
Peculiarities of the Acetylcholine Action on the Contractile Function of Cardiomyocytes from the Left and Right Atria in Rats

 ,
,  , , and
, , and 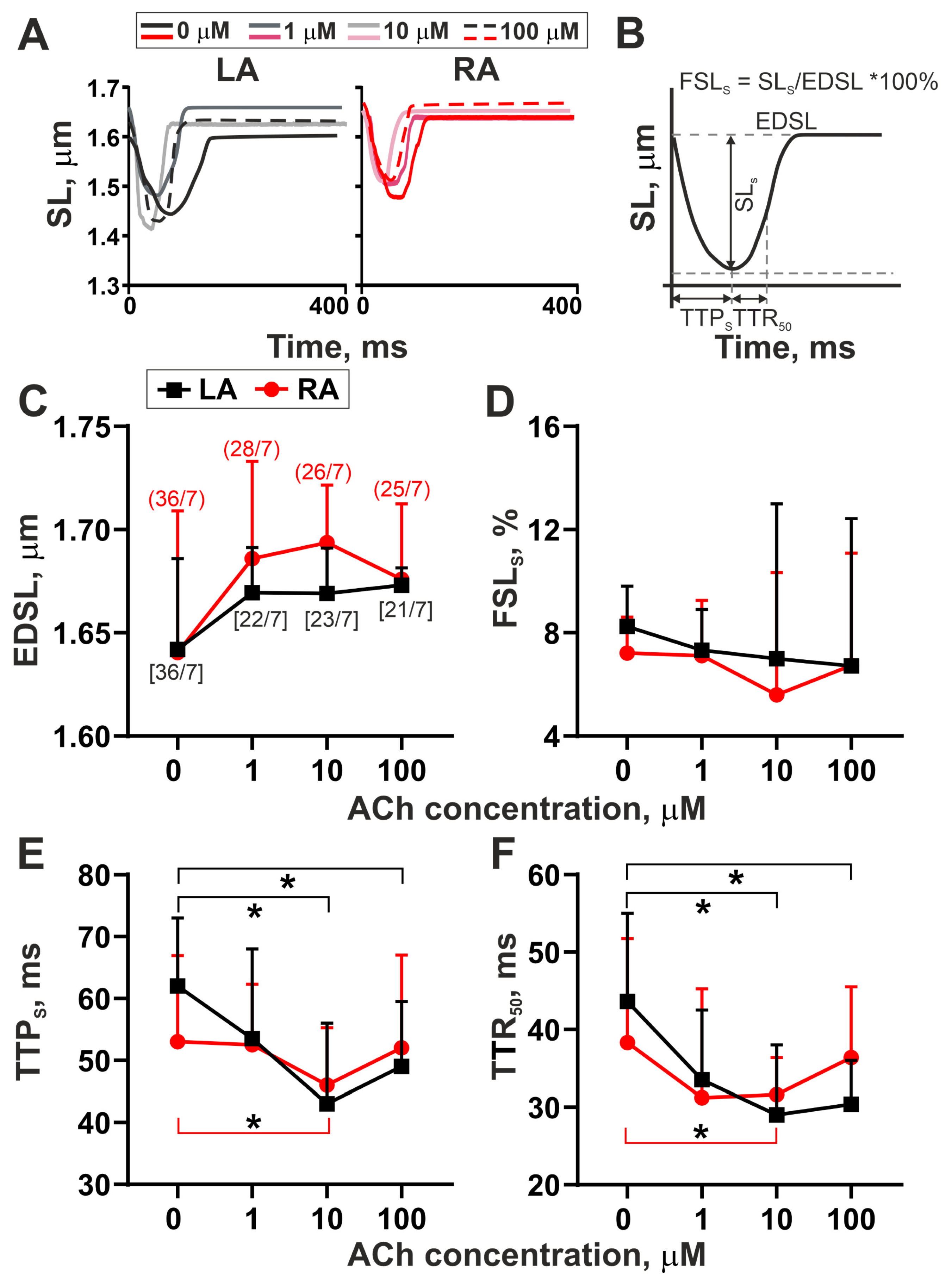{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Atrial Cardiomyocyte Isolation
2.2. Incubation of Atrial Cardiomyocytes with Acetylcholine
2.3. Measurements of Sarcomere Shortening–Relengthening in Single LA and RA Cardiomyocytes
2.4. Measurements of [Ca2+]i Transients in Single LA and RA Cardiomyocytes
2.5. Extraction of Sarcomeric Proteins and Phosphorylation Analysis
2.6. In Vitro Motility Assay
2.7. Statistical Analysis
3. Results
3.1. The Action of Acetylcholine on Sarcomere Shortening–Relengthening in Single LA and RA Cardiomyocytes
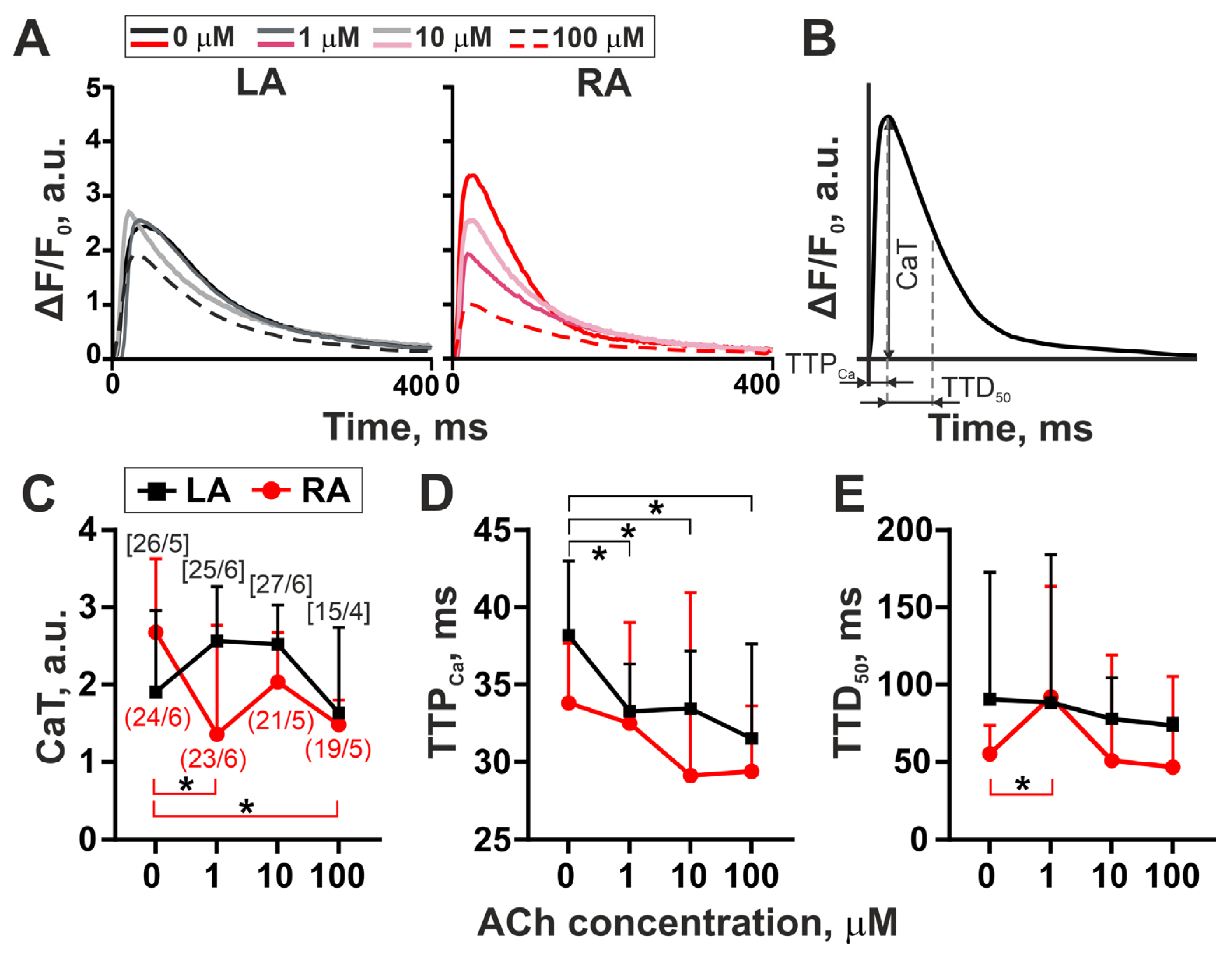
3.2. The Action of Acetylcholine on [Ca2+]i Transients in Single LA and RA Cardiomyocytes
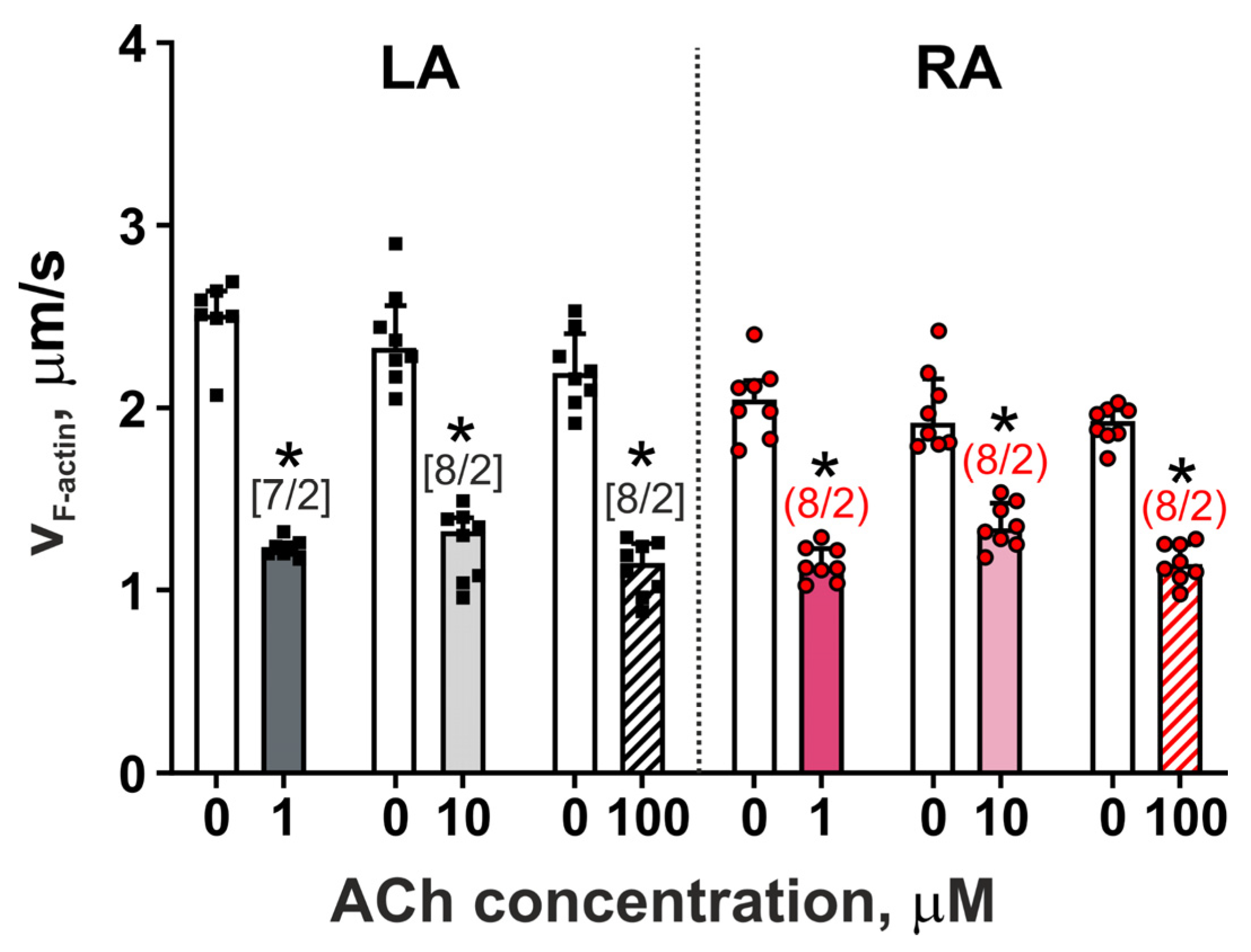
3.3. The Action of Acetylcholine on the Actin–Myosin Interaction in LA and RA Cardiomyocytes
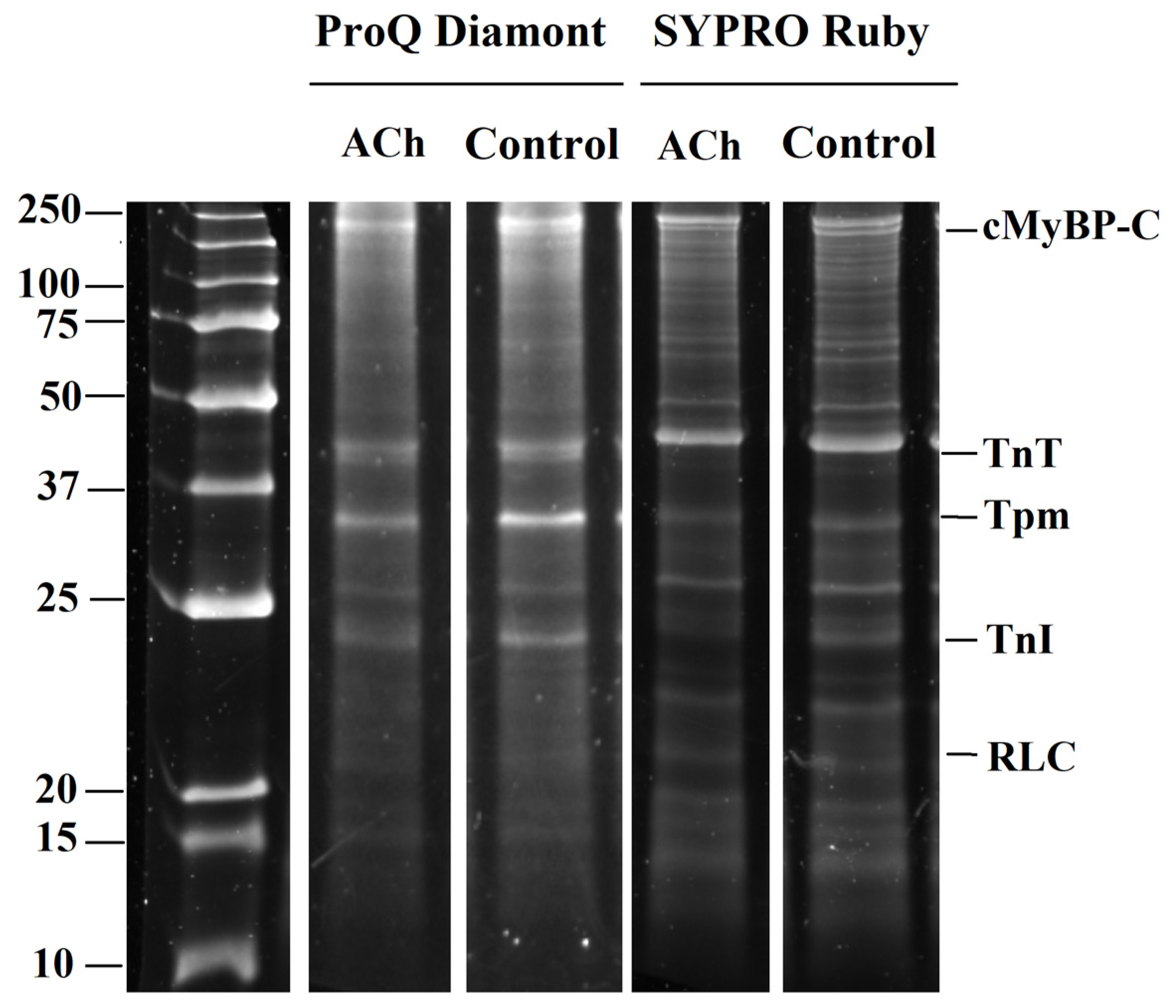
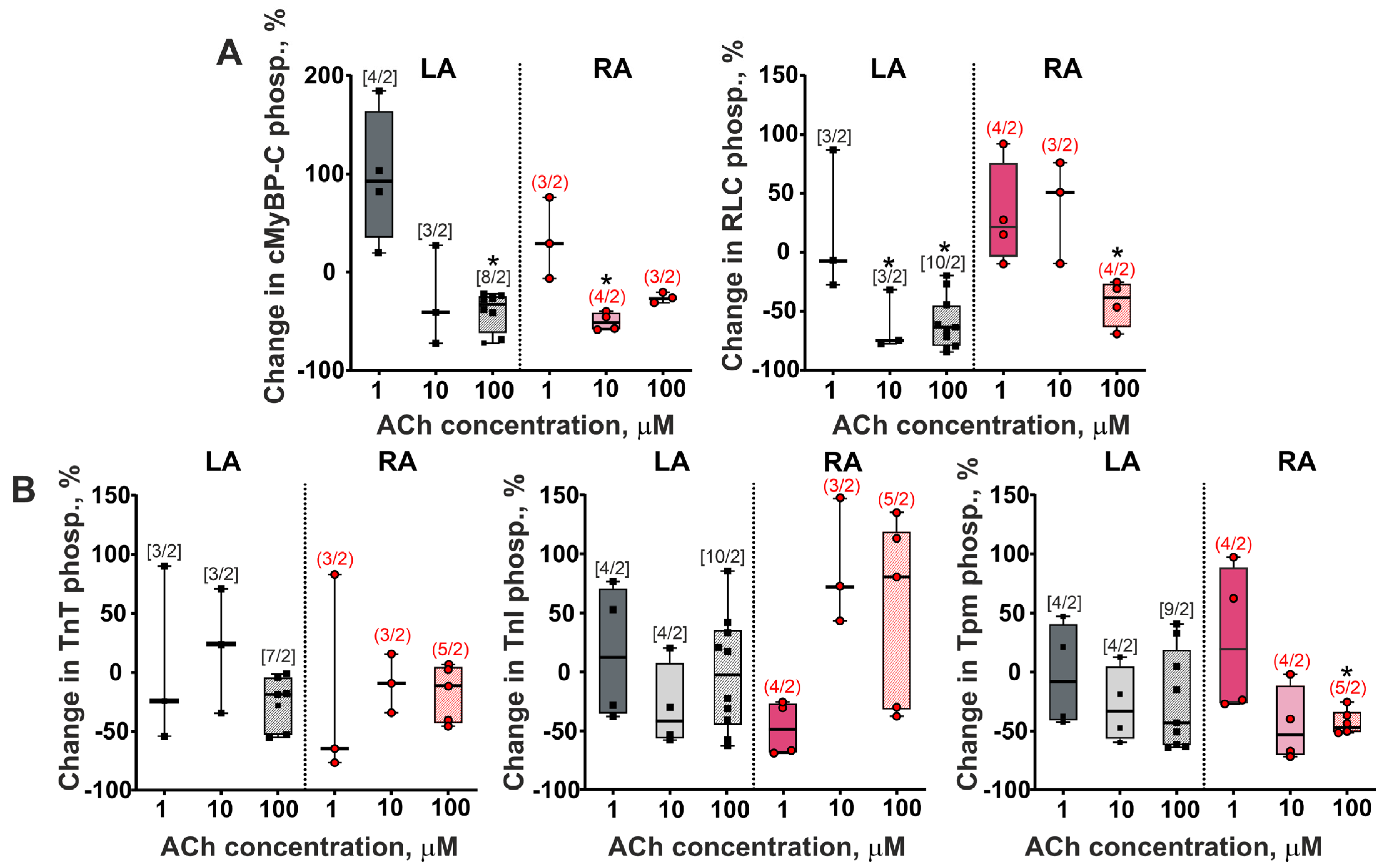
3.4. The Action of Acetylcholine on Sarcomeric Protein Phosphorylation in LA and RA Cardiomyocytes
4. Discussion
4.1. The Effects of ACh on Sarcomere Dynamics in Atrial Cardiomyocytes
4.2. Differing Sensitivity of LA and RA Cardiomyocytes to Acute ACh Application
5. Conclusions
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, P.-S.; Chen, L.S.; Fishbein, M.C.; Lin, S.-F.; Nattel, S. Role of the autonomic nervous system in atrial fibrillation: Pathophysiology and therapy. Circ. Res. 2014, 114, 1500–1515. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Cora, N.; Maning, J.; Brill, A.R.; Sizova, A. Signaling and function of cardiac autonomic nervous system receptors: Insights from the GPCR signalling universe. FEBS J. 2021, 288, 2645–2659. [Google Scholar] [CrossRef] [PubMed]
- Rana, O.R.; Schauerte, P.; Kluttig, R.; Schröder, J.W.; Koenen, R.R.; Weber, C.; Nolte, K.W.; Weis, J.; Hoffmann, R.; Marx, N. Acetylcholine as an age-dependent non-neuronal source in the heart. Auton. Neurosci. 2010, 156, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Kakinuma, Y.; Akiyama, T.; Sato, T. Cholinoceptive and cholinergic properties of cardiomyocytes involving an amplification mechanism for vagal efferent effects in sparsely innervated ventricular myocardium. FEBS J. 2009, 276, 5111–5125. [Google Scholar] [CrossRef] [PubMed]
- Zimerman, L.I.; Liberman, A.; Castro, R.; Ribeiro, J.P.; Nóbrega, A. Acute electrophysiologic consequences of pyridostigmine inhibition of cholinesterase in humans. Braz. J. Med. Biol. Res. 2010, 43, 211–216. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Verkerk, A.O.; Geuzebroek, G.S.; Veldkamp, M.W.; Wilders, R. Effects of acetylcholine and noradrenalin on action potentials of isolated rabbit sinoatrial and atrial myocytes. Front. Physiol. 2012, 3, 174. [Google Scholar] [CrossRef]
- Roy, A.; Fields, W.C.; Rocha-Resende, C.; Resende, R.R.; Guatimosim, S.; Prado, V.F.; Gros, R.; Prado, M.A. Cardiomyocyte-secreted acetylcholine is required for maintenance of homeostasis in the heart. FASEB J. 2013, 27, 5072–5082. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Akiyama, T.; Kawada, T.; Shishido, T.; Yamazaki, T.; Kamiya, A.; Mizuno, M.; Sano, S.; Sugimachi, M. In vivo direct monitoring of vagal acetylcholine release to the sinoatrial node. Auton. Neurosci. 2009, 148, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Schuessler, R.B.; Grayson, T.M.; Bromberg, B.I.; Cox, J.L.; Boineau, J.P. Cholinergically mediated tachyarrhythmias induced by a single extrastimulus in the isolated canine right atrium. Circ. Res. 1992, 71, 1254–1267. [Google Scholar] [CrossRef] [PubMed]
- Sharifov, O.F.; Fedorov, V.V.; Beloshapko, G.G.; Glukhov, A.V.; Yushmanova, A.V.; Rosenshtraukh, L.V. Roles of adrenergic and cholinergic stimulation in spontaneous atrial fibrillation in dogs. J. Am. Coll. Cardiol. 2004, 43, 483–490. [Google Scholar] [CrossRef]
- Bayer, J.D.; Boukens, B.J.; Krul, S.P.; Roney, C.H.; Driessen, A.H.; Berger, W.R.; Van den Berg, N.W.; Verkerk, A.O.; Vigmond, E.J.; Coronel, R. Acetylcholine delays atrial activation to facilitate atrial fibrillation. Front. Physiol. 2019, 10, 1105. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, R.K.; van der Does, W.F.; van Staveren, L.N.; Taverne, Y.J.; Bogers, A.J.; de Groot, N.M. Vagus nerve stimulation and atrial fibrillation: Revealing the paradox. Neuromodul. Technol. Neural Interface 2022, 25, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Linz, D.; Elliott, A.D.; Hohl, M.; Malik, V.; Schotten, U.; Dobrev, D.; Nattel, S.; Böhm, M.; Floras, J.; Lau, D.H. Role of autonomic nervous system in atrial fibrillation. Int. J. Cardiol. 2019, 287, 181–188. [Google Scholar] [CrossRef]
- Sarmast, F.; Kolli, A.; Zaitsev, A.; Parisian, K.; Dhamoon, A.S.; Guha, P.K.; Warren, M.; Anumonwo, J.M.; Taffet, S.M.; Berenfeld, O. Cholinergic atrial fibrillation: IK, ACh gradients determine unequal left/right atrial frequencies and rotor dynamics. Cardiovasc. Res. 2003, 59, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-X.; Zhao, Q.-Y.; Liang, J.-J.; Chen, H.; Yang, B.; Jiang, H.; Li, G.-S. Differential densities of muscarinic acetylcholine receptor and IK, ACh in canine supraventricular tissues and the effect of amiodarone on cholinergic atrial fibrillation and IK, ACh. Cardiology 2006, 106, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Zang, W.; Chen, L.; Yu, X.; Fang, P.; Lu, J.; Sun, Q. Comparison of effects of acetylcholine on electromechanical characteristics in guinea-pig atrium and ventricle. Exp. Physiol. 2005, 90, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Calloe, K.; Goodrow, R.; Olesen, S.-P.; Antzelevitch, C.; Cordeiro, J.M. Tissue-specific effects of acetylcholine in the canine heart. Am. J. Physiol.-Heart Circ. Physiol. 2013, 305, H66–H75. [Google Scholar] [CrossRef] [PubMed]
- Du, X.Y.; Schoemaker, R.G.; Bos, E.; Saxena, P.R. Characterization of the positive and negative inotropic effects of acetylcholine in the human myocardium. Eur. J. Pharmacol. 1995, 284, 119–127. [Google Scholar] [CrossRef]
- Mansour, M.; Mandapati, R.; Berenfeld, O.; Chen, J.; Samie, F.H.; Jalife, J. Left-to-right gradient of atrial frequencies during acute atrial fibrillation in the isolated sheep heart. Circulation 2001, 103, 2631–2636. [Google Scholar] [CrossRef]
- Dedkova, E.N.; Ji, X.; Wang, Y.G.; Blatter, L.A.; Lipsius, S.L. Signaling mechanisms that mediate nitric oxide production induced by acetylcholine exposure and withdrawal in cat atrial myocytes. Circ. Res. 2003, 93, 1233–1240. [Google Scholar] [CrossRef]
- Kuwabara, M.; Kakinuma, Y.; Ando, M.; Katare, R.G.; Yamasaki, F.; Doi, Y.; Sato, T. Nitric oxide stimulates vascular endothelial growth factor production in cardiomyocytes involved in angiogenesis. J. Physiol. Sci. 2006, 56, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Butova, X.; Myachina, T.; Khokhlova, A. A combined Langendorff-injection technique for simultaneous isolation of single cardiomyocytes from atria and ventricles of the rat heart. MethodsX 2021, 8, 101189. [Google Scholar] [CrossRef] [PubMed]
- Myachina, T.; Butova, K.; Lookin, O. Development and program implementation of an algorithm to estimate the mean sarcomere length of a cardiomyocyte. Biophysics 2019, 64, 732–737. [Google Scholar] [CrossRef]
- Margossian, S.S.; Lowey, S. Preparation of myosin and its subfragments from rabbit skeletal muscle. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1982; Volume 85, pp. 55–71. [Google Scholar]
- Pardee, J.D.; Spudich, J.A. Purification of muscle actin. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1982; Volume 85, pp. 164–181. [Google Scholar]
- Matyushenko, A.M.; Shchepkin, D.V.; Kopylova, G.V.; Popruga, K.E.; Artemova, N.V.; Pivovarova, A.V.; Bershitsky, S.Y.; Levitsky, D.I. Structural and functional effects of cardiomyopathy-causing mutations in the troponin T-binding region of cardiac tropomyosin. Biochemistry 2017, 56, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Mashanov, G.; Molloy, J. Automatic detection of single fluorophores in live cells. Biophys. J. 2007, 92, 2199–2211. [Google Scholar] [CrossRef] [PubMed]
- Vaseghi, M.; Shivkumar, K. The role of the autonomic nervous system in sudden cardiac death. Prog. Cardiovasc. Dis. 2008, 50, 404–419. [Google Scholar] [CrossRef] [PubMed]
- Trejo, J.; Brown, J.H. c-fos and c-jun are induced by muscarinic receptor activation of protein kinase C but are differentially regulated by intracellular calcium. J. Biol. Chem. 1991, 266, 7876–7882. [Google Scholar] [CrossRef] [PubMed]
- Boycott, H.E.; Nguyen, M.-N.; Vrellaku, B.; Gehmlich, K.; Robinson, P. Nitric oxide and mechano-electrical transduction in cardiomyocytes. Front. Physiol. 2020, 11, 606740. [Google Scholar] [CrossRef] [PubMed]
- Ganzinelli, S.; Joensen, L.; Borda, E.; Bernabeo, G.; Sterin-Borda, L. Mechanisms involved in the regulation of mRNA for M2 muscarinic acetylcholine receptors and endothelial and neuronal NO synthases in rat atria. Br. J. Pharmacol. 2007, 151, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Krieg, T.; Qin, Q.; Philipp, S.; Alexeyev, M.F.; Cohen, M.V.; Downey, J.M. Acetylcholine and bradykinin trigger preconditioning in the heart through a pathway that includes Akt and NOS. Am. J. Physiol.-Heart Circ. Physiol. 2004, 287, H2606–H2611. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cuello, F.; Wittig, I.; Lorenz, K.; Eaton, P. Oxidation of cardiac myofilament proteins: Priming for dysfunction? Mol. Asp. Med. 2018, 63, 47–58. [Google Scholar] [CrossRef]
- Toepfer, C.; Caorsi, V.; Kampourakis, T.; Sikkel, M.B.; West, T.G.; Leung, M.-C.; Al-Saud, S.A.; MacLeod, K.T.; Lyon, A.R.; Marston, S.B. Myosin regulatory light chain (RLC) phosphorylation change as a modulator of cardiac muscle contraction in disease. J. Biol. Chem. 2013, 288, 13446–13454. [Google Scholar] [CrossRef]
- Kumar, M.; Haghighi, K.; Kranias, E.G.; Sadayappan, S. Phosphorylation of cardiac myosin–binding protein-C contributes to calcium homeostasis. J. Biol. Chem. 2020, 295, 11275–11291. [Google Scholar] [CrossRef] [PubMed]
- Bokník, P.; Unkel, C.; Kirchhefer, U.; Kleideiter, U.; Klein-Wiele, O.; Knapp, J.; Linck, B.; Lüss, H.; Ulrich Müller, F.; Schmitz, W. Regional expression of phospholamban in the human heart. Cardiovasc. Res. 1999, 43, 67–76. [Google Scholar] [CrossRef]
- van Borren, M.M.; Verkerk, A.O.; Wilders, R.; Hajji, N.; Zegers, J.G.; Bourier, J.; Tan, H.L.; Verheijck, E.E.; Peters, S.L.; Alewijnse, A.E. Effects of muscarinic receptor stimulation on Ca2+ transient, cAMP production and pacemaker frequency of rabbit sinoatrial node cells. Basic Res. Cardiol. 2010, 105, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Sutanto, H.; Lyon, A.; Lumens, J.; Schotten, U.; Dobrev, D.; Heijman, J. Cardiomyocyte calcium handling in health and disease: Insights from in vitro and in silico studies. Prog. Biophys. Mol. Biol. 2020, 157, 54–75. [Google Scholar] [CrossRef]
- Karabina, A.; Kazmierczak, K.; Szczesna-Cordary, D.; Moore, J.R. Myosin regulatory light chain phosphorylation enhances cardiac β-myosin in vitro motility under load. Arch. Biochem. Biophys. 2015, 580, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Shiels, H.A.; White, E. The Frank–Starling mechanism in vertebrate cardiac myocytes. J. Exp. Biol. 2008, 211, 2005–2013. [Google Scholar] [CrossRef]
- Schuttler, D.; Bapat, A.; Kaab, S.; Lee, K.; Tomsits, P.; Clauss, S.; Hucker, W.J. Animal Models of Atrial Fibrillation. Circ. Res. 2020, 127, 91–110. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butova, X.; Myachina, T.; Simonova, R.; Kochurova, A.; Bozhko, Y.; Arkhipov, M.; Solovyova, O.; Kopylova, G.; Shchepkin, D.; Khokhlova, A. Peculiarities of the Acetylcholine Action on the Contractile Function of Cardiomyocytes from the Left and Right Atria in Rats. Cells 2022, 11, 3809. https://doi.org/10.3390/cells11233809
Butova X, Myachina T, Simonova R, Kochurova A, Bozhko Y, Arkhipov M, Solovyova O, Kopylova G, Shchepkin D, Khokhlova A. Peculiarities of the Acetylcholine Action on the Contractile Function of Cardiomyocytes from the Left and Right Atria in Rats. Cells. 2022; 11(23):3809. https://doi.org/10.3390/cells11233809
Chicago/Turabian StyleButova, Xenia, Tatiana Myachina, Raisa Simonova, Anastasia Kochurova, Yakov Bozhko, Michael Arkhipov, Olga Solovyova, Galina Kopylova, Daniil Shchepkin, and Anastasia Khokhlova. 2022. "Peculiarities of the Acetylcholine Action on the Contractile Function of Cardiomyocytes from the Left and Right Atria in Rats" Cells 11, no. 23: 3809. https://doi.org/10.3390/cells11233809
APA StyleButova, X., Myachina, T., Simonova, R., Kochurova, A., Bozhko, Y., Arkhipov, M., Solovyova, O., Kopylova, G., Shchepkin, D., & Khokhlova, A. (2022). Peculiarities of the Acetylcholine Action on the Contractile Function of Cardiomyocytes from the Left and Right Atria in Rats. Cells, 11(23), 3809. https://doi.org/10.3390/cells11233809