
Novel Roles of Nanog in Cancer Cells and Their Extracellular Vesicles

Abstract
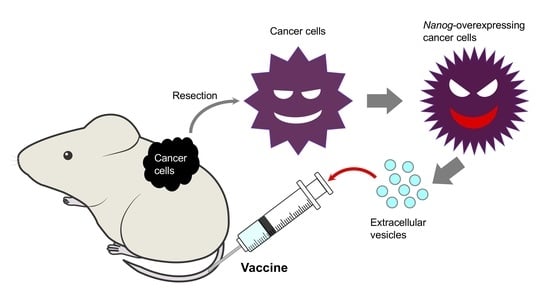
1. Introduction
2. Why Nanog?
3. Roles of Nanog in Cancer Cells
3.1. Breast Cancer
3.2. Cervical Cancer
3.3. Colon Cancer/Colorectal Cancer
3.4. Embryonic Carcinoma
3.5. Somatic Cancer
3.6. Hepatocellular Cancer
3.7. Melanoma
3.8. Ovarian Cancer
3.9. Pancreatic Cancer
3.10. Prostate Cancer
3.11. Squamous Cell Carcinoma
3.12. Cancer Stem Cells
3.13. PD-1-Treated Patients and Their Model Mice
3.14. Summary of NANOG Roles
- (a)
- High levels of Nanog expression are associated with increased malignancy, which has been observed in many types of cancers. The only exception is the case of HNSC.
- (b)
- Nanog targeting, alone, does not necessarily lead to cancer cytocide.
- (c)
- The degree of malignancy of cancer cells is not solely governed by Nanog.
- (d)
- Cancer cells with high levels of Nanog expression have high metastatic potential. It shows potential as a marker of malignant prognosis. Indeed, Nanog has shown promise as a marker for predicting the efficacy of PD-1 therapy.
- (e)
- Molecular mechanisms, leading to malignant transformations, greatly differ depending on the type of cancer. There are almost no research reports about why NANOG signaling differs depending on cancer types. This point should be clarified for the use of Nanog as a therapeutic target.
- (f)
- From a therapeutic perspective, the enhancement of immune functions is essential and, therefore, novel ideas are required to combine Nanog-targeting therapy with immunotherapy.
4. Nanog Overexpression in Melanoma
4.1. Transcriptome Analysis
4.2. Experimental Analyses
5. Properties of EVs Secreted from Cancer Cells
5.1. Tumor-Promoting Effect
5.2. Metastasis-Inhibitory Effect
6. Suppression of Cancer Metastasis by Melanoma-Derived EV
6.1. Comparison of Metastatic Potential between B16-F10 and Nanog+F10
6.2. Comparison of the Effect of B16-F10-EV and Nanog+F10-EV on Melanoma Metastasis
6.3. Role of Tgf-β1 in the Anti-Metastasis Effect
6.4. Role of Immune Cells in Preventing Metastasis
6.5. Quantitative Analysis of the Effects of EVs Taken Up by Macrophages
6.6. A Mechanism of Metastasis-Suppressive Effects by Nanog+F10-EV
7. Prospects for EV Cancer Vaccines
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fernández-Delgado, I.; Calzada-Fraile, D.; Sánchez-Madrid, S. Immune regulation by dendritic cell extracellular vesicles in cancer immunotherapy and vaccines. Cancers 2020, 12, 3558. [Google Scholar] [CrossRef]
- Tay, B.Q.; Wright, Q.; Ladwa, R.; Perry, C.; Leggatt, G.; Simpson, F.; Wells, J.W.; Panizza, B.J.; Frazer, I.H.; Cruz, J.L.G. Evolution of cancer vaccines—Challenges, achievements, and future directions. Vaccines 2021, 9, 535. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Oskarsson, T.; Acharyya, S.; Nguyen, D.X.; Zhang, X.H.; Norton, L.; Massagué, J. Tumor self-seeding by circulating cancer cells. Cell 2009, 139, 1315–1326. [Google Scholar] [CrossRef]
- Sinha, D.; Roy, S.; Saha, P.; Chatterjee, N.; Bishayee, A. Trends in research on exosomes in cancer progression and anticancer therapy. Cancers 2021, 13, 326. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Yi, X.; Chen, X.; Wu, Z.; You, H.; Chen, X.; Zhang, G.; Sun, Y.; Bu, X.; Wu, X.; et al. Warburg effect-promoted exosomal circ_0072083 releasing up-regulates NANGO expression through multiple pathways and enhances temozolomide resistance in glioma. J. Exp. Clin. Cancer Res. 2021, 40, 164. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Mazur, S.J.; Lin, T.; Appella, E.; Xu, Y. The pluripotency factor Nanog promotes breast cancer tumorigenesis and metastasis. Oncogene 2014, 33, 2655–2664. [Google Scholar] [CrossRef]
- Jewer, M.; Lee, L.; Leibovitch, M.; Zhang, G.; Liu, J.; Findlay, S.D.; Vincent, K.M.; Tandoc, K.; Dieters-Castator, D.; Quail, D.F.; et al. Translational control of breast cancer plasticity. Nat. Commun. 2020, 11, 2498. [Google Scholar] [CrossRef]
- Son, S.W.; Cho, E.; Cho, H.; Woo, S.R.; Lee, H.J.; Oh, S.J.; Kim, S.; Kim, J.H.; Chung, E.J.; Chung, J.Y.; et al. NANOG confers resistance to complement-dependent cytotoxicity in immune-edited tumor cells through up-regulating CD59. Sci. Rep. 2022, 12, 8652. [Google Scholar] [CrossRef]
- Noh, K.H.; Lee, Y.H.; Jeon, J.H.; Kang, T.H.; Mao, C.P.; Wu, T.C.; Kim, T.W. Cancer vaccination drives Nanog-dependent evolution of tumor cells towards an immune-resistant and stem-like phenotype. Cancer Res. 2012, 72, 1717–1727. [Google Scholar] [CrossRef]
- Descarpentrie, J.; Araúzo-Bravo, M.J.; He, Z.; François, A.; González, Á.; Garcia-Gallastegi, P.; Badiola, I.; Evrard, S.; Pernot, S.; Creemers, J.W.M.; et al. Role of furin in colon cancer stem cells malignant phenotype and expression of LGR5 and NANOG in KRAS and BRAF-mutated colon tumors. Cancers 2022, 14, 1195. [Google Scholar] [CrossRef]
- Zhang, M.; Peng, R.; Wang, H.; Yang, Z.; Zhang, H.; Zhang, Y.; Wang, M.; Wang, H.; Lin, J.; Zhao, Q.; et al. Nanog mediated by FAO/ACLY signaling induces cellular dormancy in colorectal cancer cells. Cell Death Dis. 2022, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.K.; Hung, L.M.; Hou, C.W.; Chen, J.K. MicroRNA 630 represses NANOG expression through transcriptional and post-transcriptional regulation in human embryonal carcinoma cells. Int. J. Mol. Sci. 2021, 21, 46. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Wang, J.; Wu, Y.; Li, Q.; Dong, M.; Liu, C.; He, Q.; Wang, R.; Wang, D.; Jiang, S.; et al. Identification of Nanog as a novel inhibitor of Rad51. Cell Death Dis. 2022, 13, 193. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Sun, L.; Jiang, K.; Gao, D.M.; Kang, X.N.; Wang, C.; Zhang, S.; Huang, S.; Qin, X.; Li, Y.; et al. NANOG promotes liver cancer cell invasion by inducing epithelial–mesenchymal transition through NODAL/SMAD3 signaling pathway. Int. J. Biochem. Cell Biol. 2013, 45, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Kumar, D.B.U.; Punj, V.; Xu, J.; Sher, L.; Tahara, S.M.; Hess, S.; Machida, K. NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab. 2016, 23, 206–219. [Google Scholar] [CrossRef]
- Yin, X.; Zhang, B.H.; Zheng, S.S.; Gao, D.M.; Qiu, S.J.; Wu, W.Z.; Ren, Z.G. Coexpression of gene Oct4 and Nanog initiates stem cell characteristics in hepatocellular carcinoma and promotes epithelial-mesenchymal transition through activation of Stat3/Snail signaling. J. Hematol. Oncol. 2015, 8, 23. [Google Scholar] [CrossRef]
- Borrull, A.; Ghislin, S.; Deshayes, F.; Lauriol, J.; Alcaide-Loridan, C.; Middendorp, S. Nanog and Oct4 overexpression increases motility and transmigration of melanoma cells. J. Cancer Res. Clin. Oncol. 2012, 138, 1145–1154. [Google Scholar] [CrossRef]
- Hasmim, M.; Noman, M.Z.; Messai, Y.; Bordereaux, D.; Gros, G.; Baud, V.; Chouaib, S. Cutting edge: Hypoxia-induced Nanog favors the intratumoral infiltration of regulatory T cells and macrophages via direct regulation of TGF-β1. J. Immunol. 2013, 191, 5802–5806. [Google Scholar] [CrossRef]
- Saito, M.; Kishi, R.; Sasai, T.; Hatakenaka, T.; Matsuki, N.; Minagawa, S. Effect of Nanog overexpression on the metastatic potential of a mouse melanoma cell line B16-BL6. Mol. Cell. Biochem. 2021, 476, 2651–2661. [Google Scholar] [CrossRef]
- Hatakenaka, T.; Matsuki, N.; Minagawa, S.; Khoo, C.S.M.; Saito, M. Anti-metastatic function of extracellular vesicles derived from Nanog-overexpressing melanoma. Curr. Oncol. 2022, 29, 1029–1046. [Google Scholar] [CrossRef]
- Siu, M.K.Y.; Jiang, Y.X.; Wang, J.J.; Leung, T.H.; Han, C.Y.; Tsang, B.K.; Cheung, A.N.; Ngan, H.Y.S.; Chan, K.K.L. Hexokinase 2 regulates ovarian cancer cell migration, invasion and stemness via FAK/ERK1/2/MMP9/NANOG/SOX9 signaling cascades. Cancers 2019, 11, 813. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yu, W.; Shrivastava, A.; Srivastava, R.K.; Shankar, S. Inhibition of pancreatic cancer stem cell characteristics by α-Mangostin: Molecular mechanisms involving Sonic hedgehog and Nanog. J. Cell. Mol. Med. 2019, 23, 2719–2730. [Google Scholar] [CrossRef] [PubMed]
- Saga, K.; Park, J.; Nimura, K.; Kawamura, N.; Ishibashi, A.; Nonomura, N.; Kaneda, Y. NANOG helps cancer cells escape NK cell attack by downregulating ICAM1 during tumorigenesis. J. Exp. Clin. Cancer Res. 2019, 38, 416. [Google Scholar] [CrossRef] [PubMed]
- Jeter, C.R.; Liu, B.; Lu, Y.; Chao, H.P.; Zhang, D.; Liu, X.; Chen, X.; Li, Q.; Rycaj, K.; Calhoun-Davis, T.; et al. NANOG reprograms prostate cancer cells to castration resistance via dynamically repressing and engaging the AR/FOXA1 signaling axis. Cell Discov. 2016, 2, 16041. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Zhang, X.; Xiang, X.; Xiong, R.; Xiao, D.; Chen, Z.; Liu, K.; Feng, G. NANOG promotes cell proliferation, invasion, and stemness via IL-6/STAT3 signaling in esophageal squamous carcinoma. Technol. Cancer Res. Treat. 2021, 20, 15330338211038492. [Google Scholar] [CrossRef]
- Huang, C.; Yoon, C.; Zhou, W.H.; Zhou, Y.C.; Zhou, W.W.; Liu, H.; Yang, X.; Lu, J.; Lee, S.Y.; Huang, K. ERK1/2-Nanog signaling pathway enhances CD44(+) cancer stem-like cell phenotypes and epithelial-to-mesenchymal transition in head and neck squamous cell carcinomas. Cell Death Dis. 2020, 11, 266. [Google Scholar] [CrossRef]
- Pedregal-Mallo, D.; Hermida-Prado, F.; Granda-Díaz, R.; Montoro-Jiménez, I.; Allonca, E.; Pozo-Agundo, E.; Álvarez-Fernández, M.; Álvarez-Marcos, C.; García-Pedrero, J.M.; Rodrigo, J.P. Prognostic significance of the pluripotency factors NANOG, SOX2, and OCT4 in head and neck squamous cell carcinomas. Cancers 2020, 12, 1794. [Google Scholar] [CrossRef]
- Luo, W.; Li, S.; Peng, B.; Ye, Y.; Deng, X.; Yao, K. Embryonic stem cells markers SOX2, OCT4 and Nanog expression and their correlations with epithelial-mesenchymal transition in nasopharyngeal carcinoma. PLoS ONE 2013, 8, e56324. [Google Scholar] [CrossRef]
- Grubelnik, G.; Boštjančič, E.; Grošelj, A.; Zidar, N. Expression of NANOG and its regulation in oral squamous cell carcinoma. Biomed Res. Int. 2020, 2020, 8573793. [Google Scholar] [CrossRef]
- Gawlik-Rzemieniewska, N.; Bednarek, I. The role of NANOG transcriptional factor in the development of malignant phenotype of cancer cells. Cancer Biol. Ther. 2016, 17, 1–10. [Google Scholar] [CrossRef]
- Oh, S.J.; Lee, H.J.; Song, K.H.; Kim, S.; Cho, E.; Lee, J.; Bosenberg, M.W.; Kim, T.W. Targeting the NANOG/HDAC1 axis reverses resistance to PD-1 blockade by reinvigorating the antitumor immunity cycle. J. Clin. Investig. 2022, 15, 132. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Bear, J.E.; Haugh, J.M. Directed migration of mesenchymal cells: Where signaling and the cytoskeleton meet. Curr. Opin. Cell Biol. 2014, 30, 74–82. [Google Scholar] [CrossRef]
- Peer, E.; Tesanovic, S.; Aberger, F. Next-generation Hedgehog/GLI pathway inhibitors for cancer therapy. Cancers 2019, 11, 538. [Google Scholar] [CrossRef]
- Seoane, J.; Gomis, R. TGF-β family signaling in tumor suppression and cancer progression. Cold Spring Harbor Perspect. Biol. 2017, 9, a022277. [Google Scholar] [CrossRef]
- Fabregat, I.; Caballero-Díaz, D. Transforming growth factor-β-induced cell plasticity in liver fibrosis and hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef]
- Suriyamurthy, S.; Baker, D.; Dijke, P.T.; Iyengar, P.V. Epigenetic reprogramming of TGF-β signaling in breast cancer. Cancers 2019, 11, 726. [Google Scholar] [CrossRef]
- Yamada, K.; Saito, M.; Matsuoka, H.; Inagaki, N. A real-time method of imaging glucose uptake in single, living mammalian cells. Nat. Protoc. 2007, 2, 753–762. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nishiuchi, Y.; Teshima, T.; Matsuoka, H.; Yamada, K. Synthesis of 2-NBDLG, a fluorescent derivative of L-glucosamine; the antipode of D-glucose tracer 2-NBDG. Tetrahedron Lett. 2008, 49, 6876–6878. [Google Scholar] [CrossRef]
- Gehrmann, U.; Näslund, T.I.; Hiltbrunner, S.; Larssen, P.; Gabrislsson, S. Harnessing the exosome-induced immune response for cancer immunotherapy. Semin. Cancer Biol. 2014, 28, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Czernek, L.; Düchler, M. Functions of cancer-derived extracellular vesicles in immunosuppression. Arch. Immunol. Ther. Exp. 2017, 65, 311–323. [Google Scholar] [CrossRef]
- Andreola, G.; Rivoltini, L.; Castelli, C.; Huber, V.; Perego, P.; Deho, P.; Squarcina, P.; Accornero, P.; Lozupone, F.; Lugini, L. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J. Exp. Med. 2002, 195, 1303–1316. [Google Scholar] [CrossRef]
- Kurywchak, P.; Tavormina, J.; Kalluri, R. The emerging roles of exosomes in the modulation of immune responses in cancer. Genome Med. 2018, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Lai, X.; Yu, S.; Chen, S.; Ma, Y.; Zhang, Y.; Li, H.; Zhu, X.; Yao, L.; Zhang, J. Exosomal miR-221/222 enhances tamoxifen resistance in recipient ER-positive breast cancer cells. Breast Cancer Res. Treat. 2014, 147, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.G.; Yang, M.F.; Ren, Y.Q.; Wu, C.H.; Wang, L.Q. Exosomes mediated transfer of lncRNA UCA1 results in increased tamoxifen resistance in breast cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4362–4368. [Google Scholar]
- Qin, X.; Yu, S.; Zhou, L.; Shi, M.; Hu, Y.; Xu, X.; Shen, B.; Liu, S.; Yan, D.; Feng, J. Cisplatin-resistant lung cancer cell–derived exosomes increase cisplatin resistance of recipient cells in exosomal miR-100–5p-dependent manner. Int. J. Nanomed. 2017, 12, 3721–3733. [Google Scholar] [CrossRef]
- Wang, J.; Lv, B.; Su, Y.; Wang, X.; Bu, J.; Yao, L. Exosome-mediated transfer of lncRNA HOTTIP promotes cisplatin resistance in gastric cancer cells by regulating HMGA1/miR-218 axis. OncoTargets Ther. 2019, 12, 11325–11338. [Google Scholar] [CrossRef]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef]
- Huang, H.; Hou, J.; Liu, K.; Liu, Q.; Shen, L.; Liu, B.; Lu, Q.; Zhang, N.; Che, L.; Li, J.; et al. RAB27A-dependent release of exosomes by liver cancer stem cells induces Nanog expression in their differentiated progenies and confers regorafenib resistance. J. Gastroenterol. Hepatol. 2021, 36, 3429–3437. [Google Scholar] [CrossRef]
- Zhao, L.-J.; Li, Y.-Y.; Zhang, Y.-T.; Fan, Q.-Q.; Ren, H.-M.; Zhang, C.; Mardinoglu, A.; Chen, W.-C.; Pang, J.-R.; Shen, D.-D.; et al. Lysine demethylase LSD1 delivered via small extracellular vesicles promotes gastric cancer cell stemness. EMBO Rep. 2021, 22, e50922. [Google Scholar] [CrossRef]
- Kitai, Y.; Kawasaki, T.; Sueyoshi, T.; Kobiyama, K.; Ishii, K.J.; Zou, J.; Akira, S.; Matsuda, T.; Kawai, T. DNA-containing exosomes derived from cancer cells treated with topotecan activate a STING-dependent pathway and reinforce antitumor immunity. J. Immunol. 2017, 198, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J.M.; Vanpouille-Box, C.; Spada, S.; Rudqvist, N.P.; Chapman, J.R.; Ueberheide, B.M.; Pilones, K.A.; Sarfraz, Y.; Formenti, S.C.; Demaria, S. Exosomes shuttle TREX1-sensitive IFN-stimulatory dsDNA from irradiated cancer cells to DCs. Cancer Immunol. Res. 2018, 6, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Düchler, M.; Czernek, L.; Peczek, L.; Cypryk, W.; Sztiller-Sikorska, M.; Czyz, M. Melanoma-derived extracellular vesicles bear the potential for the induction of antigen-specific tolerance. Cells 2019, 8, 665. [Google Scholar] [CrossRef] [PubMed]
- Rossowska, J.; Anger, N.; Wegierek, K.; Szczygieł, A.; Mierzejewska, J.; Milczarek, M.; Szermer-Olearnik, B.; Pajtasz-Piasecka, E. Antitumor potential of extracellular vesicles released by genetically modified murine colon carcinoma cells with overexpression of interleukin-12 and shRNA for TGF-β1. Front. Immunol. 2019, 10, 211. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Yeh, H.W.; Lee, S.S.; Chang, C.Y.; Lang, Y.D.; Jou, Y.S. A new switch for TGFβ in cancer. Cancer Res. 2019, 79, 3797–3805. [Google Scholar] [CrossRef]
- Fujimura, T.; Aiba, S. Significance of immunosuppressive cells as a target for immunotherapies in melanoma and non-melanoma skin cancers. Biomolecules 2020, 10, 1087. [Google Scholar] [CrossRef]
- Guo, F.; Feng, Y.C.; Zhao, G.; Zhang, R.; Cheng, Z.Z.; Kong, W.N.; Wu, H.L.; Xu, B.; Lv, X.; Ma, X.M. Tumor-associated CD163+ M2 macrophage infiltration is highly associated with PD-L1 expression in cervical cancer. Cancer Manag. Res. 2020, 12, 5831–5843. [Google Scholar] [CrossRef]
- Kubota, K.; Moriyama, M.; Furukawa, S.; Rafiul, H.A.S.M.; Maruse, Y.; Jinno, T.; Tanaka, A.; Ohta, M.; Ishiguro, N.; Yamauchi, M.; et al. CD163+ CD204+ tumor-associated macrophages contribute to T cell regulation via interleukin-10 and PD-L1 production in oral squamous cell carcinoma. Sci. Rep. 2017, 7, 1755. [Google Scholar] [CrossRef]
- Harada, K.; Dong, X.; Estrella, J.S.; Correa, A.M.; Xu, Y.; Hofstetter, W.L.; Sudo, K.; Onodera, H.; Suzuki, K.; Suzuki, A.; et al. Tumor-associated macrophage infiltration is highly associated with PD-L1 expression in gastric adenocarcinoma. Gastric Cancer 2018, 21, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Obeid, J.M.; Erdag, G.; Smolkin, M.E.; Deacon, D.H.; Patterson, J.W.; Chen, L.; Bullock, T.N.; Slingluff, C.L. PD-L1, PD-L2 and PD-1 expression in metastatic melanoma: Correlation with tumor-infiltrating immune cells and clinical outcome. Oncoimmunology 2016, 5, e1235107. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Nishikawa, H. Roles of regulatory T cells in cancer immunity. Int. Immunol. 2016, 28, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, J.; Yamazaki, S.; Sakaguchi, S. Induction of tumor immunity by removing CD25+CD4+ T cells: A common basis between tumor immunity and autoimmunity. J. Immunol. 1999, 163, 5211–5218. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Cancer [Cell Line] | NEL *1 Altering Factor [NEL] | Effects on Cancer Cell Functions | P or S *2 | Signaling Factors Downstream of Nanog [Role as a Marker of Cell Function] | Ref. | |
|---|---|---|---|---|---|---|
| Breast cancer [MCF7] | OE *3 [High] | Cell adhesion ↑ | P | Itga9 ↑, Cldn11 ↑, Cldn10 ↑, Cldn6 ↑ | Array analysis | [6] |
| Ca signaling ↑ | Atp2a1 ↑, Cacna1h ↑, Gna14 ↑, Cacna1g ↑ | |||||
| Focal adhesion ↑ | Pdgfrα ↑, Pdgfrβ ↑, Itga11 ↑, Itgb5 ↑, Rasgrf1 ↑ | |||||
| p53 signaling (tumor suppressor gene) ↓ | Gadd45a ↓, Gadd45g ↓, Pten ↓, Bax ↓ | |||||
| Tumorigenesis by co-expression with Wnt-1 | ||||||
| Breast cancer [T47D, MCF7, MDA-MB-231] | mTOR inhibitors, Chemotherapeutics [Up] | Stem cell-like phenotype ↑ | P | NANOG ↑, SNAIL ↑, NODAL ↑ | [7] | |
| Cervical carcinoma [CaSki] | OE [High] | Resistance to complement-dependent cytotoxicity ↑ | P | CD59 ↑ through promoter occupancy, CD59 binds C8 and C9 and inhibits MAC (membrane attack complex) formation | [8] | |
| Cervical cancer [TC-1 (P0~P3)] | Vaccination-induced cancer evolution (VICE) [Up] | Resistance to cytotoxic T lymphocyte lysis ↑ Stem-like phenotype ↑ | P | Stemness markers (CD133, CD44, ALDH) ↑ in TC-1(P3) | [9] | |
| Colon cancer [SW480, SW620, HT29, CT26] | Furin [Down by furin repression] | NANOG ↓, LGR5 ↓, Calcium transportation ↓ | S by NANOG ↓ | [10] | ||
| Colon/Colorectal cancer [HCT116, HT29] | OE [High] | Dormancy ↑, Chemoresistance ↑ | P | Cell cycle regulating P21 ↑, P27 ↑ | [11] | |
| Embryonal carcinoma [NT2/D1] | MIR630 [Down] | Differentiation ↑ | S by NANOG ↓ | PKC ⇒ miR630 ↑ ⇒ NANOG 3′UTR ⇒ NANOG ↓ | [12] | |
| Somatic cancer [HeLa, HCT116] | Nanog-C/CD2 as a Rad51 inhibitor [High] | Inhibition of chemo-/radiotherapy-generated DNA damages of cancer cells mediated by Rad51 | S by Nanog-C/CD2 | Interaction of C/CD2 domain with Rad51 | [13] | |
| Hepatocellular carcinoma (HCC) [MHCC97H, -L, MHCCLM3, etc.] | OE [High] | EMT ↑, Invasion ↑ | P | Nodal ⇒ Smad3 | [14] | |
| Hepatocellular carcinoma [CD133+/CD49f+ TICs from HCC of alcoholic patients, HEK239T] | Obesity, Alcohol, Virus [Up] | Self-renewal ↑, Chemoresistance ↑ | P | Obesity, Alcohol ⇒ LPS ⇒ TLR4 ↑ ⇒ NANOG ↑ ⇒ FAO ↑, NANOG ⇒ OXPHOS ↓ | [15] | |
| Hepatocellular carcinoma [MHCC97-L] | Co-expression of Oct4 and Nanog [High] | Stemness ↑, EMT ↑ | P | Stat3 ↑, Snail ↑ | [16] | |
| Melanoma (human) [A375] | OE *3 [High] | Amoeboid migration ↑ | P | ARHGAP22 ↑, DOCK10 ↑, EPHA2 ↑ | [17] | |
| Melanoma (mouse) [B16-F10] | Hypoxia [Up] | Spheroid formation ↑, Proliferation ↑ | P | Tgf-β1 ↑ | [18] | |
| Melanoma (mouse) [B16-BL6] | OE [High] | Proliferation ↑, Migration ↑, Invasion ↑ | P | Tgf-β1 ↓ | [19] | |
| Melanoma (mouse) [B16-F10] | OE [High] | Proliferation ↑, Migration ↑, Invasion ↑ | P | Tgf-β1 ↓ | [20] | |
| Ovarian [6 cell lines, Ascites of patients] | HK2 [Up] | Migration ↑, Invasion ↑, Metastasis ↑ | P | HK2 ⇒ FAK ↑ ⇒ ERK1/2 ↑ ⇒ MMP9 ↑ | [21] | |
| Stemness ↑ | P | HK2 ⇒ FAK ↑ ⇒ ERK1/2 ↑ ⇒ NANOG · SOX9 ↑ | ||||
| Pancreatic cancer stem cell (CSC) [AsPC-1, PANC-1, CSCs from primary tumors] | α-Mangostin [Down] | EMT ↓ | S by Nanog ↓ | Gli signaling (Nanog, Oct4, c-Myc, Sox-2, KLF4) ↓ [human pancreatic CSCs marker: CD133+ /CD44+/CD24+/ ESA+] | [22] | |
| Prostate cancer [DU145, PC3, 22Rv1] | OE [High] | Escape from NK cell attack ↑ | P | ICAM1 ↓ | [23] | |
| Prostate cancer [Xenograft models (LAPC4, LAPC9)] | Endogenous NANOG [Varied] | Castration resistance ↓ | P | NANOG co-occupies FOXA1 and androgen receptor loci ⇒ pro-differentiation genes ↓ | [24] | |
| Esophageal squamous carcinoma [Eca109] | OE [High] | Proliferation ↑, Invasion ↑, Stemness ↑ | P | IL6 ↑, STAT3 ↑, CCL5 ↑, VEGFA ↑, CCND1 ↑, Bcl-xL ↑ | [25] | |
| Head and neck squamous cell carcinomas (HNSC) [QLL1, SCC15, SCC25] | CD44(+) [Higher than in CD44(-) cells] | Migration ↑, Invasion ↑, Radiotherapy resistance ↑, EMT ↑ | P | NANOG and ERK1/2 synergistic effects | [26] | |
| HNSC | Patients [Varied] | S | [NANOG and SOX2 ⇒ Better prognosis] | [27] | ||
| Nasopharyngeal Carcinoma | Patients [Varied] | P | High frequency of Nanog, OCT4 at tumor invasive front, lymph node metastasis | [28] | ||
| Oral squamous cell carcinoma | Patients with/without lymph node metastasis [Varied] | P | Positive-correlation [Nanog] vs [OCT4, NOTCH1, AGR2, KLF4] at mRNA [NANOG protein/mRNA:Poor prognosis marker?] | [29] | ||
| Cancer stem cell (CSC) models [HNSC, non-small lung cancer, colon cancer, A549] | NANOG overexpressed or varied [High] | Migration ↑, Invasion ↑, EMT ↑ | P | BMI1 ⇒ E-Cadherin ↓ | [NANOG as a CSC marker (proposed)] | [30] |
| SNAIL2 ⇒ E-Cadherin ↓ | ||||||
| BMI1 ⇒ SNAIL1 ⇒ E-Cadherin ↓ | ||||||
| SNAIL2, BMI1 ⇒ SNAIL1 | ||||||
| Proliferation ↑, Self-renewal ↑, Chemoresistance ↑ | P | BMI1 | ||||
| Anti-apoptosis ↑, Chemoresistance ↑ | P | NANOG · STAT3 ⇒ miR21 ⇒ PDCD4(cancer resistance gene) ↓ | ||||
| Hedgehog signal (Hh), FAK [Varied] | Stemness ↑ | P | Hh ⇒ PATCH1/2 ↓ ⇒ SMO ↑ ⇒ GLI1 ↑ ⇒ NANOG ↑ ⇒ GLI1 ↑ (Positive feedback) | |||
| Anti-apoptosis ↑ | P | FAK ⇒ NANOG ↑ ⇒ p53 ↓ ⇒ NANOG ↓ (Negative feedback) | ||||
| PD-1 therapy treated patients, Anti–PD-1 therapy model mice | Patients with 19 types of cancer [High] | T-cell invasion ↓, Resistance to cytotoxic T-cell ↑ | P | NANOG ⇒ HDAC1 ⇒ Cxcl10 ↓, MCL1 ↑ [Antitumor immunity marker] | [31] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saito, M. Novel Roles of Nanog in Cancer Cells and Their Extracellular Vesicles. Cells 2022, 11, 3881. https://doi.org/10.3390/cells11233881
Saito M. Novel Roles of Nanog in Cancer Cells and Their Extracellular Vesicles. Cells. 2022; 11(23):3881. https://doi.org/10.3390/cells11233881
Chicago/Turabian StyleSaito, Mikako. 2022. "Novel Roles of Nanog in Cancer Cells and Their Extracellular Vesicles" Cells 11, no. 23: 3881. https://doi.org/10.3390/cells11233881
APA StyleSaito, M. (2022). Novel Roles of Nanog in Cancer Cells and Their Extracellular Vesicles. Cells, 11(23), 3881. https://doi.org/10.3390/cells11233881