] [CrossRef]
- Stroup, T.S.; Byerly, M.J.; Nasrallah, H.A.; Ray, N.; Khan, A.Y.; Lamberti, J.S.; Glick, I.D.; Steinbook, R.M.; McEvoy, J.P.; Hamer, R.M. Effects of switching from olanzapine, quetiapine, and risperidone to aripiprazole on 10-year coronary heart disease risk and metabolic syndrome status: Results from a randomized controlled trial. Schizophr. Res. 2013, 146, 190–195. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.L.; Tan, Q.Y.; Jiang, P.; Dang, R.L.; Xue, Y.; Tang, M.M.; Xu, P.; Deng, Y.; Li, H.D.; Yao, J.K. A potential mechanism underlying atypical antipsychotics-induced lipid disturbances. Transl. Psychiatry 2015, 5, e661. [Google Scholar] [CrossRef] [PubMed]
- Le Hellard, S.; Theisen, F.M.; Haberhausen, M.; Raeder, M.B.; Fernø, J.; Gebhardt, S.; Hinney, A.; Remschmidt, H.; Krieg, J.C.; Mehler-Wex, C.; et al. Association between the insulin-induced gene 2 (INSIG2) and weight gain in a German sample of antipsychotic-treated schizophrenic patients: Perturbation of SREBP-controlled lipogenesis in drug-related metabolic adverse effects? Mol. Psychiatry 2009, 14, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Sammalkorpi, K.; Valtonen, V.; Kerttula, Y.; Nikkilä, E.; Taskinen, M.R. Changes in serum lipoprotein pattern induced by acute infections. Metabolism 1988, 37, 859–865. [Google Scholar] [CrossRef]
- Deniz, O.; Gumus, S.; Yaman, H.; Ciftci, F.; Ors, F.; Cakir, E.; Tozkoparan, E.; Bilgic, H.; Ekiz, K. Serum total cholesterol, HDL-C and LDL-C concentrations significantly correlate with the radiological extent of disease and the degree of smear positivity in patients with pulmonary tuberculosis. Clin. Biochem. 2007, 40, 162–166. [Google Scholar] [CrossRef]
- Georgiadis, A.N.; Papavasiliou, E.C.; Lourida, E.S.; Alamanos, Y.; Kostara, C.; Tselepis, A.D.; Drosos, A.A. Atherogenic lipid profile is a feature characteristic of patients with early rheumatoid arthritis: Effect of early treatment—A prospective, controlled study. Arthritis Res. Ther. 2006, 8, R82. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.K.; Seeger, J.D. Lipid profiles among US elderly with untreated rheumatoid arthritis—The Third National Health and Nutrition Examination Survey. J. Rheumatol. 2005, 32, 2311–2316. [Google Scholar]
- Robertson, J.; Peters, M.J.; McInnes, I.B.; Sattar, N. Changes in lipid levels with inflammation and therapy in RA: A maturing paradigm. Nat. Rev. Rheumatol. 2013, 9, 513–523. [Google Scholar] [CrossRef]
- Heslinga, S.C.; Peters, M.J.; Ter Wee, M.M.; Van Der Horst-Bruinsma, I.E.; Van Sijl, A.M.; Smulders, Y.M.; Nurmohamed, M.T. Reduction of inflammation drives lipid changes in ankylosing spondylitis. J. Rheumatol. 2015, 42, 1842–1845. [Google Scholar] [CrossRef]
- Holdaas, H.; Potena, L.; Saliba, F. MTOR inhibitors and dyslipidemia in transplant recipients: A cause for concern? Transplant. Rev. 2015, 29, 93–102. [Google Scholar] [CrossRef]
- Kockx, M.; Kritharides, L. Cyclosporin A-Induced Hyperlipidemia. In Lipoproteins—Role in Health and Diseases; InTech: Pittsburgh, PA, USA, 2012. [Google Scholar]
- Hüsing, A.; Kabar, I.; Schmidt, H.H. Lipids in liver transplant recipients. World J. Gastroenterol. 2016, 22, 3315–3324. [Google Scholar] [CrossRef]
- Agarwal, A.; Prasad, G.V.R. Post-transplant dyslipidemia: Mechanisms, diagnosis and management. World J. Transplant. 2016, 6, 125. [Google Scholar] [CrossRef] [PubMed]
- Kockx, M.; Glaros, E.; Leung, B.; Ng, T.W.; Berbée, J.F.P.; Deswaerte, V.; Nawara, D.; Quinn, C.; Rye, K.A.; Jessup, W.; et al. Low-Density Lipoprotein Receptor-Dependent and Low-Density Lipoprotein Receptor-Independent Mechanisms of Cyclosporin A-Induced Dyslipidemia. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1338–1349. [Google Scholar] [CrossRef] [Green Version]
- Bakan, I.; Laplante, M. Connecting mTORC1 signaling to SREBP-1 activation. Curr. Opin. Lipidol. 2012, 23, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Hemkens, L.G.; Bucher, H.C. HIV infection and cardiovascular disease. Eur. Heart J. 2014, 35, 1373–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triant, V.A.; Grinspoon, S.K. Epidemiology of ischemic heart disease in HIV. Curr. Opin. HIV Aids 2017, 12, 540–547. [Google Scholar] [CrossRef]
- Feeney, E.R.; Mallon, P.W.G. HIV and HAART-Associated Dyslipidemia. Open Cardiovasc. Med. J. 2011, 5, 49–63. [Google Scholar] [CrossRef]
- Wei, Y.; Tang, C.; Sant, V.; Li, S.; Poloyac, S.M.; Xie, W. A Molecular Aspect in the Regulation of Drug Metabolism: Does PXR-Induced Enzyme Expression Always Lead to Functional Changes in Drug Metabolism? Curr. Pharmacol. Rep. 2016, 2, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Sberna, A.L.; Assem, M.; Xiao, R.; Ayers, S.; Gautier, T.; Guiu, B.; Deckert, V.; Chevriaux, A.; Grober, J.; Le Guern, N.; et al. Constitutive androstane receptor activation decreases plasma apolipoprotein B-containing lipoproteins and atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2232–2239. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| Drug Class | Drug | Mechanism | Increases | PXR Agonist | CYP3A4 Inducer | Clinically Relevant CYP3A4 Inducer |
|---|---|---|---|---|---|---|
| Androgen | Methyl testosterone | Androgen receptor activation | LDL | |||
| Antiarrhythmic | Amiodarone | Blocking of voltage gated K+ and Ca2+ channels | LDL | |||
| Antibiotic | Rifampicin | Bacterial RNA synthesis inhibition | CHOL, LDL | Yes [22,84] | Yes [22] | Yes [22] |
| Anticonvulsant | Carbamazepine | Blocking of central Na+ channel | CHOL, LDL | Yes [85] | Yes [86,87,88] | Yes [86,87,88] |
| Tiagabine | GABA reuptake inhibition | CHOL | ||||
| Antidepressant | Paroxetine | Selective serotonin reuptake inhibition | CHOL | |||
| Sertraline | Selective serotonin reuptake inhibition | CHOL | ||||
| Duloxetine | Serotonin and noradrenaline reuptake inhibition | CHOL | ||||
| Venlafaxin | Serotonin and noradrenaline reuptake inhibition | CHOL | ||||
| Antigonadotropic | Danazol | Androgen receptor activation | CHOL, LDL | |||
| Antigout | Febuxostat | Xanthine-oxidase inhibition | CHOL | |||
| Antihyperglycemic | Ertugliflozin | SGLT-2 inhibition | CHOL, LDL | |||
| Sotagliflozin | SGLT1/2 inhibition | CHOL, LDL | ||||
| Antihypertensive | Lacidipine | Ca2+ channel blocker | LDL | Yes [89] | ||
| Furosemide | Diuretic | CHOL, LDL | ||||
| Indapamide | Diuretic | CHOL, LDL | ||||
| Propranolol | Beta-blocker | CHOL | ||||
| Antimycotic | Fluconazole | Ergosterol synthesis inhibition | CHOL | |||
| Voriconazole | Ergosterol synthesis inhibition | CHOL | ||||
| Antineoplastic | Apalutamide | Antiandrogen | CHOL, LDL | Possible [90] | Yes [90] | Yes [90] |
| Anastrozole | Aromatase inhibition | CHOL | ||||
| Letrozole | Aromatase inhibition | CHOL | ||||
| Mitotane | Adrenal cortex inhibition | CHOL, LDL | Yes [32] | Yes [32,91] | Yes [92] | |
| Asparaginase | Depletion of circulating asparagine | CHOL | ||||
| Histrelin | GnRH agonist | CHOL | ||||
| Degarelix | GnRH blocker | CHOL | ||||
| Pegvisomant | IGF1 inhibition | CHOL | ||||
| Ruxolitinib | JAK inhibition | CHOL | Possible **** | |||
| Rucaparib | PARP inhibition | CHOL | ||||
| Verteporfin | Phototherapy sensitizer | CHOL | ||||
| Cladribine | Purine analogue | CHOL | ||||
| Tegafur | Pyrimidine analogue | CHOL, LDL | ||||
| Padeliporfin | Radiation therapy sensitizer | CHOL, LDL | ||||
| Brigatinib | Tyrosine kinase inhibition | CHOL, LDL | Possible * | Yes * | ||
| Cabozantinib | Tyrosine kinase inhibition | CHOL, LDL | ||||
| Dasatinib | Tyrosine kinase inhibition | CHOL, LDL | Yes [89] | |||
| Lenvatinib | Tyrosine kinase inhibition | CHOL, LDL | Yes | |||
| Lorlatinib | Tyrosine kinase inhibition | CHOL, LDL | Yes ** | Yes **,*** | ||
| Nilotinib | Tyrosine kinase inhibition | CHOL, LDL | Yes [93] | |||
| Pazopanib | Tyrosine kinase inhibition | CHOL | ||||
| Antipsychotic, atypical | Amisulpride | Inhibition of D2 and 5-HT2A receptors | CHOL | |||
| Aripiprazole | Inhibition of D2 and 5-HT2A receptors | LDL | ||||
| Cariprazine | Inhibition of D2 and 5-HT2A receptors | CHOL, LDL | ||||
| Clozapine | Inhibition of D2 and 5-HT2A receptors | CHOL, LDL | Yes [94] | |||
| Olanzapine | Inhibition of D2 and 5-HT2A receptors | CHOL, LDL | ||||
| Paliperidone | Inhibition of D2 and 5-HT2A receptors | CHOL, LDL | ||||
| Quetiapine | Inhibition of D2 and 5-HT2A receptors | CHOL, LDL | Yes [95] | Yes [95] | ||
| Risperidone | Inhibition of D2 and 5-HT2A receptors | CHOL, LDL | ||||
| Antipsychotic, typical | Fluphenazine | Inhibition of D2 receptors | CHOL | |||
| Zuclopenthixol | Inhibition of D2 receptors | CHOL, LDL | ||||
| Antiretroviral | Cobicistat | CYP3A inhibition | CHOL | |||
| Raltegravir | Integrase inhibition | CHOL, LDL | ||||
| Efavirenz | Non-nucleoside reverse transcriptase inhibition | CHOL, LDL | Yes [50] | Yes [50,96,97] | Yes [97,98] | |
| Etravirine | Non-nucleoside reverse transcriptase inhibition | CHOL, LDL | Yes [99] | Yes [100,101,102] | Yes [100,101] | |
| Nevirapine | Non-nucleoside reverse transcriptase inhibition | CHOL, LDL | Yes [103,104,105] | Yes [103,104,105] | ||
| Rilpivirine | Non-nucleoside reverse transcriptase inhibition | CHOL, LDL | Yes [99] | |||
| Darunavir | Protease inhibition | CHOL | Yes [50] | |||
| Fosamprenavir | Protease inhibition | CHOL | Yes [106] | Yes [106] | Yes [106] | |
| Indinavir | Protease inhibition | CHOL | ||||
| Lopinavir | Protease inhibition | CHOL | Yes [50] | |||
| Ritonavir | Protease inhibition | CHOL | Yes [107] | Yes [108,109] | Yes [108,109] | |
| Saquinavir | Protease inhibition | CHOL, LDL | Yes [107] | |||
| Tipranavir | Protease inhibition | CHOL | Yes ***** | Yes ***** | ||
| Antithyroid | Methimazole | Thyroperoxidase inhibition | LDL | |||
| Antiviral | Boceprevir | Protease inhibition | CHOL | |||
| Barbiturate | Phenobarbital | GABA stimulation | LDL | Yes [110] | Yes [111] | Yes [111] |
| Emergency contraception | Ulipristal | Progesterone receptor modulation | CHOL | |||
| Immunosuppressant | Cyclosporin | Calcineurin inhibition | CHOL, LDL | Yes [112] | ||
| Tacrolimus | Calcineurin inhibition | CHOL | Yes [113] | |||
| Rituximab | CD20 inhibition | CHOL, LDL | ||||
| Beclomethasone | Glucocorticoid receptor activation | CHOL | Yes [114] | |||
| Dexamethasone | Glucocorticoid receptor activation | CHOL | Yes [115] | Yes [116,117,118,119] | Yes [117,118,119] | |
| Prednisolone | Glucocorticoid receptor activation | CHOL | Yes [120,121] | Yes [120,121] | ||
| Prednisone | Glucocorticoid receptor activation | CHOL | Yes [116,122] | Yes [122] | ||
| Anakinra | IL-1 inhibition | CHOL | ||||
| Rilonacept | IL-1 inhibition | CHOL, LDL | ||||
| Basiliximab | IL-2 inhibition | CHOL, LDL | ||||
| Sarilumab | IL-6 inhibition | CHOL, LDL | ||||
| Siltuximab | IL-6 inhibition | CHOL | ||||
| Tocilizumab | IL-6 inhibition | CHOL, LDL | ||||
| Baricitinib | JAK inhibition | CHOL, LDL | ||||
| Tofacitinib | JAK inhibition | CHOL, LDL | ||||
| Everolimus | mTOR inhibition | CHOL, LDL | ||||
| Sirolimus | mTOR inhibition | CHOL | ||||
| Temsirolimus | mTOR inhibition | CHOL, LDL | ||||
| Leflunomide | Pyrimidine synthesis inhibition | CHOL, LDL | ||||
| Mycophenolate mofetil | Purine synthesis inhibition | CHOL | ||||
| Adalimumab | TNF inhibition | CHOL, LDL | ||||
| Certolizumab Pegol | TNF inhibition | CHOL, LDL | ||||
| Golimumab | TNF inhibition | CHOL, LDL | ||||
| Infliximab | TNF inhibition | CHOL, LDL | ||||
| Non-steroidal anti-inflammatory drug | Acetylsalisylic acid | COX inhibition | CHOL | Yes [123] | ||
| Diclofenac | COX inhibition | CHOL | ||||
| Ibuprofen | COX inhibition | CHOL | ||||
| Other | Ataluren | Ribosome function modulation | CHOL | |||
| Human normal immunoglobulin | Improved pathogen removal | CHOL | ||||
| Leuprorelin | GnRH analogue | CHOL, LDL | ||||
| Proton pump inhibitor | Lansoprazole | Stomach acid reduction | CHOL | Yes [124] | Yes [124] | |
| Pantoprazole | Stomach acid reduction | CHOL | ||||
| Antioxidant | Idebenone | Mitochondrial electron transport chain stimulation | CHOL | |||
| Retinoid | Alitretinoin | Retinoid receptor activation | CHOL | Yes [125] | ||
| Bexarotene | Retinoid receptor activation | CHOL, LDL | Yes [126,127] | Yes [126,127] | ||
| Isotretinoin | Retinoid receptor activation | CHOL | Yes [125] | |||
| Tretinoin | Retinoid receptor activation | CHOL | Yes [125] | |||
| Stimulant | Modafinil | Dopaminergic modulation | CHOL | Yes [128] | Yes [129] | |
| Vitamin | Cholecalciferol | Vitamin D receptor activation | CHOL, LDL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karpale, M.; Hukkanen, J.; Hakkola, J. Nuclear Receptor PXR in Drug-Induced Hypercholesterolemia. Cells 2022, 11, 313. https://doi.org/10.3390/cells11030313
Karpale M, Hukkanen J, Hakkola J. Nuclear Receptor PXR in Drug-Induced Hypercholesterolemia. Cells. 2022; 11(3):313. https://doi.org/10.3390/cells11030313
Chicago/Turabian StyleKarpale, Mikko, Janne Hukkanen, and Jukka Hakkola. 2022. "Nuclear Receptor PXR in Drug-Induced Hypercholesterolemia" Cells 11, no. 3: 313. https://doi.org/10.3390/cells11030313
APA StyleKarpale, M., Hukkanen, J., & Hakkola, J. (2022). Nuclear Receptor PXR in Drug-Induced Hypercholesterolemia. Cells, 11(3), 313. https://doi.org/10.3390/cells11030313