

Translation Potential and Challenges of In Vitro and Murine Models in Cancer Clinic

Abstract
1. Introduction
2. Syngeneic Tumor Models
3. Genetically Engineered Mouse Models (GEMM)
4. Cell Lines and Cell Line-Derived Xenograft (CDX)
5. Patient-Derived Xenografts (PDX)
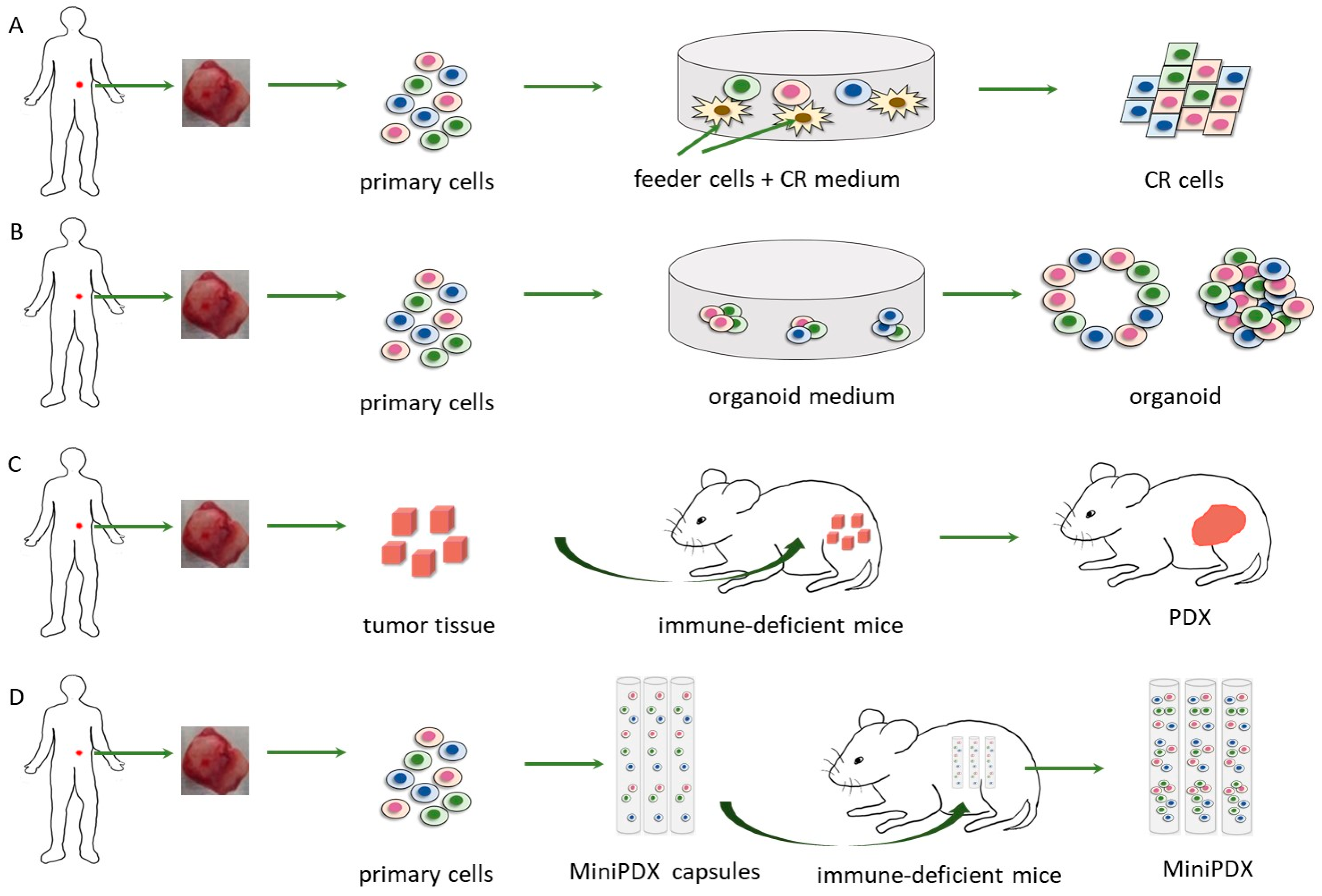
6. Conditionally Reprogrammed (CR) Cells
7. Patient-Derived Organoid
8. MiniPDX
9. Conclusions and Perspective
10. Disclosure
Funding
Acknowledgments
Conflicts of Interest
References
- Sengupta, R.; Zaidi, S.K. Discovery Science Driving Clinical Breakthroughs. Clin. Cancer Res. 2021, 27, 5757–5759. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. GLO-BOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Rubin, E.H.; Gilliland, D.G. Drug development and clinical trials—The path to an approved cancer drug. Nat. Rev. Clin. Oncol. 2012, 9, 215–222. [Google Scholar] [CrossRef]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Boehm, J.; Golub, T.R. An ecosystem of cancer cell line factories to support a cancer dependency map. Nat. Rev. Genet. 2015, 16, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Codenotti, S.; Mansoury, W.; Pinardi, L.; Monti, E.; Marampon, F.; Fanzani, A. Animal models of well-differentiated/dedifferentiated liposarcoma: Utility and limitations. OncoTargets Ther. 2019, 12, 5257–5268. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, M.; Guais, A.; Sanders, E.; Campion, F.; Fichtner, I.; Bonte, J.; Baronzio, G.; Fiorentini, G.; Israël, M.; Schwartz, L. Screening of well-established drugs targeting cancer metabolism: Reproducibility of the efficacy of a highly effective drug combination in mice. Investig. New Drugs 2011, 30, 1331–1342. [Google Scholar] [CrossRef]
- Vallespi, M.G.; Pimentel, G.; Cabrales-Rico, A.; Garza, J.; Oliva, B.; Mendoza, O.; Gomez, Y.; Basaco, T.; Sánchez, I.; Calderon, C.; et al. Antitumor efficacy, pharmacokinetic and biodistribution studies of the anticancer peptide CIGB-552 in mouse models. J. Pept. Sci. 2014, 20, 850–859. [Google Scholar] [CrossRef]
- Behrens, D.; Walther, W.; Fichtner, I. Pancreatic cancer models for translational research. Pharmacol. Ther. 2017, 173, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Mosely, S.I.; Prime, J.E.; Sainson, R.C.; Koopmann, J.-O.; Wang, D.Y.; Greenawalt, D.M.; Ahdesmaki, M.J.; Leyland, R.; Mullins, S.; Pacelli, L.; et al. Rational Selection of Syngeneic Preclinical Tumor Models for Immunotherapeutic Drug Discovery. Cancer Immunol. Res. 2017, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, N.M.; DeMayo, F.; Finegold, M.J.; Medina, D.; Tilley, W.D.; Aspinall, J.O.; Cunha, G.R.; Donjacour, A.A.; Matusik, R.J.; Rosen, J.M. Prostate cancer in a transgenic mouse. Proc. Natl. Acad. Sci. USA 1995, 92, 3439–3443. [Google Scholar] [CrossRef]
- Sinn, E.; Muller, W.; Pattengale, P.; Tepler, I.; Wallace, R.; Leder, P. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: Synergistic action of oncogenes in vivo. Cell 1987, 49, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Toyama, K.; Shioya, H.; Ito, M.; Hirota, M.; Hasegawa, S.; Matsumoto, H.; Takano, H.; Akiyama, T.; Toyoshima, K.; et al. Rapid Colorectal Adenoma Formation Initiated by Conditional Targeting of the Apc Gene. Science 1997, 278, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Hooijkaas, A.I.; Gadiot, J.; van der Valk, M.; Mooi, W.J.; Blank, C.U. Targeting BRAF in an Inducible Murine Model of Mela-noma. Am. J. Pathol. 2012, 181, 785–794. [Google Scholar] [CrossRef]
- Pitteri, S.J.; JeBailey, L.; Faça, V.M.; Thorpe, J.D.; Silva, M.A.; Ireton, R.C.; Horton, M.B.; Wang, H.; Pruitt, L.C.; Zhang, Q.; et al. Integrated Proteomic Analysis of Human Cancer Cells and Plasma from Tumor Bearing Mice for Ovarian Cancer Biomarker Discovery. PLoS ONE 2009, 4, e7916. [Google Scholar] [CrossRef]
- Fijneman, R.J.; de Wit, M.; Pourghiasian, M.; Piersma, S.R.; Pham, T.V.; Warmoes, M.O.; Lavaei, M.; Piso, C.; Smit, F.; Diemen, P.M.D.-V.; et al. Proximal Fluid Proteome Profiling of Mouse Colon Tumors Reveals Biomarkers for Early Diagnosis of Human Colorectal Cancer. Clin. Cancer Res. 2012, 18, 2613–2624. [Google Scholar] [CrossRef]
- Kaplan-Lefko, P.J.; Chen, T.-M.; Ittmann, M.M.; Barrios, R.J.; Ayala, G.E.; Huss, W.J.; Maddison, L.A.; Foster, B.A.; Greenberg, N.M. Pathobiology of autochthonous prostate cancer in a pre-clinical transgenic mouse model. Prostate 2003, 55, 219–237. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adeno-carcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Kucherlapati, R. Genetically Modified Mouse Models for Biomarker Discovery and Preclinical Drug Testing. Clin. Cancer Res. 2012, 18, 625–630. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sharma, S.V.; Haber, D.A.; Settleman, J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat. Cancer 2010, 10, 241–253. [Google Scholar] [CrossRef]
- Huo, K.-G.; D’Arcangelo, E.; Tsao, M.-S. Patient-derived cell line, xenograft and organoid models in lung cancer therapy. Transl. Lung Cancer Res. 2020, 9, 2214–2232. [Google Scholar] [CrossRef] [PubMed]
- Suzawa, K.; Toyooka, S.; Sakaguchi, M.; Morita, M.; Yamamoto, H.; Tomida, S.; Ohtsuka, T.; Watanabe, M.; Hashida, S.; Maki, Y.; et al. Antitumor effect of afatinib, as a human epidermal growth factor receptor 2-targeted therapy, in lung cancers har-boring HER 2 oncogene alterations. Cancer Sci. 2015, 107, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Kasiri, S.; Shao, C.; Chen, B.; Wilson, A.N.; Yenerall, P.; Timmons, B.C.; Girard, L.; Tian, H.; Behrens, C.; Wistuba, I.I.; et al. GLI1 Blockade Potentiates the Antitumor Activity of PI3K Antagonists in Lung Squamous Cell Carcinoma. Cancer Res. 2017, 77, 4448–4459. [Google Scholar] [CrossRef] [PubMed]
- Otto, R.; Sers, C.; Leser, U. Robust in-silico identification of cancer cell lines based on next generation sequencing. Oncotarget 2017, 8, 34310–34320. [Google Scholar] [CrossRef][Green Version]
- Vaughan, L.; Glänzel, W.; Korch, C.; Capes-Davis, A. Widespread Use of Misidentified Cell Line KB (HeLa): Incorrect At-tribution and Its Impact Revealed through Mining the Scientific Literature. Cancer Res. 2017, 77, 2784–2788. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.J.; Beal, K.M.; DeGruttola, H.S.; Brennan, S.; Marzilli, L.A.; Anderson, K. Utilization of sequence variants as biomarkers to analyze population dynamics in cloned cell lines. Biotechnol. Bioeng. 2017, 114, 1744–1752. [Google Scholar] [CrossRef]
- Horbach, S.P.J.M.; Halffman, W. The ghosts of HeLa: How cell line misidentification contaminates the scientific literature. PLoS ONE 2017, 12, e0186281. [Google Scholar] [CrossRef]
- Reiter, J.G.; Makohon-Moore, A.P.; Gerold, J.M.; Heyde, A.; Attiyeh, M.A.; Kohutek, Z.A.; Tokheim, C.J.; Brown, A.; DeBlasio, R.M.; Niyazov, J.; et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 2018, 361, 1033–1037. [Google Scholar] [CrossRef]
- Perales-Patón, J.; Piñeiro-Yáñez, E.; Tejero, H.; Lopez-Casas, P.P.; Hidalgo, M.; López, G.G.; Al-Shahrour, F. Pancreas Cancer Precision Treatment Using Avatar Mice from a Bioinformatics Perspective. Public Heal. Genom. 2017, 20, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.M.; Stokes, J.B.; Adair, S.J.; Stelow, E.B.; Borgman, C.A.; Lowrey, B.T.; Xin, W.; Blais, E.M.; Lee, J.K.; Papin, J.A.; et al. Clinical, Molecular and Genetic Validation of a Murine Orthotopic Xenograft Model of Pancreatic Adenocarcinoma Using Fresh Human Specimens. PLoS ONE 2013, 8, e77065. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Q.; Ijiri, M.; Rodriguez, R.; Gandour-Edwards, R.; Lee, J.; Tepper, C.G.; Li, Y.; Beckett, L.; Lam, K.; Goodwin, N.; et al. Novel Patient Metastatic Pleural Effusion-Derived Xenograft Model of Renal Medullary Carcinoma Demonstrates Therapeutic Efficacy of Sunitinib. Front. Oncol. 2021, 11, 648097. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Stossel, C.; Schvimer, M.; Atias, D.; Halperin, S.; Buzhor, E.; Raitses-Gurevich, M.; Cohen, K.; Pri-Chen, S.; Wilson, J.; et al. Pancreatic cancer ascites xenograft—An expeditious model mirroring advanced therapeutic resistant disease. Oncotarget 2017, 8, 40778–40790. [Google Scholar] [CrossRef]
- Hodgkinson, C.L.; Morrow, C.J.; Li, Y.; Metcalf, R.L.; Rothwell, D.G.; Trapani, F.; Polanski, R.; Burt, D.J.; Simpson, K.L.; Morris, K.; et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat. Med. 2014, 20, 897–903. [Google Scholar] [CrossRef]
- Cho, S.-Y.; Kang, W.; Han, J.Y.; Min, S.; Kang, J.; Lee, A.; Kwon, J.Y.; Lee, C.; Park, H. An Integrative Approach to Precision Cancer Medicine Using Patient-Derived Xenografts. Mol. Cells 2016, 39, 77–86. [Google Scholar] [CrossRef]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Pa-tient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef]
- Fichtner, I.; Rolff, J.; Soong, R.; Hoffmann, J.; Hammer, S.; Sommer, A.; Becker, M.; Merk, J. Establishment of Patient-Derived Non-Small Cell Lung Cancer Xenografts as Models for the Identification of Predictive Biomarkers. Clin. Cancer Res. 2008, 14, 6456–6468. [Google Scholar] [CrossRef]
- Fleming, J.M.; Miller, T.C.; Meyer, M.J.; Ginsburg, E.; Vonderhaar, B.K. Local regulation of human breast xenograft models. J. Cell. Physiol. 2010, 224, 795–806. [Google Scholar] [CrossRef]
- Petrillo, L.A.; Wolf, D.M.; Kapoun, A.M.; Wang, N.J.; Barczak, A.; Xiao, Y.; Korkaya, H.; Baehner, F.; Lewicki, J.; Wicha, M.; et al. Xenografts faithfully recapitulate breast cancer-specific gene expression patterns of parent primary breast tumors. Breast Cancer Res. Treat. 2012, 135, 913–922. [Google Scholar] [CrossRef]
- Kim, M.P.; Evans, D.B.; Wang, H.; Abbruzzese, J.L.; Fleming, J.B.; Gallick, G.E. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat. Protoc. 2009, 4, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Guenot, D.; Guerin, E.; Aguillon-Romain, S.; Pencreach, E.; Schneider, A.; Neuville, A.; Chenard, M.-P.; Duluc, I.; du Manoir, S.; Brigand, C.; et al. Primary tumour genetic alterations and intra-tumoral heterogeneity are maintained in xenografts of human colon cancers showing chromosome instability. J. Pathol. 2006, 208, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Seol, H.S.; Kang, H.; Lee, S.-I.; Kim, N.E.; Kim, T.I.; Chun, S.M.; Yu, C.S.; Suh, Y.-A.; Singh, S.R.; Chang, S.; et al. Development and characterization of a colon PDX model that reproduces drug responsiveness and the mutation profiles of its original tumor. Cancer Lett. 2014, 345, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Corso, S.; Cargnelutti, M.; Durando, S.; Menegon, S.; Apicella, M.; Migliore, C.; Capeloa, T.; Ughetto, S.; Isella, C.; Medico, E.; et al. Rituximab Treatment Prevents Lymphoma Onset in Gastric Cancer Patient-Derived Xenografts. Neoplasia 2018, 20, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Dobbin, Z.C.; Katre, A.A.; Steg, A.D.; Erickson, B.K.; Shah, M.M.; Alvarez, R.D.; Conner, M.G.; Schneider, D.; Chen, D.; Landen, C.N. Using heterogeneity of the patient-derived xenograft model to identify the chemoresistant population in ovarian cancer. Oncotarget 2014, 5, 8750–8764. [Google Scholar] [CrossRef]
- Gu, Q.; Zhang, B.; Sun, H.; Xu, Q.; Tan, Y.; Wang, G.; Luo, Q.; Xu, W.; Yang, S.; Li, J.; et al. Genomic characterization of a large panel of patient-derived hepatocellular carcinoma xenograft tumor models for preclinical development. Oncotarget 2015, 6, 20160–20176. [Google Scholar] [CrossRef]
- Beshiri, M.L.; Tice, C.M.; Tran, C.; Nguyen, H.M.; Sowalsky, A.G.; Agarwal, S.; Jansson, K.H.; Yang, Q.; McGowen, K.M.; Yin, J.; et al. A PDX/Organoid Biobank of Advanced Prostate Cancers Captures Genomic and Phenotypic Heterogeneity for Disease Modeling and Therapeutic Screening. Clin. Cancer Res. 2018, 24, 4332–4345. [Google Scholar] [CrossRef] [PubMed]
- Tsoli, M.; Shen, H.; Mayoh, C.; Franshaw, L.; Ehteda, A.; Upton, D.; Carvalho, D.; Vinci, M.; Meel, M.H.; van Vuurden, D.; et al. International experience in the development of patient-derived xenograft models of diffuse intrinsic pontine glioma. J. Neuro-Oncology 2018, 141, 253–263. [Google Scholar] [CrossRef]
- Xiao, M.; Rebecca, V.W.; Herlyn, M. A Melanoma Patient-Derived Xenograft Model. J. Vis. Exp. 2019, 147, e59508. [Google Scholar] [CrossRef]
- Li, H.; Wheeler, S.; Park, Y.; Ju, Z.; Thomas, S.M.; Fichera, M.; Egloff, A.M.; Lui, V.W.; Duvvuri, U.; Bauman, J.E.; et al. Pro-teomic Characterization of Head and Neck Cancer Patient-Derived Xenografts. Mol. Cancer Res. 2016, 14, 278–286. [Google Scholar] [CrossRef]
- Jung, J.; Lee, C.H.; Seol, H.S.; Choi, Y.S.; Kim, E.; Lee, E.J.; Rhee, J.-K.; Singh, S.R.; Jun, E.S.; Han, B.; et al. Generation and molecular characterization of pancreatic cancer patient-derived xenografts reveals their heterologous nature. Oncotarget 2016, 7, 62533–62546. [Google Scholar] [CrossRef]
- Williams, S.A.; Anderson, W.C.; Santaguida, M.T.; Dylla, S.J. Patient-derived xenografts, the cancer stem cell paradigm, and cancer pathobiology in the 21st century. Lab. Investig. 2013, 93, 970–982. [Google Scholar] [CrossRef]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Corà, D.; Di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A Molecularly Annotated Platform of Patient-Derived Xenografts (“Xenopatients”) Identifies HER2 as an Effective Therapeutic Target in Cetuximab-Resistant Colorectal Cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Leto, S.M.; Sassi, F.; Catalano, I.; Torri, V.; Migliardi, G.; Zanella, E.R.; Throsby, M.; Bertotti, A.; Trusolino, L. Sustained Inhi-bition of HER3 and EGFR Is Necessary to Induce Regression of HER2-Amplified Gastrointestinal Carcinomas. Clin. Cancer Res. 2015, 21, 5519–5531. [Google Scholar] [CrossRef]
- Rubio-Viqueira, B.; Jimeno, A.; Cusatis, G.; Zhang, X.; Iacobuzio-Donahue, C.; Karikari, C.; Shi, C.; Danenberg, K.; Danenberg, P.V.; Kuramochi, H.; et al. An In vivo Platform for Translational Drug Development in Pancreatic Cancer. Clin. Cancer Res. 2006, 12, 4652–4661. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Bruckheimer, E.; Rajeshkumar, N.; Garrido-Laguna, I.; De Oliveira, E.; Rubio-Viqueira, B.; Strawn, S.; Wick, M.J.; Martell, J.; Sidransky, D. A Pilot Clinical Study of Treatment Guided by Personalized Tumorgrafts in Patients with Advanced Cancer. Mol. Cancer Ther. 2011, 10, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Stebbing, J.; Paz, K.; Schwartz, G.K.; Wexler, L.H.; Maki, R.G.; Pollock, R.E.; Morris, R.; Cohen, R.; Shankar, A.; Blackman, G.; et al. Patient-derived xenografts for individualized care in advanced sarcoma. Cancer 2014, 120, 2006–2015. [Google Scholar] [CrossRef] [PubMed]
- Damhofer, H.; Ebbing, E.A.; Steins, A.; Welling, L.; Tol, J.A.; Krishnadath, K.K.; Van Leusden, T.; Van De Vijver, M.J.; Besselink, M.G.; Busch, O.R.; et al. Establishment of patient-derived xenograft models and cell lines for malignancies of the upper gas-trointestinal tract. J. Transl. Med. 2015, 13, 115. [Google Scholar] [CrossRef]
- Sugaya, M.; Takenoyama, M.; Osaki, T.; Yasuda, M.; Nagashima, A.; Sugio, K.; Yasumoto, K. Establishment of 15 Cancer Cell Lines from Patients with Lung Cancer and the Potential Tools for Immunotherapy. Chest 2002, 122, 282–288. [Google Scholar] [CrossRef][Green Version]
- Meijer, T.G.; Naipal, K.A.; Jager, A.; van Gent, D.C. Ex vivo tumor culture systems for functional drug testing and therapy response prediction. Futur. Sci. OA 2017, 3, FSO190. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ory, V.; Chapman, S.; Yuan, H.; Albanese, C.; Kallakury, B.; Timofeeva, O.A.; Nealon, C.; Dakic, A.; Simic, V.; et al. ROCK Inhibitor and Feeder Cells Induce the Conditional Reprogramming of Epithelial Cells. Am. J. Pathol. 2012, 180, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Suprynowicz, F.A.; Upadhyay, G.; Krawczyk, E.; Kramer, S.C.; Hebert, J.D.; Liu, X.; Yuan, H.; Cheluvaraju, C.; Clapp, P.W.; Boucher, R.C., Jr.; et al. Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, 20035–20040. [Google Scholar] [CrossRef]
- Liu, X.; Krawczyk, E.; Suprynowicz, F.A.; Palechor-Ceron, N.; Yuan, H.; Dakic, A.; Simic, V.; Zheng, Y.-L.; Sripadhan, P.; Chen, C.; et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat. Protoc. 2017, 12, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Choudhury, S.; Wangsa, D.; Lescott, C.J.; Wilkins, D.J.; Sripadhan, P.; Liu, X.; Wangsa, D.; Ried, T.; Moskaluk, C.; et al. A multiplex preclinical model for adenoid cystic carcinoma of the salivary gland identifies regorafenib as a potential ther-apeutic drug. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Mahajan, A.S.; Sugita, B.; Duttargi, A.N.; Saenz, F.R.; Krawczyk, E.; McCutcheon, J.N.; Fonseca, A.S.; Kallakury, B.; Pohl-mann, P.; Gusev, Y.; et al. Genomic comparison of early-passage conditionally reprogrammed breast cancer cells to their cor-responding primary tumors. PLoS ONE 2017, 12, e0186190. [Google Scholar] [CrossRef]
- Timofeeva, O.A.; Palechor-Ceron, N.; Li, G.; Yuan, H.; Krawczyk, E.; Zhong, X.; Liu, G.; Upadhyay, G.; Dakic, A.; Yu, S.; et al. Conditionally reprogrammed normal and primary tumor prostate epithelial cells: A novel patient-derived cell model for studies of human prostate cancer. Oncotarget 2016, 8, 22741–22758. [Google Scholar] [CrossRef]
- Saeed, K.; Rahkama, V.; Eldfors, S.; Bychkov, D.; Mpindi, J.P.; Yadav, B.; Paavolainen, L.; Aittokallio, T.; Heckman, C.; Wennerberg, K.; et al. Comprehensive Drug Testing of Patient-derived Conditionally Reprogrammed Cells from Castra-tion-resistant Prostate Cancer. Eur. Urol. 2017, 71, 319–327. [Google Scholar] [CrossRef]
- Beglyarova, N.; Banina, E.; Zhou, Y.; Mukhamadeeva, R.; Andrianov, G.; Bobrov, E.; Lysenko, E.; Skobeleva, N.; Gabitova, L.; Restifo, D.; et al. Screening of Conditionally Reprogrammed Patient-Derived Carcinoma Cells Identifies ERCC3–MYC Inter-actions as a Target in Pancreatic Cancer. Clin. Cancer Res. 2016, 22, 6153–6163. [Google Scholar] [CrossRef]
- Li, Y.; Guo, D.; Zhang, Y.; Wang, L.; Sun, T.; Li, Z.; Zhang, X.; Wang, S.; Chen, Y.; Wu, A. Rapid screening for individualized chemotherapy optimization of colorectal cancer: A novel conditional reprogramming technology-based functional diagnostic assay. Transl. Oncol. 2020, 14, 100935. [Google Scholar] [CrossRef]
- Wu, M.; Hong, G.; Chen, Y.; Ye, L.; Zhang, K.; Cai, K.; Yang, H.; Long, X.; Gao, W.; Li, H. Personalized drug testing in a patient with non-small-cell lung cancer using cultured cancer cells from pleural effusion. J. Int. Med. Res. 2020, 48. [Google Scholar] [CrossRef] [PubMed]
- Correa, B.R.S.; Hu, J.; Penalva, L.; Schlegel, R.; Rimm, D.L.; Galante, P.A.F.; Agarwal, S. Patient-derived conditionally repro-grammed cells maintain intra-tumor genetic heterogeneity. Sci. Rep. 2018, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Krawczyk, E.; Blancato, J.; Albanese, C.; Zhou, D.; Wang, N.; Paul, S.; Alkhilaiwi, F.; Palechor-Ceron, N.; Dakic, A.; et al. HPV positive neuroendocrine cervical cancer cells are dependent on Myc but not E6/E7 viral oncogenes. Sci. Rep. 2017, 7, 45617. [Google Scholar] [CrossRef] [PubMed]
- Alamri, A.M.; Kang, K.; Groeneveld, S.; Wang, W.; Zhong, X.; Kallakury, B.; Hennighausen, L.; Liu, X.; Furth, P.A. Primary cancer cell culture: Mammary-optimized vs conditional reprogramming. Endocrine-Related Cancer 2016, 23, 535–554. [Google Scholar] [CrossRef]
- Borodovsky, A.; McQuiston, T.J.; Stetson, D.; Ahmed, A.; Whitston, D.; Zhang, J.; Grondine, M.; Lawson, D.; Challberg, S.S.; Zinda, M.; et al. Generation of stable PDX derived cell lines using conditional reprogramming. Mol. Cancer 2017, 16, 177. [Google Scholar] [CrossRef]
- Mondal, A.M.; Ma, A.-H.; Li, G.; Krawczyk, E.; Yuan, R.; Lu, J.; Schlegel, R.; Stamatakis, L.; Kowalczyk, K.J.; Philips, G.K.; et al. Fidelity of a PDX-CR model for bladder cancer. Biochem. Biophys. Res. Commun. 2019, 517, 49–56. [Google Scholar] [CrossRef]
- Choudhary, S.; Ramasundaram, P.; Dziopa, E.; Mannion, C.; Kissin, Y.; Tricoli, L.; Albanese, C.; Lee, W.; Zilberberg, J. Human ex vivo 3D bone model recapitulates osteocyte response to metastatic prostate cancer. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Palechor-Ceron, N.; Krawczyk, E.; Dakic, A.; Simic, V.; Yuan, H.; Blancato, J.; Wang, W.; Hubbard, F.; Zheng, Y.-L.; Dan, H.; et al. Conditional Reprogramming for Patient-Derived Cancer Models and Next-Generation Living Biobanks. Cells 2019, 8, 1327. [Google Scholar] [CrossRef]
- Luo, Y.; Ju, L.; Wang, G.; Chen, C.; Wang, Y.; Chen, L.; Zhang, Y.; Xiao, Y.; Wang, X. Comprehensive genomic profiling of urothelial carcinoma cell lines reveals hidden research bias and caveats. Clin. Transl. Med. 2020, 10, 294–296. [Google Scholar] [CrossRef]
- Mimoto, R.; Yogosawa, S.; Saijo, H.; Fushimi, A.; Nogi, H.; Asakura, T.; Yoshida, K.; Takeyama, H. Clinical implications of drug-screening assay for recurrent metastatic hormone receptor-positive, human epidermal receptor 2-negative breast cancer using conditionally reprogrammed cells. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Alamri, A.M.; Liu, X.; Blancato, J.K.; Haddad, B.R.; Wang, W.; Zhong, X.; Choudhary, S.; Krawczyk, E.; Kallakury, B.V.; Davidson, B.J.; et al. Expanding primary cells from mucoepidermoid and other salivary gland neoplasms for genetic and chemosensitivity testing. Dis. Model. Mech. 2018, 11, dmm031716. [Google Scholar] [CrossRef]
- Kurihara, M. Clinical Experience with UFT in Japan. Adv. Exp. Med. Biol. 1993, 339, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Tsushima, T.; Hironaka, S.; Boku, N.; Machida, N.; Yamazaki, K.; Yasui, H.; Taku, K.; Fukutomi, A.; Onozawa, Y. Safety and efficacy of S-1 monotherapy in elderly patients with advanced gastric cancer. Gastric Cancer 2010, 13, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; You, C.; Lu, X.; Zhang, L. Phase II trial of concurrent chemoradiotherapy with S-1 versus weekly cisplatin for lo-coregionally advanced nasopharyngeal carcinoma. Mol. Clin. Oncol. 2015, 3, 687–691. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 Stem Cells Build Crypt-Villus Structures in Vitro without a Mesenchymal Niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Driehuis, E.; Kretzschmar, K.; Clevers, H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat. Protoc. 2020, 15, 3380–3409, Erratum in Nat. Protoc. 2021, 16, 5739. [Google Scholar] [CrossRef]
- Drost, J.; Karthaus, W.R.; Gao, D.; Driehuis, E.; Sawyers, C.L.; Chen, Y.; Clevers, H. Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc. 2016, 11, 347–358. [Google Scholar] [CrossRef]
- Kopper, O.; de Witte, C.J.; Lõhmussaar, K.; Valle-Inclan, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med. 2019, 25, 838–849. [Google Scholar] [CrossRef]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Per-sonalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef]
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.M.J.; Kandoth, C.; Williams, A.B.; et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018, 173, 515–528.e17. [Google Scholar] [CrossRef]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef] [PubMed]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschenes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Xu, X.; Yang, L.; Zhu, J.; Wan, J.; Shen, L.; Xia, F.; Fu, G.; Deng, Y.; Pan, M.; et al. Patient-Derived Organoids Predict Chemoradiation Responses of Locally Advanced Rectal Cancer. Cell Stem Cell 2020, 26, 17–26.E16. [Google Scholar] [CrossRef]
- Friedman, A.A.; Letai, A.; Fisher, D.E.; Flaherty, K.T. Precision medicine for cancer with next-generation functional diagnostics. Nat. Rev. Cancer 2015, 15, 747–756. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, W.; Long, Y.; Liu, H.; Cheng, J.; Guo, L.; Li, R.; Meng, C.; Yu, S.; Zhao, Q.; et al. Characterization of drug responses of mini patient-derived xenografts in mice for predicting cancer patient clinical therapeutic response. Cancer Commun. 2018, 38, 60–72. [Google Scholar] [CrossRef]
- Zhan, M.; Yang, R.-M.; Wang, H.; He, M.; Chen, W.; Xu, S.-W.; Yang, L.-H.; Liu, Q.; Long, M.-M.; Wang, J. Guided chemo-therapy based on patient-derived mini-xenograft models improves survival of gallbladder carcinoma patients. Cancer Commun. 2018, 38, 48–49. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Chen, H.; Wen, D.; Mou, S.; Zhang, F.; Zheng, S. Personalized treatment based on mini patient-derived xenografts and WES/RNA sequencing in a patient with metastatic duodenal adenocarcinoma. Cancer Commun. 2018, 38, 54–57. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, J.; Li, K.; Wang, J.; Dai, Y.; Kang, Y. A Novel, Personalized Drug-Screening System for Platinum-Resistant Ovarian Cancer Patients: A Preliminary Clinical Report. Cancer Manag. Res. 2021, 13, 2849–2867. [Google Scholar] [CrossRef]
- Wang, X.; Sun, Y.; Xu, Y.; Wen, D.; An, N.; Leng, X.; Fu, G.; Lu, S.; Chen, Z. Mini-patient-derived xenograft assay based on microfluidic technology promises to be an effective tool for screening individualized chemotherapy regimens for advanced non-small cell lung cancer. Cell Biol. Int. 2021, 45, 1887–1896. [Google Scholar] [CrossRef]
- Li, C.; Sun, Y.-D.; Yu, G.-Y.; Cui, J.-R.; Lou, Z.; Zhang, H.; Huang, Y.; Bai, C.-G.; Deng, L.-L.; Liu, P.; et al. Integrated Omics of Metastatic Colorectal Cancer. Cancer Cell 2020, 38, 734–747.e9. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yuan, Z.; Zhang, Y.; Cui, Z.; Li, Y.; Hou, J.; Liu, X.; Liu, Z.; Shi, R.; Tian, Q.; et al. MiniPDX-guided postoperative anticancer treatment can effectively prolong the survival of patients with hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2020, 87, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, T.; Meng, Z.; Wang, L.; Li, M.; Chen, J.; Qin, T.; Yu, J.; Zhang, M.; Bie, Z.; et al. XPO1 inhibition synergizes with PARP1 inhibition in small cell lung cancer by targeting nuclear transport of FOXO3a. Cancer Lett. 2021, 503, 197–212. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Feature | PDX | MiniPDX | Organoid | Conditional Reprogramming | CDX | GEMM | Syngeneic Model |
|---|---|---|---|---|---|---|---|
| Success rate of initiation | Low | High | Medium | Medium | High | High | High |
| Humanization | Yes | Yes | Yes | Yes | Yes | No | No |
| Initial sample source | Fresh clinical specimens | Fresh clinical specimens | Fresh clinical specimens | Fresh clinical specimens | Human cancer cell line | - | Mouse cancer cell line |
| Medium-dependency | No | No | Yes | Yes | No | No | No |
| Administration approaches | Systemically | Systemically | Through medium | Through medium | Systemi-cally | System-ically | Systemica-lly |
| Numbers of animals needed | High | Low | - | - | High | High | High |
| Facility requirements | High | High | Low | Low | High | Medium | Medium |
| Cost | High | Medium | Low | Low | Medium to high | High | Medium to high |
| High-throughput drug screening | No | No | Yes | Yes | No | No | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, Y.; Xie, B.; Shen, H.C.; Wen, D. Translation Potential and Challenges of In Vitro and Murine Models in Cancer Clinic. Cells 2022, 11, 3868. https://doi.org/10.3390/cells11233868
Long Y, Xie B, Shen HC, Wen D. Translation Potential and Challenges of In Vitro and Murine Models in Cancer Clinic. Cells. 2022; 11(23):3868. https://doi.org/10.3390/cells11233868
Chicago/Turabian StyleLong, Yuan, Bin Xie, Hong C. Shen, and Danyi Wen. 2022. "Translation Potential and Challenges of In Vitro and Murine Models in Cancer Clinic" Cells 11, no. 23: 3868. https://doi.org/10.3390/cells11233868
APA StyleLong, Y., Xie, B., Shen, H. C., & Wen, D. (2022). Translation Potential and Challenges of In Vitro and Murine Models in Cancer Clinic. Cells, 11(23), 3868. https://doi.org/10.3390/cells11233868