
Genomic Amplification of UBQLN4 Is a Prognostic and Treatment Resistance Factor

 , , ,
, , ,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Analysis of Public Datasets
2.2. Immunohistochemistry
2.3. DNA Copy Number Analysis
2.4. Statistical Evaluation of DNA Amplification Recurrence
2.5. CRISPR Screen Datasets
2.6. Identification of Common Essential Genes
2.7. Gene Set Enrichment Analysis
2.8. Drug Sensitivity Analysis
2.9. Identification of Candidate miRNAs That Bind to UBQLN4 mRNA
2.10. Biostatistics Analysis
3. Results
3.1. UBQLN4 Is Upregulated in Various Types of Cancer
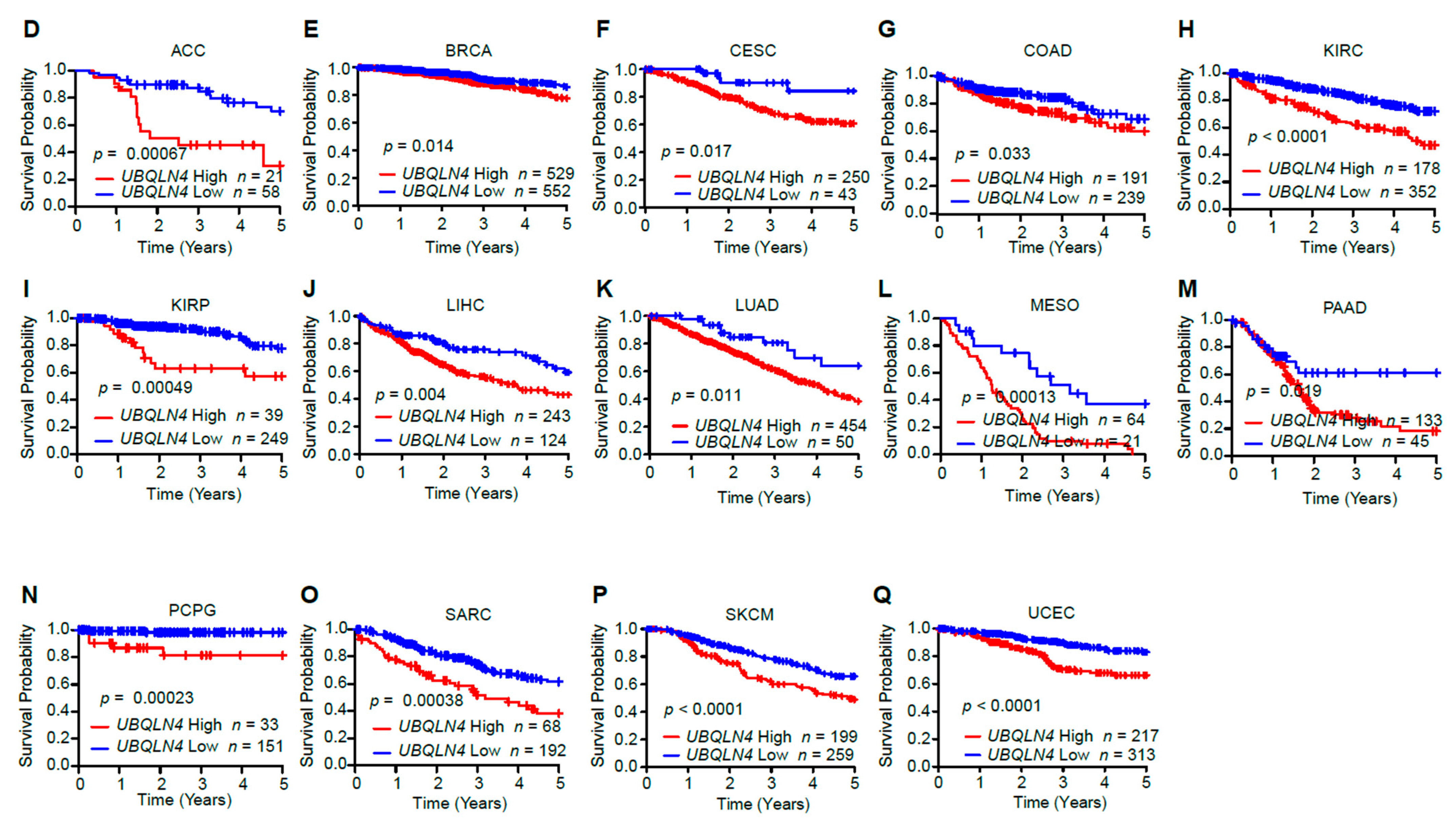
3.2. UBQLN4 Upregulation Is Associated with Poor Prognosis
3.3. MiRNA and DNA Methylation Do Not Significantly Control UBQLN4 Gene Expression
3.4. Copy Number Amplification in The Early Phases of Cancer Induces UBQLN4 Upregulation
3.5. UBQLN4 Is an Essential Gene for Pan-Cancer Cells
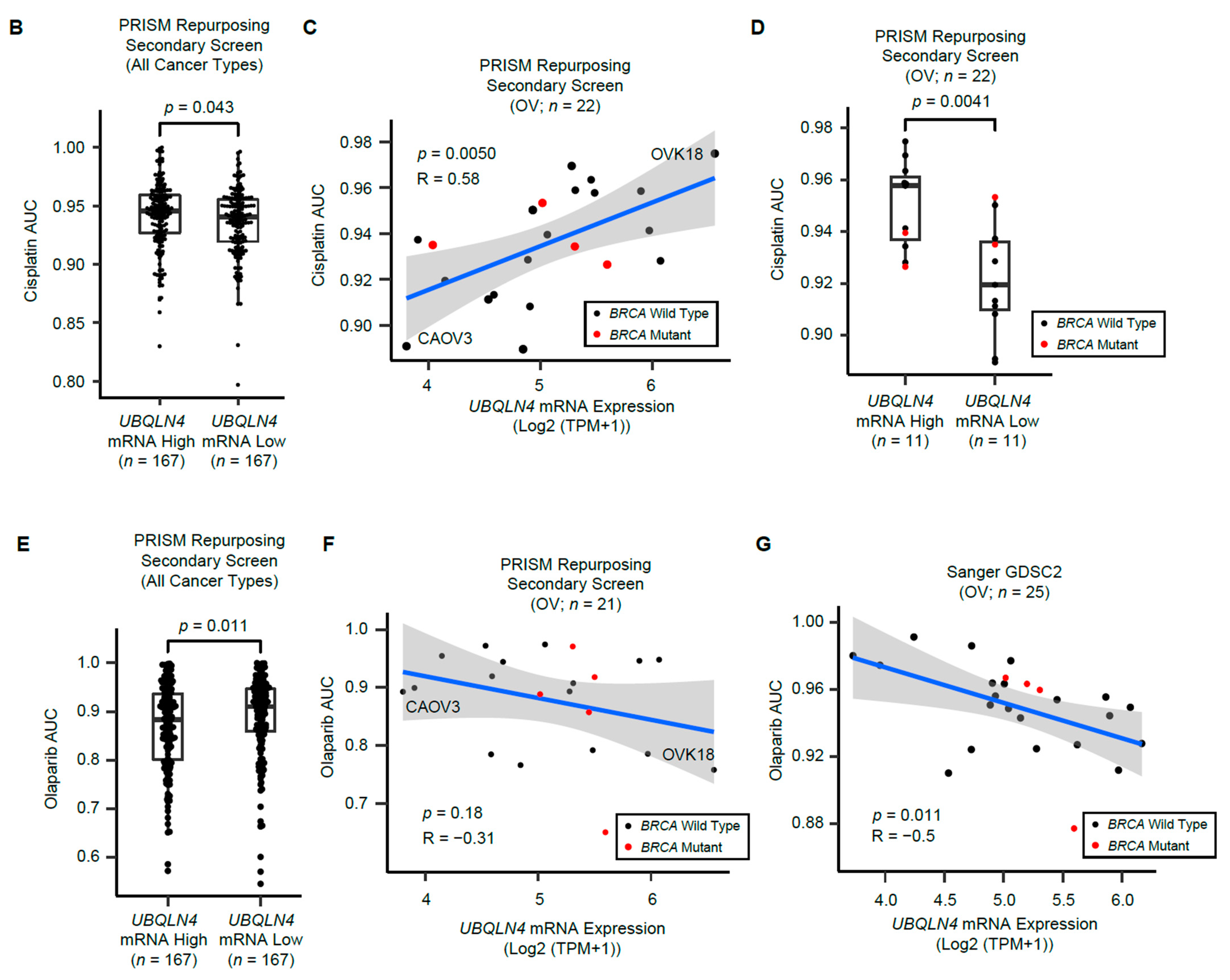
3.6. UBQLN4 mRNA Levels Predict Cisplatin and Olaparib Responses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ait Saada, A.; Lambert, S.A.E.; Carr, A.M. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 2018, 71, 135–147. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D'Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Dietlein, F.; Thelen, L.; Reinhardt, H.C. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 2014, 30, 326–339. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef]
- Marín, I. The ubiquilin gene family: Evolutionary patterns and functional insights. BMC Evol. Biol. 2014, 14, 63. [Google Scholar] [CrossRef]
- Jachimowicz, R.D.; Beleggia, F.; Isensee, J.; Velpula, B.B.; Goergens, J.; Bustos, M.A.; Doll, M.A.; Shenoy, A.; Checa-Rodriguez, C.; Wiederstein, J.L.; et al. UBQLN4 Represses Homologous Recombination and Is Overexpressed in Aggressive Tumors. Cell 2019, 176, 505–519. [Google Scholar] [CrossRef]
- Murakami, T.; Shoji, Y.; Nishi, T.; Chang, S.C.; Jachimowicz, R.D.; Hoshimoto, S.; Ono, S.; Shiloh, Y.; Takeuchi, H.; Kitagawa, Y.; et al. Regulation of MRE11A by UBQLN4 leads to cisplatin resistance in patients with esophageal squamous cell carcinoma. Mol. Oncol. 2021, 15, 1069–1087. [Google Scholar] [CrossRef]
- Shoji, Y.; Yokoe, T.; Kobayashi, Y.; Murakami, T.; Bostick, P.J.; Shiloh, Y.; Hoon, D.S.B.; Bustos, M.A. UBQLN4 promotes STING proteasomal degradation during cisplatin-induced DNA damage in triple-negative breast cancer. Clin. Transl. Med. 2022, 12, e985. [Google Scholar] [CrossRef]
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Kawahara, H. UBQLN4 recognizes mislocalized transmembrane domain proteins and targets these to proteasomal degradation. EMBO Rep. 2016, 17, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.D.; Riley, B.; Burright, E.N.; Duvick, L.A.; Zoghbi, H.Y.; Orr, H.T. Identification and characterization of an ataxin-1-interacting protein: A1Up, a ubiquitin-like nuclear protein. Hum. Mol. Genet. 2000, 9, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Arnott, D.; Brown, E.J. Ubiquilin4 is an adaptor protein that recruits Ubiquilin1 to the autophagy machinery. EMBO Rep. 2013, 14, 373–381. [Google Scholar] [CrossRef]
- Li, X.; Su, V.; Kurata, W.E.; Jin, C.; Lau, A.F. A novel connexin43-interacting protein, CIP75, which belongs to the UbL-UBA protein family, regulates the turnover of connexin43. J. Biol. Chem. 2008, 283, 5748–5759. [Google Scholar] [CrossRef]
- Huang, S.; Li, Y.; Yuan, X.; Zhao, M.; Wang, J.; Li, Y.; Li, Y.; Lin, H.; Zhang, Q.; Wang, W.; et al. The UbL-UBA Ubiquilin4 protein functions as a tumor suppressor in gastric cancer by p53-dependent and p53-independent regulation of p21. Cell Death Differ. 2019, 26, 516–530. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Kim, J.; Bowlby, R.; Mungall, A.J.; Robertson, A.G.; Odze, R.D.; Cherniack, A.D.; Shih, J.; Pedamallu, C.S.; Cibulskis, C.; Dunford, A.; et al. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Dempster, J.M.; Boyle, I.; Vazquez, F.; Root, D.E.; Boehm, J.S.; Hahn, W.C.; Tsherniak, A.; McFarland, J.M. Chronos: A cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome Biol. 2021, 22, 343. [Google Scholar] [CrossRef] [PubMed]
- Dempster, J.M.; Pacini, C.; Pantel, S.; Behan, F.M.; Green, T.; Krill-Burger, J.; Beaver, C.M.; Younger, S.T.; Zhivich, V.; Najgebauer, H.; et al. Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat. Commun. 2019, 10, 5817. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef]
- Jakubek, Y.A.; Chang, K.; Sivakumar, S.; Yu, Y.; Giordano, M.R.; Fowler, J.; Huff, C.D.; Kadara, H.; Vilar, E.; Scheet, P. Large-scale analysis of acquired chromosomal alterations in non-tumor samples from patients with cancer. Nat. Biotechnol. 2020, 38, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Niida, A.; Imoto, S.; Shimamura, T.; Miyano, S. Statistical model-based testing to evaluate the recurrence of genomic aberrations. Bioinformatics 2012, 28, i115–i120. [Google Scholar] [CrossRef]
- Saiki, R.; Momozawa, Y.; Nannya, Y.; Nakagawa, M.M.; Ochi, Y.; Yoshizato, T.; Terao, C.; Kuroda, Y.; Shiraishi, Y.; Chiba, K.; et al. Combined landscape of single-nucleotide variants and copy number alterations in clonal hematopoiesis. Nat. Med. 2021, 27, 1239–1249. [Google Scholar] [CrossRef]
- Madar, V.; Batista, S. FastLSU: A more practical approach for the Benjamini-Hochberg FDR controlling procedure for huge-scale testing problems. Bioinformatics 2016, 32, 1716–1723. [Google Scholar] [CrossRef]
- Pacini, C.; Dempster, J.M.; Boyle, I.; Goncalves, E.; Najgebauer, H.; Karakoc, E.; van der Meer, D.; Barthorpe, A.; Lightfoot, H.; Jaaks, P.; et al. Integrated cross-study datasets of genetic dependencies in cancer. Nat. Commun. 2021, 12, 1661. [Google Scholar] [CrossRef]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef]
- Mizuno, H.; Kitada, K.; Nakai, K.; Sarai, A. PrognoScan: A new database for meta-analysis of the prognostic value of genes. BMC Med. Genom. 2009, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xu, P.; Cui, G.; Xu, X.; Li, K.; Chen, X.; Bao, J. UBQLN4 promotes progression of HCC via activating wnt-β-catenin pathway and is regulated by miR-370. Cancer Cell Int. 2020, 20, 3. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Liang, Y.; Sun, G.; He, Q.; Qu, H.; Gao, P. UBQLN4 is activated by C/EBPβ and exerts oncogenic effects on colorectal cancer via the Wnt/β-catenin signaling pathway. Cell Death Discov. 2021, 7, 398. [Google Scholar] [CrossRef]
- Li, C.; Xu, J.; Wang, X.; Zhang, C.; Yu, Z.; Liu, J.; Tai, Z.; Luo, Z.; Yi, X.; Zhong, Z. Whole exome and transcriptome sequencing reveal clonal evolution and exhibit immune-related features in metastatic colorectal tumors. Cell Death Discov. 2021, 7, 222. [Google Scholar] [CrossRef] [PubMed]
- Amirouchene-Angelozzi, N.; Swanton, C.; Bardelli, A. Tumor Evolution as a Therapeutic Target. Cancer Discov. 2017, 7, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.D.; Liu, Y.; Bentivoglio, C.M.; Barlowe, C. Sel1p/Ubx2p participates in a distinct Cdc48p-dependent endoplasmic reticulum-associated degradation pathway. Traffic 2006, 7, 1213–1223. [Google Scholar] [CrossRef]
- Minami, R.; Hayakawa, A.; Kagawa, H.; Yanagi, Y.; Yokosawa, H.; Kawahara, H. BAG-6 is essential for selective elimination of defective proteasomal substrates. J. Cell Biol. 2010, 190, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Leznicki, P.; High, S. SGTA antagonizes BAG6-mediated protein triage. Proc. Natl. Acad. Sci. USA 2012, 109, 19214–19219. [Google Scholar] [CrossRef]
- Hirayama, S.; Sugihara, M.; Morito, D.; Iemura, S.I.; Natsume, T.; Murata, S.; Nagata, K. Nuclear export of ubiquitinated proteins via the UBIN-POST system. Proc. Natl. Acad. Sci. USA 2018, 115, E4199–E4208. [Google Scholar] [CrossRef]
- Sawada, G.; Niida, A.; Uchi, R.; Hirata, H.; Shimamura, T.; Suzuki, Y.; Shiraishi, Y.; Chiba, K.; Imoto, S.; Takahashi, Y.; et al. Genomic Landscape of Esophageal Squamous Cell Carcinoma in a Japanese Population. Gastroenterology 2016, 150, 1171–1182. [Google Scholar] [CrossRef]
- Wu, X.; Yang, J.; Na, T.; Zhang, K.; Davidoff, A.M.; Yuan, B.Z.; Wang, Y. RIG-I and IL-6 are negative-feedback regulators of STING induced by double-stranded DNA. PLoS ONE 2017, 12, e0182961. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Pan, R.; Ding, H.; Gu, L.; Yang, Y.; Li, C.; Xu, Y.; Hu, R.; Chen, H.; Zhang, X.; et al. UBQLN4 is an ATM substrate that stabilizes the anti-apoptotic proteins BCL2A1 and BCL2L10 in mesothelioma. Mol. Oncol. 2021, 15, 3738–3752. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Pujade-Lauraine, E. Olaparib as maintenance treatment for patients with platinum-sensitive relapsed ovarian cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919849753. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobayashi, Y.; Bustos, M.A.; Shoji, Y.; Jachimowicz, R.D.; Shiloh, Y.; Hoon, D.S.B. Genomic Amplification of UBQLN4 Is a Prognostic and Treatment Resistance Factor. Cells 2022, 11, 3311. https://doi.org/10.3390/cells11203311
Kobayashi Y, Bustos MA, Shoji Y, Jachimowicz RD, Shiloh Y, Hoon DSB. Genomic Amplification of UBQLN4 Is a Prognostic and Treatment Resistance Factor. Cells. 2022; 11(20):3311. https://doi.org/10.3390/cells11203311
Chicago/Turabian StyleKobayashi, Yuta, Matias A. Bustos, Yoshiaki Shoji, Ron D. Jachimowicz, Yosef Shiloh, and Dave S. B. Hoon. 2022. "Genomic Amplification of UBQLN4 Is a Prognostic and Treatment Resistance Factor" Cells 11, no. 20: 3311. https://doi.org/10.3390/cells11203311
APA StyleKobayashi, Y., Bustos, M. A., Shoji, Y., Jachimowicz, R. D., Shiloh, Y., & Hoon, D. S. B. (2022). Genomic Amplification of UBQLN4 Is a Prognostic and Treatment Resistance Factor. Cells, 11(20), 3311. https://doi.org/10.3390/cells11203311