

The Power of Gene Technologies: 1001 Ways to Create a Cell Model

 , , ,
, , , 
{kind=link}
{kind=link}
Abstract
1. Introduction
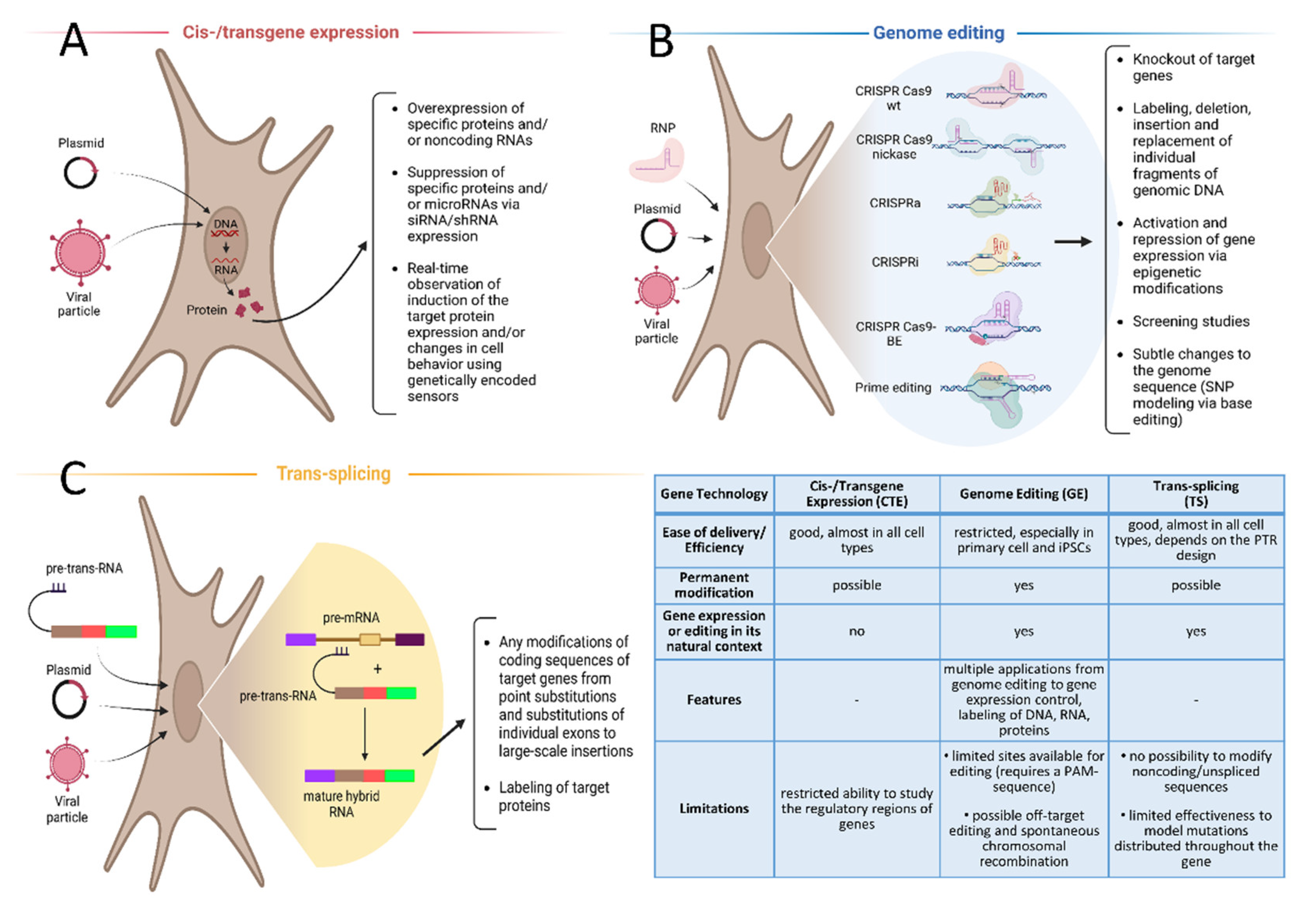
2. Cis-/Transgene Expression in Cell Lines, Genetically Encoded Sensors
3. Genome Editing
4. Trans-Splicing
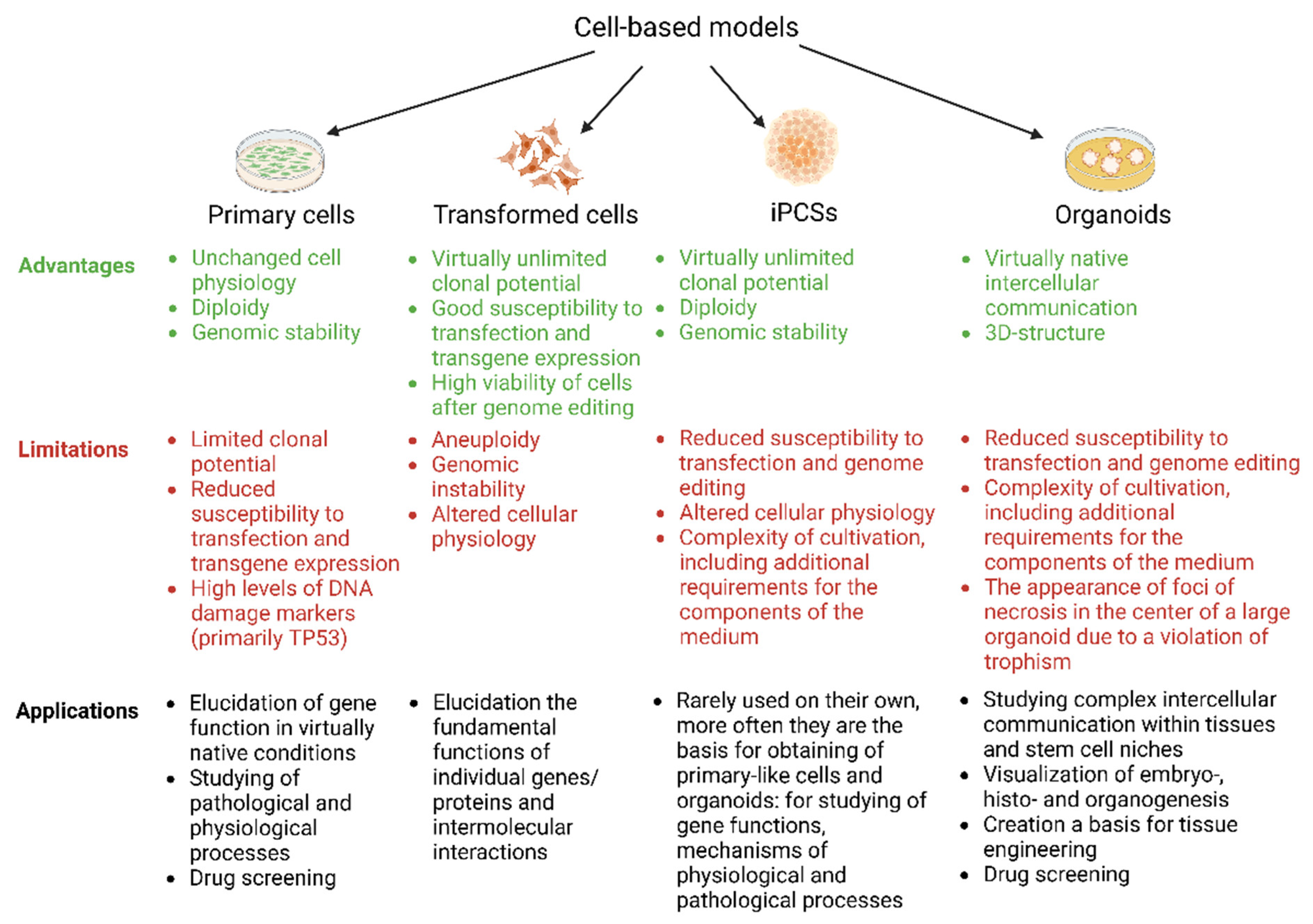
5. Creating Cell Models Based on Transformed, Primary, or Induced Pluripotent Cells
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- GBD 2019 Cancer Risk Factors Collaborators. The global burden of cancer attributable to risk factors, 2010-19: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 563–591. [Google Scholar] [CrossRef]
- Baker, R.E.; Mahmud, A.S.; Miller, I.F.; Rajeev, M.; Rasambainarivo, F.; Rice, B.L.; Takahashi, S.; Tatem, A.J.; Wagner, C.E.; Wang, L.F.; et al. Infectious disease in an era of global change. Nat. Rev. Microbiol. 2022, 20, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, U.; Ahmed, R.; Masoud, M.S.; Tariq, M.; Ashfaq, U.A.; Augustine, R.; Hasan, A. Stem cells basedin vitromodels: Trends and prospects in biomaterials cytotoxicity studies. Biomed. Mater. 2021, 16, 042003. [Google Scholar] [CrossRef]
- Varier, P.; Raju, G.; Madhusudanan, P.; Jerard, C.; Shankarappa, S.A. A Brief Review of In Vitro Models for Injury and Regeneration in the Peripheral Nervous System. Int. J. Mol. Sci. 2022, 23, 816. [Google Scholar] [CrossRef]
- Antunes, N.; Kundu, B.; Kundu, S.C.; Reis, R.L.; Correlo, V. In Vitro Cancer Models: A Closer Look at Limitations on Translation. Bioengineering 2022, 9, 166. [Google Scholar] [CrossRef]
- Bermudez-Brito, M.; Plaza-Díaz, J.; Fontana, L.; Muñoz-Quezada, S.; Gil, A. In vitro cell and tissue models for studying host-microbe interactions: A review. Br. J. Nutr. 2013, 109 (Suppl. S2), S27–S34. [Google Scholar] [CrossRef]
- Mollaki, V. Ethical Challenges in Organoid Use. BioTech 2021, 10, 12. [Google Scholar] [CrossRef]
- Lo, B.; Parham, L. Ethical issues in stem cell research. Endocr. Rev. 2009, 30, 204–213. [Google Scholar] [CrossRef]
- Haugabook, S.J.; Ferrer, M.; Ottinger, E.A. In vitro and in vivo translational models for rare liver diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1003–1018. [Google Scholar] [CrossRef]
- Li, Z.K.; Zhou, Q. Cellular models for disease exploring and drug screening. Protein Cell 2010, 1, 355–362. [Google Scholar] [CrossRef][Green Version]
- Chang, C.Y.; Ting, H.C.; Liu, C.A.; Su, H.L.; Chiou, T.W.; Lin, S.Z.; Harn, H.J.; Ho, T.J. Induced Pluripotent Stem Cell (iPSC)-Based Neurodegenerative Disease Models for Phenotype Recapitulation and Drug Screening. Molecules 2020, 25, 2000. [Google Scholar] [CrossRef]
- Segeritz, C.P.; Vallier, L. Basic Science Methods for Clinical Researchers; Academic Press: Cambridge, MA, USA, 2017; pp. 151–172. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef]
- Alnasser, S.M. Review on mechanistic strategy of gene therapy in the treatment of disease. Gene 2021, 769, 145246. [Google Scholar] [CrossRef]
- Zheng, C.; Baum, B.J. Evaluation of promoters for use in tissue-specific gene delivery. Methods Mol. Biol. 2008, 434, 205–219. [Google Scholar] [CrossRef][Green Version]
- Breger, L.; Wettergren, E.E.; Quintino, L.; Lundberg, C. Regulated Gene Therapy. Methods Mol. Biol. 2016, 1382, 57–66. [Google Scholar] [CrossRef]
- Schouten, H.J.; Krens, F.A.; Jacobsen, E. Cisgenic plants are similar to traditionally bred plants: International regulations for genetically modified organisms should be altered to exempt cisgenesis. EMBO Rep. 2006, 7, 750–753. [Google Scholar] [CrossRef]
- Haberle, V.; Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol. 2018, 19, 621–637. [Google Scholar] [CrossRef]
- Vanaja, A.; Yella, V.R. Delineation of the DNA Structural Features of Eukaryotic Core Promoter Classes. ACS Omega 2022, 7, 5657–5669. [Google Scholar] [CrossRef]
- Sloutskin, A.; Shir-Shapira, H.; Freiman, R.N.; Juven-Gershon, T. The Core Promoter Is a Regulatory Hub for Developmental Gene Expression. Front. Cell Dev. Biol. 2021, 9, 666508. [Google Scholar] [CrossRef]
- Qin, J.Y.; Zhang, L.; Clift, K.L.; Hulur, I.; Xiang, A.P.; Ren, B.Z.; Lahn, B.T. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PLoS ONE 2010, 5, e10611. [Google Scholar] [CrossRef]
- Athanasopoulos, T.; Munye, M.M.; Yáñez-Muñoz, R.J. Nonintegrating Gene Therapy Vectors. Hematol. Oncol. Clin. N. Am. 2017, 31, 753–770. [Google Scholar] [CrossRef]
- Chen, Y.H.; Keiser, M.S.; Davidson, B.L. Viral Vectors for Gene Transfer. Curr. Protoc. Mouse Biol. 2018, 8, e58. [Google Scholar] [CrossRef]
- Ono, C.; Okamoto, T.; Abe, T.; Matsuura, Y. Baculovirus as a Tool for Gene Delivery and Gene Therapy. Viruses 2018, 10, 510. [Google Scholar] [CrossRef]
- Pushparaj, P.N.; Aarthi, J.J.; Manikandan, J.; Kumar, S.D. siRNA, miRNA, and shRNA, in vivo applications. J. Dent. Res. 2008, 87, 992–1003. [Google Scholar] [CrossRef]
- Mansergh, F.C.; Daly, C.S.; Hurley, A.L.; Wride, M.A.; Hunter, S.M.; Evans, M.J. Gene expression profiles during early differentiation of mouse embryonic stem cells. BMC Dev. Biol. 2009, 9, 5. [Google Scholar] [CrossRef]
- Griesi-Oliveira, K.; Fogo, M.S.; Pinto, B.G.G.; Alves, A.Y.; Suzuki, A.M.; Morales, A.G.; Ezquina, S.; Sosa, O.J.; Sutton, G.J.; Sunaga-Franze, D.Y.; et al. Transcriptome of iPSC-derived neuronal cells reveals a module of co-expressed genes consistently associated with autism spectrum disorder. Mol. Psychiatry 2021, 26, 1589–1605. [Google Scholar] [CrossRef]
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Li, H.P.; Hung, Y.H.; Leu, Y.W.; Wu, W.H.; Wang, F.S.; Lee, K.D.; Chang, P.J.; Wu, C.S.; Lu, Y.J.; et al. Targeted methylation of CMV and E1A viral promoters. Biochem. Biophys. Res. Commun. 2010, 402, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Dahodwala, H.; Amenyah, S.D.; Nicoletti, S.; Henry, M.N.; Lees-Murdock, D.J.; Sharfstein, S.T. Evaluation of site-specific methylation of the CMV promoter and its role in CHO cell productivity of a recombinant monoclonal antibody. Antib. Ther. 2022, 5, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.Y.; Falcone, J.L.; Curci, S.; Hofer, A.M. Interrogating cyclic AMP signaling using optical approaches. Cell Calcium. 2017, 64, 47–56. [Google Scholar] [CrossRef]
- Chadney, O.M.T.; Blankvoort, S.; Grimstvedt, J.S.; Utz, A.; Kentros, C.G. Multiplexing viral approaches to the study of the neuronal circuits. J. Neurosci. Methods 2021, 357, 109142. [Google Scholar] [CrossRef]
- Fukuyasu, S.; Kayashima, H.; Moribayashi, A.; Matsuoka, S.; Nagasaki, A.; Okawa, H.; Yatani, H.; Saeki, M.; Egusa, H. Cell-Based Double-Screening Method to Identify a Reliable Candidate for Osteogenesis-Targeting Compounds. Biomedicines 2022, 10, 426. [Google Scholar] [CrossRef]
- Wada, K.; Taniguchi, A.; Kobayashi, J.; Yamato, M.; Okano, T. Live cells-based cytotoxic sensorchip fabricated in a microfluidic system. Biotechnol. Bioeng. 2008, 99, 1513–1517. [Google Scholar] [CrossRef]
- Sun, J.; Huang, J.; Bao, G.; Zheng, H.; Wang, C.; Wei, J.; Fu, Y.; Qiu, J.; Liao, Y.; Cai, J. MRI detection of the malignant transformation of stem cells through reporter gene expression driven by a tumor-specific promoter. Stem Cell Res. Ther. 2021, 12, 284. [Google Scholar] [CrossRef]
- Ouyang, M.; Sun, J.; Chien, S.; Wang, Y. Determination of hierarchical relationship of Src and Rac at subcellular locations with FRET biosensors. Proc. Natl. Acad. Sci. USA 2008, 105, 14353–14358. [Google Scholar] [CrossRef]
- Piatkevich, K.D.; Jung, E.E.; Straub, C.; Linghu, C.; Park, D.; Suk, H.J.; Hochbaum, D.R.; Goodwin, D.; Pnevmatikakis, E.; Pak, N.; et al. A robotic multidimensional directed evolution approach applied to fluorescent voltage reporters. Nat. Chem. Biol. 2018, 14, 352–360. [Google Scholar] [CrossRef]
- Nadler, D.C.; Morgan, S.A.; Flamholz, A.; Kortright, K.E.; Savage, D.F. Rapid construction of metabolite biosensors using domain-insertion profiling. Nat. Commun. 2016, 7, 12266. [Google Scholar] [CrossRef]
- Bilan, D.S.; Belousov, V.V. In Vivo Imaging of Hydrogen Peroxide with HyPer Probes. Antioxid. Redox Signal. 2018, 29, 569–584. [Google Scholar] [CrossRef]
- Lobas, M.A.; Tao, R.; Nagai, J.; Kronschläger, M.T.; Borden, P.M.; Marvin, J.S.; Looger, L.L.; Khakh, B.S. A genetically encoded single-wavelength sensor for imaging cytosolic and cell surface ATP. Nat. Commun. 2019, 10, 711. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, H.; Zhang, L.; Wu, R.; Chung, C.I.; Zhang, S.Q.; Torra, J.; Schepis, A.; Coughlin, S.R.; Kornberg, T.B.; et al. Visualizing Dynamics of Cell Signaling In Vivo with a Phase Separation-Based Kinase Reporter. Mol. Cell 2018, 69, 334–346.e4. [Google Scholar] [CrossRef] [PubMed]
- Nguyen le, X.T.; Raval, A.; Garcia, J.S.; Mitchell, B.S. Regulation of ribosomal gene expression in cancer. J. Cell Physiol. 2015, 230, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hu, Z.; Zhao, Y.; Huang, S.; He, X. Transcriptome-Wide Analysis Reveals the Landscape of Aberrant Alternative Splicing Events in Liver Cancer. Hepatology 2019, 69, 359–375. [Google Scholar] [CrossRef] [PubMed]
- Tyurin-Kuzmin, P.A.; Karagyaur, M.N.; Kulebyakin, K.Y.; Dyikanov, D.T.; Chechekhin, V.I.; Ivanova, A.M.; Skryabina, M.N.; Arbatskiy, M.S.; Sysoeva, V.Y.; Kalinina, N.I.; et al. Functional Heterogeneity of Protein Kinase A Activation in Multipotent Stromal Cells. Int. J. Mol. Sci. 2020, 21, 4442. [Google Scholar] [CrossRef]
- Yeh, H.W.; Hsu, E.C.; Lee, S.S.; Lang, Y.D.; Lin, Y.C.; Chang, C.Y.; Lee, S.Y.; Gu, D.L.; Shih, J.H.; Ho, C.M.; et al. PSPC1 mediates TGF-β1 autocrine signalling and Smad2/3 target switching to promote EMT, stemness and metastasis. Nat. Cell Biol. 2018, 20, 479–491. [Google Scholar] [CrossRef]
- Golden, R.J.; Chen, B.; Li, T.; Braun, J.; Manjunath, H.; Chen, X.; Wu, J.; Schmid, V.; Chang, T.C.; Kopp, F.; et al. An Argonaute phosphorylation cycle promotes microRNA-mediated silencing. Nature 2017, 542, 197–202. [Google Scholar] [CrossRef]
- Chen, Y.; Ou, Y.; Dong, J.; Yang, G.; Zeng, Z.; Liu, Y.; Liu, B.; Li, W.; He, X.; Lan, T. Osteopontin promotes collagen I synthesis in hepatic stellate cells by miRNA-129-5p inhibition. Exp. Cell Res. 2018, 362, 343–348. [Google Scholar] [CrossRef]
- Rysenkova, K.D.; Troyanovskiy, K.E.; Klimovich, P.S.; Bulyakova, T.R.; Shelomentseva, E.M.; Shmakova, A.A.; Tanygina, D.Y.; Ivashkina, O.I.; Anokhin, K.V.; Karagyaur, M.N.; et al. Identification of a novel small RNA encoded in the mouse urokinase receptor uPAR gene (Plaur) and its molecular target Mef2d. Front. Mol. Neurosci. 2022, 15, 865858. [Google Scholar] [CrossRef]
- Dansithong, W.; Paul, S.; Scoles, D.R.; Pulst, S.M.; Huynh, D.P. Generation of SNCA Cell Models Using Zinc Finger Nuclease (ZFN) Technology for Efficient High-Throughput Drug Screening. PLoS ONE 2015, 10, e0136930. [Google Scholar] [CrossRef]
- Malankhanova, T.B.; Malakhova, A.A.; Medvedev, S.P.; Zakian, S.M. Modern Genome Editing Technologies in Huntington’s Disease Research. J. Huntingtons Dis. 2017, 6, 19–31. [Google Scholar] [CrossRef]
- Chadwick, A.C.; Musunuru, K. Genome Editing for the Study of Cardiovascular Diseases. Curr. Cardiol. Rep. 2017, 19, 22. [Google Scholar] [CrossRef]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef]
- Kampmann, M. CRISPRi and CRISPRa Screens in Mammalian Cells for Precision Biology and Medicine. ACS Chem. Biol. 2018, 13, 406–416. [Google Scholar] [CrossRef]
- Syding, L.A.; Nickl, P.; Kasparek, P.; Sedlacek, R. CRISPR/Cas9 Epigenome Editing Potential for Rare Imprinting Diseases, A Review. Cells 2020, 9, 993. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 is a master regulator of tau uptake and spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]
- Datlinger, P.; Rendeiro, A.F.; Schmidl, C.; Krausgruber, T.; Traxler, P.; Klughammer, J.; Schuster, L.C.; Kuchler, A.; Alpar, D.; Bock, C. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 2017, 14, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Steinhart, Z.; Pavlovic, Z.; Chandrashekhar, M.; Hart, T.; Wang, X.; Zhang, X.; Robitaille, M.; Brown, K.R.; Jaksani, S.; Overmeer, R.; et al. Genome-wide CRISPR screens reveal a Wnt-FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat. Med. 2017, 23, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Lawson, K.A.; Sousa, C.M.; Zhang, X.; Kim, E.; Akthar, R.; Caumanns, J.J.; Yao, Y.; Mikolajewicz, N.; Ross, C.; Brown, K.R.; et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 2020, 586, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Crowther, M.D.; Dolton, G.; Legut, M.; Caillaud, M.E.; Lloyd, A.; Attaf, M.; Galloway, S.A.E.; Rius, C.; Farrell, C.P.; Szomolay, B.; et al. Genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat. Immunol. 2020, 21, 178–185. [Google Scholar] [CrossRef]
- Chang, J.; Tang, L.; Lei, H.; Zhang, X.G.; Zuo, Z.; Huang, W.; Fu, H. Effects of lentiviral infection of mesenchymal stem cells on the expression of octamer transcription factor 4. Mol. Med. Rep. 2014, 10, 2249–2254. [Google Scholar] [CrossRef]
- Liu, Z.; Wen, X.; Wang, H.; Zhou, J.; Zhao, M.; Lin, Q.; Wang, Y.; Li, J.; Li, D.; Du, Z.; et al. Molecular imaging of induced pluripotent stem cell immunogenicity with in vivo development in ischemic myocardium. PLoS ONE 2013, 8, e66369. [Google Scholar] [CrossRef]
- Fu, Y.W.; Dai, X.Y.; Wang, W.T.; Yang, Z.X.; Zhao, J.J.; Zhang, J.P.; Wen, W.; Zhang, F.; Oberg, K.C.; Zhang, L.; et al. Dynamics and competition of CRISPR-Cas9 ribonucleoproteins and AAV donor-mediated NHEJ, MMEJ and HDR editing. Nucleic Acids Res. 2021, 49, 969–985. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for Improving HDR Efficiency. Front. Genet. 2019, 9, 691. [Google Scholar] [CrossRef]
- Kass, E.M.; Helgadottir, H.R.; Chen, C.C.; Barbera, M.; Wang, R.; Westermark, U.K.; Ludwig, T.; Moynahan, M.E.; Jasin, M. Double-strand break repair by homologous recombination in primary mouse somatic cells requires BRCA1 but not the ATM kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 5564–5569. [Google Scholar] [CrossRef]
- Basalova, N.A.; Illarionova, M.Y.; Skryabina, M.N.; Vigovskiy, M.A.; Karagyaur, M.N.; Efimenko, A.Y. Potential of using CRISPR/Cas9 technology for knockout of non-coding RNA genes in human mesenchymal stromal cells. IJMS, 2022; under review. [Google Scholar]
- Das, A.T.; Tenenbaum, L.; Berkhout, B. Tet-On Systems for Doxycycline-inducible Gene Expression. Curr. Gene Ther. 2016, 16, 156–167. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.; Shan, L.; Han, L.; Ma, S.; Zhang, Y.; Hao, B.; Lin, Y.; Rong, Z. Gene activation in human cells using CRISPR/Cpf1-p300 and CRISPR/Cpf1-SunTag systems. Protein Cell 2018, 9, 380–383. [Google Scholar] [CrossRef]
- Karagyaur, M.N.; Dyikanov, D.T.; Tyurin-Kuzmin, P.A.; Dzhauari, S.S.; Skryabina, M.N.; Vigovskiy, M.A.; Primak, A.L.; Kalinina, N.I.; Tkachuk, V.A. A novel Cre/lox71-based system for inducible expression of recombinant proteins and genome editing. Cells 2022, 11, 2141. [Google Scholar] [CrossRef]
- Chang, H.; Yi, B.; Ma, R.; Zhang, X.; Zhao, H.; Xi, Y. CRISPR/Cas9, a novel genomic tool to knock down microRNA in vitro and in vivo. Sci. Rep. 2016, 6, 22312. [Google Scholar] [CrossRef]
- Jiang, Q.; Meng, X.; Meng, L.; Chang, N.; Xiong, J.; Cao, H.; Liang, Z. Small indels induced by CRISPR/Cas9 in the 5′ region of microRNA lead to its depletion and Drosha processing retardance. RNA Biol. 2014, 11, 1243–1249. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Z.; Wang, Y.; Guo, Y.; Xu, P.; Yuan, P.; Liu, Z.; He, Y.; Wei, W. Genome-wide screening for functional long noncoding RNAs in human cells by Cas9 targeting of splice sites. Nat. Biotechnol. 2018, 36, 1203–1210. [Google Scholar] [CrossRef]
- Aquino-Jarquin, G. Emerging Role of CRISPR/Cas9 Technology for MicroRNAs Editing in Cancer Research. Cancer Res. 2017, 77, 6812–6817. [Google Scholar] [CrossRef]
- Zhao, Y.; Dai, Z.; Liang, Y.; Yin, M.; Ma, K.; He, M.; Ouyang, H.; Teng, C.B. Sequence-specific inhibition of microRNA via CRISPR/CRISPRi system. Sci. Rep. 2014, 4, 3943. [Google Scholar] [CrossRef]
- Bowling, S.; Sritharan, D.; Osorio, F.G.; Nguyen, M.; Cheung, P.; Rodriguez-Fraticelli, A.; Patel, S.; Yuan, W.C.; Fujiwara, Y.; Li, B.E.; et al. An Engineered CRISPR-Cas9 Mouse Line for Simultaneous Readout of Lineage Histories and Gene Expression Profiles in Single Cells. Cell 2020, 181, 1410–1422. [Google Scholar] [CrossRef]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Izpisua Belmonte, J.C. In vivo genome editing via the HITI method as a tool for gene therapy. J. Hum. Genet. 2018, 63, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.J.; Saee-Marand, M.; Nyström, N.N.; Evans, M.M.; Chen, Y.; Martinez, F.M.; Hamilton, A.M.; Ronald, J.A. Safe harbor-targeted CRISPR-Cas9 homology-independent targeted integration for multimodality reporter gene-based cell tracking. Sci. Adv. 2021, 7, eabc3791. [Google Scholar] [CrossRef] [PubMed]
- Gray, D.H.; Santos, J.; Keir, A.G.; Villegas, I.; Maddock, S.; Trope, E.C.; Long, J.D.; Kuo, C.Y. A comparison of DNA repair pathways to achieve a site-specific gene modification of the Bruton’s tyrosine kinase gene. Mol. Ther. Nucleic Acids 2021, 27, 505–516. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Tyurin-Kuzmin, P.A.; Karagyaur, M.N.; Rubtsov, Y.P.; Dyikanov, D.T.; Vasiliev, P.A.; Vorotnikov, A.V. CRISPR/Cas9-mediated modification of the extreme C-terminus impairs PDGF-stimulated activity of Duox2. Biol. Chem. 2018, 399, 437–446. [Google Scholar] [CrossRef]
- Richter, M.F.; Zhao, K.T.; Eton, E.; Lapinaite, A.; Newby, G.A.; Thuronyi, B.W.; Wilson, C.; Koblan, L.W.; Zeng, J.; Bauer, D.E.; et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020, 38, 883–891. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Liu, Y.; Yang, B.; Wang, X.; Wei, J.; Lu, Z.; Zhang, Y.; Wu, J.; Huang, X.; et al. Base editing with a Cpf1-cytidine deaminase fusion. Nat. Biotechnol. 2018, 36, 324–327. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.; Li, Z.; Peterson, R.T.; Yeh, J.R. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015, 523, 481–485. [Google Scholar] [CrossRef]
- Nishimasu, H.; Shi, X.; Ishiguro, S.; Gao, L.; Hirano, S.; Okazaki, S.; Noda, T.; Abudayyeh, O.O.; Gootenberg, J.S.; Mori, H.; et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 2018, 361, 1259–1262. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Liu, P.; Liang, S.Q.; Zheng, C.; Mintzer, E.; Zhao, Y.G.; Ponnienselvan, K.; Mir, A.; Sontheimer, E.J.; Gao, G.; Flotte, T.R.; et al. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nat. Commun. 2021, 12, 2121. [Google Scholar] [CrossRef]
- Smirnikhina, S.A. Prime Editing: Making the Move to Prime Time. CRISPR J. 2020, 3, 319–321. [Google Scholar] [CrossRef]
- Li, X.; Zhou, L.; Gao, B.Q.; Li, G.; Wang, X.; Wang, Y.; Wei, J.; Han, W.; Wang, Z.; Li, J.; et al. Highly efficient prime editing by introducing same-sense mutations in pegRNA or stabilizing its structure. Nat. Commun. 2022, 13, 1669. [Google Scholar] [CrossRef]
- Chen, P.J.; Hussmann, J.A.; Yan, J.; Knipping, F.; Ravisankar, P.; Chen, P.F.; Chen, C.; Nelson, J.W.; Newby, G.A.; Sahin, M.; et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 2021, 184, 5635–5652.e29. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Gao, X.D.; Podracky, C.J.; Nelson, A.T.; Koblan, L.W.; Raguram, A.; Levy, J.M.; Mercer, J.A.M.; Liu, D.R. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat. Biotechnol. 2022, 40, 731–740. [Google Scholar] [CrossRef]
- Nakamura, M.; Gao, Y.; Dominguez, A.A.; Qi, L.S. CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 2021, 23, 11–22. [Google Scholar] [CrossRef]
- Goell, J.H.; Hilton, I.B. CRISPR/Cas-Based Epigenome Editing: Advances, Applications, and Clinical Utility. Trends Biotechnol. 2021, 39, 678–691. [Google Scholar] [CrossRef]
- Wang, H.; Xu, X.; Nguyen, C.M.; Liu, Y.; Gao, Y.; Lin, X.; Daley, T.; Kipniss, N.H.; La Russa, M.; Qi, L.S. CRISPR-Mediated Programmable 3D Genome Positioning and Nuclear Organization. Cell 2018, 175, 1405–1417.e14. [Google Scholar] [CrossRef]
- Schultzhaus, Z.; Wang, Z.; Stenger, D. CRISPR-based enrichment strategies for targeted sequencing. Biotechnol. Adv. 2021, 46, 107672. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Li, J.; Zhu, X.; Wang, X.; Fan, L.; Sun, W.; Liao, L.; Zhang, J.; Li, X.; Ye, J.; et al. CRISPR-assisted detection of RNA-protein interactions in living cells. Nat. Methods 2020, 17, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Sharma, A.R.; Bhattacharya, M.; Lee, S.S.; Chakraborty, C. CRISPR-Cas9, A Preclinical and Clinical Perspective for the Treatment of Human Diseases. Mol. Ther. 2021, 29, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Sutton, R.E.; Boothroyd, J.C. Evidence for trans splicing in trypanosomes. Cell 1986, 47, 527–535. [Google Scholar] [CrossRef]
- Michaeli, S. Trans-splicing in trypanosomes, machinery and its impact on the parasite transcriptome. Future Microbiol. 2011, 6, 459–474. [Google Scholar] [CrossRef]
- Flouriot, G.; Brand, H.; Seraphin, B.; Gannon, F. Natural trans-spliced mRNAs are generated from the human estrogen receptor-alpha (hER alpha) gene. J. Biol. Chem. 2002, 277, 26244–26251. [Google Scholar] [CrossRef]
- Hong, E.M.; Ingemarsdotter, C.K.; Lever, A.M.L. Therapeutic applications of trans-splicing. Br. Med. Bull. 2020, 136, 4–20. [Google Scholar] [CrossRef]
- Azibani, F.; Brull, A.; Arandel, L.; Beuvin, M.; Nelson, I.; Jollet, A.; Ziat, E.; Prudhon, B.; Benkhelifa-Ziyyat, S.; Bitoun, M.; et al. Gene Therapy via Trans-Splicing for LMNA-Related Congenital Muscular Dystrophy. Mol. Ther. Nucleic Acids 2018, 10, 376–386. [Google Scholar] [CrossRef]
- Gruber, C.; Koller, U.; Murauer, E.M.; Hainzl, S.; Hüttner, C.; Kocher, T.; South, A.P.; Hintner, H.; Bauer, J.W. The design and optimization of RNA trans-splicing molecules for skin cancer therapy. Mol. Oncol. 2013, 7, 1056–1068. [Google Scholar] [CrossRef]
- Dooley, S.J.; McDougald, D.S.; Fisher, K.J.; Bennicelli, J.L.; Mitchell, L.G.; Bennett, J. Spliceosome-Mediated Pre-mRNA trans-Splicing Can Repair CEP290 mRNA. Mol. Ther. Nucleic Acids 2018, 12, 294–308. [Google Scholar] [CrossRef]
- Gupta, S.K.; Jea, J.D.; Yen, L. Validating Gene Fusion as the Source of Chimeric RNAs. Methods Mol. Biol. 2020, 2079, 187–207. [Google Scholar] [CrossRef]
- Havens, M.A.; Duelli, D.M.; Hastings, M.L. Targeting RNA splicing for disease therapy. Wiley Interdiscip. Rev. RNA 2013, 4, 247–266. [Google Scholar] [CrossRef]
- Poddar, S.; Loh, P.S.; Ooi, Z.H.; Osman, F.; Eul, J.; Patzel, V. RNA Structure Design Improves Activity and Specificity of trans-Splicing-Triggered Cell Death in a Suicide Gene Therapy Approach. Mol. Ther. Nucleic Acids 2018, 11, 41–56. [Google Scholar] [CrossRef]
- Hannon, G.J.; Maroney, P.A.; Yu, Y.T.; Hannon, G.E.; Nilsen, T.W. Interaction of U6 snRNA with a sequence required for function of the nematode SL RNA in trans-splicing. Science 1992, 258, 1775–1780. [Google Scholar] [CrossRef]
- Landry, J.J.; Pyl, P.T.; Rausch, T.; Zichner, T.; Tekkedil, M.M.; Stütz, A.M.; Jauch, A.; Aiyar, R.S.; Pau, G.; Delhomme, N.; et al. The genomic and transcriptomic landscape of a HeLa cell line. G3 2013, 3, 1213–1224. [Google Scholar] [CrossRef]
- Stepanenko, A.A.; Dmitrenko, V.V. HEK293 in cell biology and cancer research, phenotype, karyotype, tumorigenicity, and stress-induced genome-phenotype evolution. Gene 2015, 569, 182–190. [Google Scholar] [CrossRef]
- Kulebyakin, K.; Tyurin-Kuzmin, P.; Efimenko, A.; Voloshin, N.; Kartoshkin, A.; Karagyaur, M.; Grigorieva, O.; Novoseletskaya, E.; Sysoeva, V.; Makarevich, P.; et al. Decreased Insulin Sensitivity in Telomerase-Immortalized Mesenchymal Stem Cells Affects Efficacy and Outcome of Adipogenic Differentiation in vitro. Front. Cell Dev. Biol. 2022, 9, 1–11. [Google Scholar] [CrossRef]
- Dyikanov, D.T.; Vasiluev, P.A.; Rysenkova, K.D.; Aleksandrushkina, N.A.; Tyurin-Kuzmin, P.A.; Kulebyakin, K.Y.; Rubtsov, Y.P.; Shmakova, A.A.; Evseeva, M.N.; Balatskiy, A.V.; et al. Optimization of CRISPR/Cas9 Technology to Knock Out Genes of Interest in Aneuploid Cell Lines. Tissue Eng. Part C Methods 2019, 25, 168–175. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef]
- Xue, C.; Greene, E.C. DNA Repair Pathway Choices in CRISPR-Cas9-Mediated Genome Editing. Trends Genet. 2021, 37, 639–656. [Google Scholar] [CrossRef]
- Seki, A.; Rutz, S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J. Exp. Med. 2018, 215, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shen, J.; Li, D.; Cheng, Y. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics 2021, 11, 614–648. [Google Scholar] [CrossRef] [PubMed]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Lee, C.; Bonifant, C.L.; Ressom, H.; Waldman, T. Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol. Cell Biol. 2007, 27, 662–677. [Google Scholar] [CrossRef]
- Ben Jehuda, R.; Shemer, Y.; Binah, O. Genome Editing in Induced Pluripotent Stem Cells using CRISPR/Cas9. Stem Cell Rev. Rep. 2018, 14, 323–336. [Google Scholar] [CrossRef]
- Petazzi, P.; Menéndez, P.; Sevilla, A. CRISPR/Cas9-Mediated Gene Knockout and Knockin Human iPSCs. Methods Mol. Biol. 2022, 2454, 559–574. [Google Scholar] [CrossRef]
- Karagyaur, M.N.; Rubtsov, Y.P.; Vasiliev, P.A.; Tkachuk, V.A. Practical Recommendations for Improving Efficiency and Accuracy of the CRISPR/Cas9 Genome Editing System. Biochemistry 2018, 83, 629–642. [Google Scholar] [CrossRef]
- Turhan, A.G.; Hwang, J.W.; Chaker, D.; Tasteyre, A.; Latsis, T.; Griscelli, F.; Desterke, C.; Bennaceur-Griscelli, A. iPSC-Derived Organoids as Therapeutic Models in Regenerative Medicine and Oncology. Front. Med. 2021, 8, 728543. [Google Scholar] [CrossRef]
- Nicholson, M.W.; Ting, C.Y.; Chan, D.Z.H.; Cheng, Y.C.; Lee, Y.C.; Hsu, C.C.; Huang, C.Y.; Hsieh, P.C.H. Utility of iPSC-Derived Cells for Disease Modeling, Drug Development, and Cell Therapy. Cells 2022, 11, 1853. [Google Scholar] [CrossRef]
- Gurwitz, D. Human iPSC-derived neurons and lymphoblastoid cells for personalized medicine research in neuropsychiatric disorders. Dialogues Clin. Neurosci. 2016, 18, 267–276. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Boucherit, N.; Gorvel, L.; Olive, D. 3D Tumor Models and Their Use for the Testing of Immunotherapies. Front. Immunol. 2020, 11, 603640. [Google Scholar] [CrossRef]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef]
- Gaebler, A.M.; Hennig, A.; Buczolich, K.; Weitz, J.; Welsch, T.; Stange, D.E.; Pape, K. Universal and Efficient Electroporation Protocol for Genetic Engineering of Gastrointestinal Organoids. J. Vis. Exp. 2020, 156, e60704. [Google Scholar] [CrossRef]
- Maru, Y.; Orihashi, K.; Hippo, Y. Lentivirus-Based Stable Gene Delivery into Intestinal Organoids. Methods Mol. Biol. 2016, 1422, 13–21. [Google Scholar] [CrossRef]
- Shiri, Z.; Simorgh, S.; Naderi, S.; Baharvand, H. Optogenetics in the Era of Cerebral Organoids. Trends Biotechnol. 2019, 37, 1282–1294. [Google Scholar] [CrossRef]
- Shnaider, T.A.; Pristyazhnyuk, I.E. CLARITY and Light-Sheet microscopy sample preparation in application to human cerebral organoids. Vavilovskii Zhurnal Genet. Sel. 2021, 25, 889–895. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karagyaur, M.; Primak, A.; Efimenko, A.; Skryabina, M.; Tkachuk, V. The Power of Gene Technologies: 1001 Ways to Create a Cell Model. Cells 2022, 11, 3235. https://doi.org/10.3390/cells11203235
Karagyaur M, Primak A, Efimenko A, Skryabina M, Tkachuk V. The Power of Gene Technologies: 1001 Ways to Create a Cell Model. Cells. 2022; 11(20):3235. https://doi.org/10.3390/cells11203235
Chicago/Turabian StyleKaragyaur, Maxim, Alexandra Primak, Anastasia Efimenko, Mariya Skryabina, and Vsevolod Tkachuk. 2022. "The Power of Gene Technologies: 1001 Ways to Create a Cell Model" Cells 11, no. 20: 3235. https://doi.org/10.3390/cells11203235
APA StyleKaragyaur, M., Primak, A., Efimenko, A., Skryabina, M., & Tkachuk, V. (2022). The Power of Gene Technologies: 1001 Ways to Create a Cell Model. Cells, 11(20), 3235. https://doi.org/10.3390/cells11203235