


Anticoagulants: A Short History, Their Mechanism of Action, Pharmacology, and Indications

 , , ,
, , ,
Abstract
1. Introduction
2. A Short History of Anticoagulants: From Serendipity to Molecular Design
2.1. Unfractionated Heparin
2.2. Vitamin K Antagonists
2.3. Low-Molecular-Weight Heparin
2.4. Fondaparinux, Argatroban, and Bivalirudin
2.5. Direct Oral Anticoagulants
3. Pharmacokinetics
3.1. UFH, LMWHs, and Fondaparinux
3.2. Vitamin K Antagonists
3.3. Bivalirudin and Argatroban
3.4. DOACs
4. Medical Indications
5. Drug–Drug Interactions
6. Introducing DOACs in Complex Patient Populations
7. A New Generation of Anticoagulants
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fang, J.; Alderman, M.H. Dissociation of hospitalization and mortality trends for myocardial infarction in the United States from 1988 to 1997. Am. J. Med. 2002, 113, 208–214. [Google Scholar] [CrossRef]
- Feigin, V.L.; Lawes, C.M.; Bennett, D.A.; Anderson, C.S. Stroke epidemiology: A review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. 2003, 2, 43–53. [Google Scholar] [CrossRef]
- White, R.H. The Epidemiology of Venous Thromboembolism. Circulation 2003, 107, I-4–I-8. [Google Scholar] [CrossRef]
- Mackman, N.; Bergmeier, W.; Stouffer, G.A.; Weitz, J.I. Therapeutic strategies for thrombosis: New targets and approaches. Nat. Rev. Drug Discov. 2020, 19, 333–352. [Google Scholar] [CrossRef]
- Ten Cate, H.; Hackeng, T.M.; de Frutos, P.G. Coagulation factor and protease pathways in thrombosis and cardiovascular disease. Thromb. Haemost. 2017, 117, 1265–1271. [Google Scholar] [CrossRef]
- Mclean, J. The Discovery of Heparin. Circulation 1959, 19, 75–78. [Google Scholar] [CrossRef]
- Last, J.A. The Missing Link: The Story of Karl Paul Link. Toxicol. Sci. 2002, 66, 4–6. [Google Scholar] [CrossRef]
- Weitz, J.I.; Jaffer, I.H.; Fredenburgh, J.C. Recent advances in the treatment of venous thromboembolism in the era of the direct oral anticoagulants. F1000Research 2017, 6, 985. [Google Scholar] [CrossRef]
- Yeh, C.H.; Gross, P.L.; Weitz, J.I. Evolving use of new oral anticoagulants for treatment of venous thromboembolism. Blood 2014, 124, 1020–1028. [Google Scholar] [CrossRef]
- Pollack, C.V.; Peacock, F.W.; Bernstein, R.A.; Clark, C.L.; Douketis, J.; Fermann, G.J.; Fiore, G.J.; Frost, A.; Jahromi, B.; Johnson, C.; et al. The safety of oral anticoagulants registry (SOAR): A national, ED-based study of the evaluation and management of bleeding and bleeding concerns due to the use of oral anticoagulants. Am. J. Emerg. Med. 2020, 38, 1163–1170. [Google Scholar] [CrossRef]
- Oates, J.A.; Wood, A.J.J.; Hirsh, J. Heparin. N. Engl. J. Med. 1991, 324, 1565–1574. [Google Scholar] [CrossRef]
- Couch, N.P. About heparin, or … Whatever happened to Jay McLean? J. Vasc. Surg. 1989, 10, 1–8. [Google Scholar]
- Abildgaard, U. Highly Purified Antithrombin III with Heparin Cofactor Activity Prepared by Disc Electrophoresis. Scand. J. Clin. Lab. Investig. 1968, 21, 89–91. [Google Scholar] [CrossRef]
- Rosenberg, R.D.; Rosenberg, J.S. Natural anticoagulant mechanisms. J. Clin. Investig. 1984, 74, 1–6. [Google Scholar] [CrossRef]
- Rezaie, A.R.; Giri, H. Anticoagulant and signaling functions of antithrombin. J. Thromb. Haemost. 2020, 18, 3142–3153. [Google Scholar] [CrossRef]
- Huntington, J.A. Serpin structure, function and dysfunction: Serpin structure, function and dysfunction. J. Thromb. Haemost. 2011, 9, 26–34. [Google Scholar] [CrossRef]
- Casu, B.; Oreste, P.; Torri, G.; Zoppetti, G.; Choay, J.; Lormeau, J.C.; Petitou, M.; Sinäy, P. The structure of heparin oligosaccharide fragments with high anti-(factor Xa) activity containing the minimal antithrombin III-binding sequence. Chemical and 13C nuclear-magnetic-resonance studies. Biochem. J. 1981, 197, 599–609. [Google Scholar] [CrossRef]
- Damus, P.S.; Hicks, M.; Rosenberg, R.D. Anticoagulant Action of Heparin. Nature 1973, 246, 355–357. [Google Scholar] [CrossRef]
- Link, K.P. The Discovery of Dicumarol and Its Sequels. Circulation 1959, 19, 97–107. [Google Scholar] [CrossRef]
- Whitlon, D.S.; Sadowski, J.A.; Suttie, J.W. Mechanism of coumarin action: Significance of vitamin K epoxide reductase inhibition. Biochemistry 1978, 17, 1371–1377. [Google Scholar] [CrossRef]
- Bell, R.G. Metabolism of vitamin K and prothrombin synthesis: Anticoagulants and the vitamin K--epoxide cycle. Fed. Proc. 1978, 37, 2599–2604. [Google Scholar]
- Stenflo, J.; Fernlund, P.; Egan, W.; Roepstorff, P. Vitamin K Dependent Modifications of Glutamic Acid Residues in Prothrombin. Proc. Natl. Acad. Sci. USA 1974, 71, 2730–2733. [Google Scholar] [CrossRef]
- Sackett, D.L. Clinical epidemiology. J. Clin. Epidemiol. 2002, 55, 1161–1166. [Google Scholar] [CrossRef]
- Johnson, E.A.; Kirkwood, T.B.; Stirling, Y.; Perez-Requejo, J.L.; Ingram, G.I.; Bangham, D.R.; Brozović, M. Four heparin preparations: Anti-Xa potentiating effect of heparin after subcutaneous injection. Thromb. Haemost. 1976, 35, 586–591. [Google Scholar] [CrossRef]
- Fareed, J.; Kumar, A.; Walenga, J.M.; Emanuele, R.M.; Williamson, K.; Hoppensteadt, D. Antithrombotic actions and pharmacokinetics of heparin fractions and fragments. Nouv. Rev. Fr. d’Hematol. 1984, 26, 267–275. [Google Scholar]
- Colombus Investigators. Low-Molecular-Weight Heparin in the Treatment of Patients with Venous Thromboembolism. N. Engl. J. Med. 1997, 337, 657–662. [Google Scholar] [CrossRef]
- Simonneau, G.; Sors, H.; Charbonnier, B.; Page, Y.; Laaban, J.P.; Azarian, R.; Laurent, M.; Hirsch, J.L.; Ferrari, E.; Bosson, J.L.; et al. A Comparison of Low-Molecular-Weight Heparin with Unfractionated Heparin for Acute Pulmonary Embolism. N. Engl. J. Med. 1997, 337, 663–669. [Google Scholar] [CrossRef]
- Levine, M.; Gent, M.; Hirsh, J.; Leclerc, J.; Anderson, D.; Weitz, J.; Ginsberg, J.; Turpie, A.G.; Demers, C.; Kovacs, M. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N. Engl. J. Med. 1996, 334, 677–681. [Google Scholar] [CrossRef]
- Koopman, M.M.; Prandoni, P.; Piovella, F.; Ockelford, P.A.; Brandjes, D.P.; van der Meer, J.; Gallus, A.S.; Simonneau, G.; Chesterman, C.H.; Prins, M.H. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. The Tasman Study Group. N. Engl. J. Med. 1996, 334, 682–687. [Google Scholar] [CrossRef]
- Zhou, S.-F.; Zhong, W.-Z. Drug Design and Discovery: Principles and Applications. Molecules 2017, 22, 279. [Google Scholar] [CrossRef]
- Anderson, A.C. The Process of Structure-Based Drug Design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Walenga, J.M.; Jeske, W.P.; Samama, M.M.; Frapaise, X.F.; Bick, R.L.; Fareed, J. Fondaparinux: A synthetic heparin pentasaccharide as a new antithrombotic agent. Expert Opin. Investig. Drugs 2002, 11, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, D.; Murphy, N. Argatroban: A synthetic thrombin inhibitor of low relative molecular mass. Coron. Artery Dis. 1996, 7, 455–458. [Google Scholar] [CrossRef]
- Oliveira, A.L.; Viegas, M.F.; da Silva, S.L.; Soares, A.M.; Ramos, M.J.; Fernandes, P.A. The chemistry of snake venom and its medicinal potential. Nat. Rev. Chem. 2022, 6, 451–469. [Google Scholar] [CrossRef] [PubMed]
- Nowak, G. Pharmacology of Recombinant Hirudin. Semin. Thromb. Hemost. 2002, 28, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Fenton, J.W. Leeches to hirulogs and other thrombin-directed antithrombotics. Hematol. Oncol. Clin. N. Am. 1992, 6, 1121–1129. [Google Scholar] [CrossRef]
- Bain, J.; Meyer, A. Comparison of bivalirudin to lepirudin and argatroban in patients with heparin-induced thrombocytopenia. Am. J. Health-Syst. Pharm. 2015, 72, S104–S109. [Google Scholar] [CrossRef]
- Siegal, D.; Yudin, J.; Kaatz, S.; Douketis, J.D.; Lim, W.; Spyropoulos, A.C. Periprocedural heparin bridging in patients receiving vitamin K antagonists: Systematic review and meta-analysis of bleeding and thromboembolic rates. Circulation 2012, 126, 1630–1639. [Google Scholar] [CrossRef]
- Chen, A.; Stecker, E.; Warden, B.A. Direct Oral Anticoagulant Use: A Practical Guide to Common Clinical Challenges. J. Am. Heart Assoc. 2020, 9, e017559. [Google Scholar] [CrossRef]
- Rose, D.K.; Bar, B. Direct Oral Anticoagulant Agents: Pharmacologic Profile, Indications, Coagulation Monitoring, and Reversal Agents. J. Stroke Cerebrovasc. Dis. 2018, 27, 2049–2058. [Google Scholar] [CrossRef]
- Gómez-Outes, A.; Suárez-Gea, M.L.; Lecumberri, R.; Terleira-Fernández, A.I.; Vargas-Castrillón, E. Direct-acting oral anticoagulants: Pharmacology, indications, management, and future perspectives. Eur. J. Haematol. 2015, 95, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, H.; Wåhlander, K.; Gustafsson, D.; Welin, L.T.; Frison, L.; Schulman, S.; Thrive Investigators. A randomized, controlled, dose-guiding study of the oral direct thrombin inhibitor ximelagatran compared with standard therapy for the treatment of acute deep vein thrombosis: THRIVE I. J. Thromb. Haemost. 2003, 1, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Francis, C.W.; Berkowitz, S.D.; Comp, P.C.; Lieberman, J.R.; Ginsberg, J.S.; Paiement, G.; Peters, G.R.; Roth, A.W.; McElhattan, J.; Colwell, C.W., Jr.; et al. Comparison of Ximelagatran with Warfarin for the Prevention of Venous Thromboembolism after Total Knee Replacement. N. Engl. J. Med. 2003, 349, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Schulman, S.; Wåhlander, K.; Lundström, T.; Clason, S.B.; Eriksson, H.; THRIVE III Investigators. Secondary prevention of venous thromboembolism with the oral direct thrombin inhibitor ximelagatran. N. Engl. J. Med. 2003, 349, 1713–1721. [Google Scholar] [CrossRef]
- Cully, M. Ximelagatran sets the stage for NOACs. Nat. Rev. Cardiol. 2017. [Google Scholar] [CrossRef]
- Svendsen, L.; Brogli, M.; Lindeberg, G.; Stocker, K. Differentiation of thrombin- and factor Xa-related amidolytic activity in plasma by means of a synthetic thrombin inhibitor. Thromb. Res. 1984, 34, 457–462. [Google Scholar] [CrossRef]
- Hauel, N.H.; Nar, H.; Priepke, H.; Ries, U.; Stassen, J.; Wienen, W. Structure-Based Design of Novel Potent Nonpeptide Thrombin Inhibitors. J. Med. Chem. 2002, 45, 1757–1766. [Google Scholar] [CrossRef]
- Milling, T.J.; Ziebell, C.M. A review of oral anticoagulants, old and new, in major bleeding and the need for urgent surgery. Trends Cardiovasc. Med. 2020, 30, 86–90. [Google Scholar] [CrossRef]
- Perzborn, E.; Roehrig, S.; Straub, A.; Kubitza, D.; Misselwitz, F. The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor. Nat. Rev. Drug Discov. 2011, 10, 61–75. [Google Scholar] [CrossRef]
- Nutt, E.; Gasic, T.; Rodkey, J.; Gasic, G.J.; Jacobs, J.W.; Friedman, P.A.; Simpson, E. The amino acid sequence of antistasin. A potent inhibitor of factor Xa reveals a repeated internal structure. J. Biol. Chem. 1988, 263, 10162–10167. [Google Scholar] [CrossRef]
- Mavrakanas, T.; Bounameaux, H. The potential role of new oral anticoagulants in the prevention and treatment of thromboembolism. Pharmacol. Ther. 2011, 130, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, L.; Gusto, G.; Khachatryan, A.; Quignot, N.; Chaves, J.; Moniot, A.; Mokgokong, R. Effectiveness and Safety of Oral Anticoagulants in the Treatment of Acute Venous Thromboembolism: A Nationwide Comparative Cohort Study in France. Thromb. Haemost. 2022, 122, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, J.; Warkentin, T.E.; Shaughnessy, S.G.; Anand, S.S.; Halperin, J.J.; Raschke, R.; Granger, C.; Ohman, E.M.; Dalen, J.E. Heparin and low-molecular-weight heparin: Mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy, and safety. Chest 2001, 119, 64S–94S. [Google Scholar] [CrossRef]
- Weitz, J.I. Low-molecular-weight heparins. N. Engl. J. Med. 1997, 337, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, E.; Kalaska, B.; Miklosz, J.; Mogielnicki, A. The toxicology of heparin reversal with protamine: Past, present and future. Expert Opin. Drug Metab. Toxicol. 2016, 12, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Howell, W.H.; Holt, E. Two New Factors in Blood Coagulation—Heparin and Pro-Antithrombin. Am. J. Physiol. -Leg. Content 1918, 47, 328–341. [Google Scholar] [CrossRef]
- Hirsh, J.; van Aken, W.G.; Gallus, A.S.; Dollery, C.T.; Cade, J.F.; Yung, W.L. Heparin kinetics in venous thrombosis and pulmonary embolism. Circulation 1976, 53, 691–695. [Google Scholar] [CrossRef]
- Young, E.; Prins, M.; Levine, M.N.; Hirsh, J. Heparin binding to plasma proteins, an important mechanism for heparin resistance. Thromb. Haemost. 1992, 67, 639–643. [Google Scholar] [CrossRef]
- Bârzu, T.; Molho, P.; Tobelem, G.; Petitou, M.; Caen, J. Binding and endocytosis of heparin by human endothelial cells in culture. Biochim. Biophys. Acta BBA -Mol. Cell Res. 1985, 845, 196–203. [Google Scholar] [CrossRef]
- Young, E.; Wells, P.; Holloway, S.; Weitz, J.; Hirsh, J. Ex-vivo and in-vitro evidence that low molecular weight heparins exhibit less binding to plasma proteins than unfractionated heparin. Thromb. Haemost. 1994, 71, 300–304. [Google Scholar] [CrossRef]
- Bloemen, A.; Testroote, M.J.G.; Janssen-Heijnen, M.L.G.; Janzing, H.M.J. Incidence and diagnosis of heparin-induced thrombocytopenia (HIT) in patients with traumatic injuries treated with unfractioned or low-molecular-weight heparin: A literature review. Injury 2012, 43, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, M.; Zermatten, M.G.; Bertaggia Calderara, D.; Aliotta, A.; Alberio, L. Heparin-Induced Thrombocytopenia: A Review of New Concepts in Pathogenesis, Diagnosis, and Management. J. Clin. Med. 2021, 10, 683. [Google Scholar] [CrossRef] [PubMed]
- Amiral, J.; Bridey, F.; Dreyfus, M.; Vissoc, A.M.; Fressinaud, F.; Wolf, M.; Meyer, D. Platelet factor 4 complexed to heparin is the target for antibodies generated in heparin-induced thrombocytopenia. Thromb. Haemost. 1992, 68, 95–96. [Google Scholar] [CrossRef] [PubMed]
- Burch, M.; Cooper, B. Fondaparinux-Associated Heparin-Induced Thrombocytopenia. Bayl. Univ. Med. Cent. Proc. 2012, 25, 13–15. [Google Scholar] [CrossRef]
- Nelsestuen, G.L.; Zytkovicz, T.H.; Howard, J.B. The mode of action of vitamin K. Identification of gamma-carboxyglutamic acid as a component of prothrombin. J. Biol. Chem. 1974, 249, 6347–6350. [Google Scholar] [CrossRef]
- Hanley, J.P. Warfarin reversal. J. Clin. Pathol. 2004, 57, 1132–1139. [Google Scholar] [CrossRef]
- Hull, R.D.; Raskob, G.E.; Hirsh, J.; Jay, R.M.; Leclerc, J.R.; Geerts, W.H.; Rosenbloom, D.; Sackett, D.L.; Anderson, C.; Harrison, L. Continuous Intravenous Heparin Compared with Intermittent Subcutaneous Heparin in the Initial Treatment of Proximal-Vein Thrombosis. N. Engl. J. Med. 1986, 315, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Salzman, E.W.; Deykin, D.; Shapiro, R.M.; Rosenberg, R. Management of Heparin Therapy: Controlled Prospective Trial. N. Engl. J. Med. 1975, 292, 1046–1050. [Google Scholar] [CrossRef] [PubMed]
- Poller, L. Standardization of anticoagulant control. Ric. Clin. Lab. 1978, 8, 237–247. [Google Scholar] [CrossRef]
- Wang, M.; Zeraatkar, D.; Obeda, M.; Lee, M.; Garcia, C.; Nguyen, L.; Agarwal, A.; Al-Shalabi, F.; Benipal, H.; Ahmad, A.; et al. Drug-drug interactions with warfarin: A systematic review and meta-analysis. Br. J. Clin. Pharmacol. 2021, 87, 4051–4100. [Google Scholar] [CrossRef]
- Hirsh, J.; Dalen, J.; Anderson, D.R.; Poller, L.; Bussey, H.; Ansell, J.; Deykin, D. Oral Anticoagulants: Mechanism of Action, Clinical Effectiveness, and Optimal Therapeutic Range. Chest 2001, 119, 8S–21S. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.E.; Wallis, D.E.; Berkowitz, S.D.; Matthai, W.H.; Fareed, J.; Walenga, J.M.; Bartholomew, J.; Sham, R.; Lerner, R.G.; Zeigler, Z.R.; et al. Argatroban Anticoagulant Therapy in Patients With Heparin-Induced Thrombocytopenia. Circulation 2001, 103, 1838–1843. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E. Heparin-Induced Thrombocytopenia: Diagnosis and Management. Circulation 2004, 110, e454–e458. [Google Scholar] [CrossRef] [PubMed]
- Eichler, P.; Lubenow, N.; Strobel, U.; Greinacher, A. Antibodies against lepirudin are polyspecific and recognize epitopes on bivalirudin. Blood 2004, 103, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Shammas, N.W. Bivalirudin: Pharmacology and clinical applications. Cardiovasc. Drug Rev. 2005, 23, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Van de Car, D.A.; Rao, S.V.; Ohman, E.M. Bivalirudin: A review of the pharmacology and clinical application. Expert Rev. Cardiovasc. Ther. 2010, 8, 1673–1681. [Google Scholar] [CrossRef]
- Jeske, W.P.; Fareed, J.; Hoppensteadt, D.A.; Lewis, B.; Walenga, J.M. Pharmacology of argatroban. Expert Rev. Hematol. 2010, 3, 527–539. [Google Scholar] [CrossRef]
- Bellomo, T.R.; Jeakle, M.A.; Meyerhoff, M.E.; Bartlett, R.H.; Major, T.C. The Effects of the Combined Argatroban/Nitric Oxide-Releasing Polymer on Platelet Microparticle-Induced Thrombogenicity in Coated Extracorporeal Circuits. ASAIO J. 2021, 67, 573–582. [Google Scholar] [CrossRef]
- Major, T.C.; Handa, H.; Brisbois, E.J.; Reynolds, M.M.; Annich, G.M.; Meyerhoff, M.E.; Bartlett, R.H. The mediation of platelet quiescence by NO-releasing polymers via cGMP-induced serine 239 phosphorylation of vasodilator-stimulated phosphoprotein. Biomaterials 2013, 34, 8086–8096. [Google Scholar] [CrossRef]
- Ageno, W.; Haas, S.; Weitz, J.I.; Goldhaber, S.Z.; Turpie, A.G.C.; Goto, S.; Pantep Angchaisuksiri, P.; Dalsgaard Nielsen, J.; Kayani, G.; Pieper, K.S.; et al. Characteristics and Management of Patients with Venous Thromboembolism: The GARFIELD-VTE Registry. Thromb. Haemost. 2019, 119, 319–327. [Google Scholar] [CrossRef]
- Hauptmann, J.; Stürzebecher, J. Synthetic Inhibitors of Thrombin and Factor Xa. Thromb. Res. 1999, 93, 203–241. [Google Scholar] [PubMed]
- Pollack, C.V.; Reilly, P.A.; Eikelboom, J.; Glund, S.; Verhamme, P.; Bernstein, R.A.; Dubiel, R.; Huisman, M.V.; Hylek, E.M.; Kamphuisen, P.W.; et al. Idarucizumab for Dabigatran Reversal. N. Engl. J. Med. 2015, 373, 511–520. [Google Scholar] [CrossRef]
- Connolly, S.J.; Milling, T.J., Jr.; Eikelboom, J.W.; Gibson, M.C.; Curnutte, J.T.; Gold, A.; Bronson, M.D.; Lu, G.; Conley, P.B.; Verhamme, P.; et al. Andexanet Alfa for Acute Major Bleeding Associated with Factor Xa Inhibitors. N. Engl. J. Med. 2016, 375, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Cuker, A.; Burnett, A.; Triller, D.; Crowther, M.; Ansell, J.; Van Cott, E.M.; Wirth, D.; Kaatz, S. Reversal of direct oral anticoagulants: Guidance from the Anticoagulation Forum. Am. J. Hematol. 2019, 94, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Colombo, E.; Tenconi, M.; Baldessin, L.; Corsini, A. Drug-Drug Interactions of Direct Oral Anticoagulants (DOACs): From Pharmacological to Clinical Practice. Pharmaceutics 2022, 14, 1120. [Google Scholar] [CrossRef]
- Raymond, J.; Imbert, L.; Cousin, T.; Duflot, T.; Varin, R.; Wils, J.; Lamoureux, F. Pharmacogenetics of Direct Oral Anticoagulants: A Systematic Review. J. Pers. Med. 2021, 11, 37. [Google Scholar] [CrossRef]
- Steffel, J.; Collins, R.; Antz, M.; Cornu, P.; Desteghe, L.; Haeusler, K.G.; Oldgren, J.; Reinecke, H.; Roldan-Schilling, V.; Rowell, N.; et al. 2021 European Heart Rhythm Association Practical Guide on the Use of Non-Vitamin K Antagonist Oral Anticoagulants in Patients with Atrial Fibrillation. EP Eur. 2021, 23, 1612–1676. [Google Scholar]
- Gibbons, J.A.; de Vries, M.; Krauwinkel, W.; Ohtsu, Y.; Noukens, J.; van der Walt, J.; Mol, R.; Mordenti, J.; Ouatas, T. Pharmacokinetic Drug Interaction Studies with Enzalutamide. Clin. Pharmacokinet. 2015, 54, 1057–1069. [Google Scholar] [CrossRef]
- Li, A.; Li, M.K.; Crowther, M.; Vazquez, S.R. Drug-drug interactions with direct oral anticoagulants associated with adverse events in the real world: A systematic review. Thromb. Res. 2020, 194, 240–245. [Google Scholar] [CrossRef]
- Baillargeon, J.; Holmes, H.M.; Lin, Y.; Raji, M.A.; Sharma, G.; Kuo, Y. Concurrent use of warfarin and antibiotics and the risk of bleeding in older adults. Am. J. Med. 2012, 125, 183–189. [Google Scholar] [CrossRef]
- Forbes, H.L.; Polasek, T.M. Potential drug–drug interactions with direct oral anticoagulants in elderly hospitalized patients. Ther. Adv. Drug Saf. 2017, 8, 319–328. [Google Scholar] [CrossRef] [PubMed]
- McDonough, C.W. Pharmacogenomics in Cardiovascular Diseases. Curr. Protoc. 2021, 1, e189. [Google Scholar] [CrossRef] [PubMed]
- Padrini, R. Clinical Pharmacokinetics and Pharmacodynamics of Direct Oral Anticoagulants in Patients with Renal Failure. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ballestri, S.; Capitelli, M.; Fontana, M.C.; Arioli, D.; Romagnoli, E.; Graziosi, C.; Lonardo, A.; Marietta, M.; Dentali, F.; Cioni, G. Direct Oral Anticoagulants in Patients with Liver Disease in the Era of Non-Alcoholic Fatty Liver Disease Global Epidemic: A Narrative Review. Adv. Ther. 2020, 37, 1910–1932. [Google Scholar] [PubMed]
- Caldwell, P.H.Y.; Murphy, S.B.; Butow, P.N.; Craig, J.C. Clinical trials in children. Lancet 2004, 364, 803–811. [Google Scholar] [CrossRef]
- Male, C.; Lensing, A.W.A.; Palumbo, J.S.; Kumar, R.; Nurmeev, I.; Hege, K.; Bonnet, D.; Connor, P.; Hooimeijer, H.L.; Torres, M. Rivaroxaban compared with standard anticoagulants for the treatment of acute venous thromboembolism in children: A randomised, controlled, phase 3 trial. Lancet Haematol. 2020, 7, e18–e27. [Google Scholar] [CrossRef]
- Beyer-Westendorf, J.; Tittl, L.; Bistervels, I.; Middeldorp, S.; Schaefer, C.; Paulus, W.; Thomas, W.; Kemkes-Matthes, B.; Sandra Marten, S.; Bornhauser, M. Safety of direct oral anticoagulant exposure during pregnancy: A retrospective cohort study. Lancet Haematol. 2020, 7, e884–e891. [Google Scholar] [CrossRef]
- Wiesen, M.H.J.; Blaich, C.; Müller, C.; Streichert, T.; Pfister, R.; Michels, G. The Direct Factor Xa Inhibitor Rivaroxaban Passes Into Human Breast Milk. Chest 2016, 150, e1–e4. [Google Scholar] [CrossRef]
- Daei, M.; Khalili, H.; Heidari, Z. Direct oral anticoagulant safety during breastfeeding: A narrative review. Eur. J. Clin. Pharmacol. 2021, 77, 1465–1471. [Google Scholar] [CrossRef]
- Pengo, V.; Banzato, A.; Bison, E.; Zoppellaro, G.; Padayattil Jose, S.; Denas, G. Efficacy and safety of rivaroxaban vs. warfarin in high-risk patients with antiphospholipid syndrome: Rationale and design of the Trial on Rivaroxaban in AntiPhospholipid Syndrome (TRAPS) trial. Lupus 2016, 25, 301–306. [Google Scholar] [CrossRef]
- Pengo, V.; Denas, G.; Zoppellaro, G.; Padayattil Jose, S.; Hoxha, A.; Ruffatti, A.; Andreoli, L.; Tincani, A.; Cenci, C.; Prisco, D.; et al. Rivaroxaban vs. warfarin in high-risk patients with antiphospholipid syndrome. Blood 2018, 132, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Pengo, V.; Hoxha, A.; Andreoli, L.; Tincani, A.; Silvestri, E.; Prisco, D.; Fierro, T.; Gresele, P.; Cafolla, A.; De Micheli, V.; et al. Trial of Rivaroxaban in AntiPhospholipid Syndrome (TRAPS): Two-year outcomes after the study closure. J. Thromb. Haemost. 2021, 19, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Eikelboom, J.W.; Connolly, S.J.; Brueckmann, M.; Granger, C.B.; Kappetein, A.P.; Mack, M.J.; Blatchford, J.; Devenny, K.; Friedman, J.; Guiver, K.; et al. Dabigatran versus Warfarin in Patients with Mechanical Heart Valves. N. Engl. J. Med. 2013, 369, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.J.; Karthikeyan, G.; Ntsekhe, M.; Haileamlak, A.; El Sayed, A.; El Ghamrawy, A.; Damasceno, A.; Avezum, A.; Dans, A.M.L.; Gitura, B.; et al. Rivaroxaban in Rheumatic Heart Disease–Associated Atrial Fibrillation. N. Engl. J. Med. 2022, 387, 978–988. [Google Scholar] [CrossRef]
- Klok, F.A.; Ageno, W.; Ay, C.; Bäck, M.; Barco, S.; Bertoletti, L.; Becattini, C.; Carlsen, J.; Delcroix, M.; van Es, N.; et al. Optimal follow-up after acute pulmonary embolism: A position paper of the European Society of Cardiology Working Group on Pulmonary Circulation and Right Ventricular Function, in collaboration with the European Society of Cardiology Working Group on Atherosclerosis and Vascular Biology, endorsed by the European Respiratory Society. Eur. Heart J. 2022, 43, 183–189. [Google Scholar]
- Bertoletti, L.; Mismetti, V.; Giannakoulas, G. Use of Anticoagulants in Patients with Pulmonary Hypertension. Hämostaseologie 2020, 40, 348–355. [Google Scholar] [CrossRef]
- Margelidon-Cozzolino, V.; Delavenne, X.; Catella-Chatron, J.; De Magalhaes, E.; Bezzeghoud, S.; Humbert, M.; Montani, D.; Bertoletti, L. Indications and potential pitfalls of anticoagulants in pulmonary hypertension: Would DOACs become a better option than VKAs? Blood Rev. 2019, 37, 100579. [Google Scholar] [CrossRef]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. 2022, 60, 2200879. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Eikelboom, J.W.; Connolly, S.J.; Steg, G.P.; Anand, S.S.; Verma, S.; Branch, K.R.H.; Probstfield, J.; Bosch, J.; Shestakovska, O.; et al. Role of Combination Antiplatelet and Anticoagulation Therapy in Diabetes Mellitus and Cardiovascular Disease: Insights From the COMPASS Trial. Circulation 2020, 141, 1841–1854. [Google Scholar] [CrossRef]
- Flumignan, C.D.; Nakano, L.C.; Baptista-Silva, J.C.; Flumignan, R.L. Antiplatelet agents for the treatment of deep venous thrombosis. Cochrane Database Syst. Rev. 2022, 25, CD012369. [Google Scholar] [CrossRef]
- Giraud, M.; Catella, J.; Cognet, L.; Helfer, H.; Accassat, S.; Chapelle, C.; Mismetti, P.; Laporte, S.; Mahé, I.; Bertoletti, L. Management of acute venous thromboembolism in patients taking antiplatelet therapy. Thromb. Res. 2021, 208, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Poenou, G.; Dumitru Dumitru, T.; Lafaie, L.; Mismetti, V.; Heestermans, M.; Bertoletti, L. Factor XI Inhibition for the Prevention of Venous Thromboembolism: An Update on Current Evidence and Future perspectives. Vasc. Health Risk Manag. 2022, 18, 359–373. [Google Scholar] [CrossRef] [PubMed]
- Key, N.S.; Khorana, A.A.; Kuderer, N.M.; Bohlke, K.; Lee, A.Y.Y.; Arcelus, J.I.; Wong, S.L.; Balaban, E.P.; Flowers, C.R.; Francis, C.W.; et al. Venous Thromboembolism Prophylaxis and Treatment in Patients With Cancer: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2020, 38, 496–520. [Google Scholar] [CrossRef] [PubMed]
- Farge, D.; Frere, C.; Connors, J.M.; Ay, C.; Khorana, A.A.; Munoz, A.; Brenner, B.; Kakkar, A.; Rafii, H.; Solymoss, S.; et al. 2019 international clinical practice guidelines for the treatment and prophylaxis of venous thromboembolism in patients with cancer. Lancet Oncol. 2019, 20, e566–e581. [Google Scholar] [CrossRef]
- Khorana, A.A.; Noble, S.; Lee, A.Y.Y.; Soff, G.; Meyer, G.; O’Connell, C.; Carrier, M. Role of direct oral anticoagulants in the treatment of cancer-associated venous thromboembolism: Guidance from the SSC of the ISTH. J. Thromb. Haemost. 2018, 16, 1891–1894. [Google Scholar] [CrossRef]
- Lyman, G.H.; Carrier, M.; Ay, C.; Di Nisio, M.; Hicks, L.K.; Khorana, A.A.; Leavitt, A.D.; Lee, A.Y.Y.; Macbeth, F.; Morgan, R.L.; et al. American Society of Hematology 2021 guidelines for management of venous thromboembolism: Prevention and treatment in patients with cancer. Blood Adv. 2021, 5, 927–974. [Google Scholar] [CrossRef]


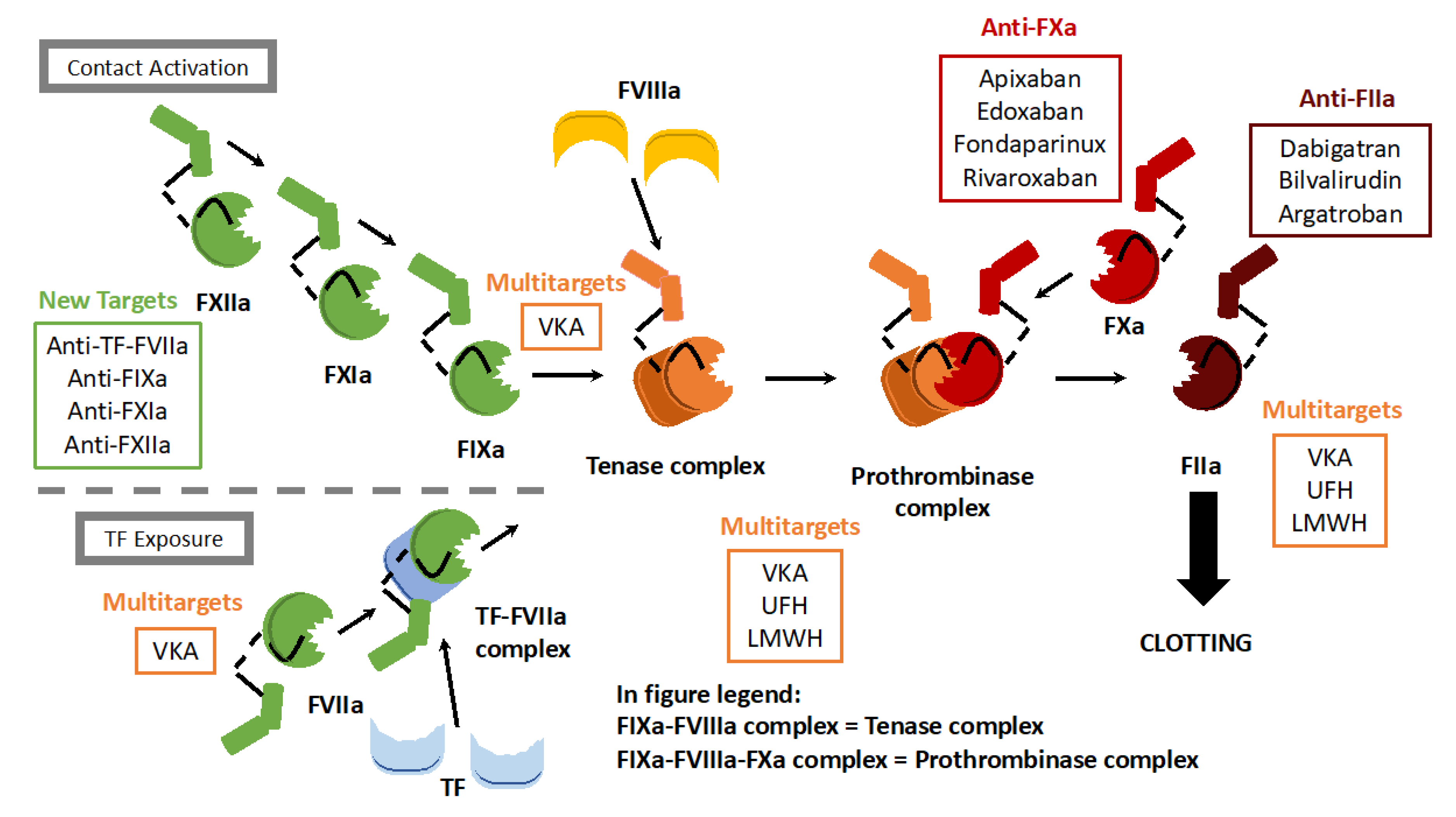{kind=link}
| UFH | Enoxaparin | Fondaparinux | |
|---|---|---|---|
| Type | Small molecule | Small molecule | Small molecule |
| Origin | Obtained from liver, lung, mast cells, and other cells of vertebrates | Obtained from liver, lung, mast cells, and other cells of vertebrates | Synthetic molecule |
| FDA approval | N/A, first used in clinical practice in 1941 | 1993 | 2001 |
| Target | Antithrombin | Antithrombin | Antithrombin |
| Available administration route | IV/SC | (IV)/SC | (IV)/SC |
| Absorption | N/A | N/A | N/A |
| Bioavailability | Unpredictable | 90%–92% | 100% |
| Vd | 40–70 mL/min | 4.3 L | 7–11 L |
| Protein binding | >90% | <UFH | >94% (specifically to antithrombin) |
| Time to peak activity (h) | Rapid after bolus, 4–6 after infusion | 3–5 | 2–3 |
| Monitoring | aXa UFH | aXa LMWH | aXa Fondaparinux |
| Half-life (h) | 1.0–1.5 | 5 | 17–21 |
| Elimination | Reticuloendothelial system + renal | Renal | Renal |
| Neutralizing agent | No hemodialysis Sulfate protamine | Hemodialysis Sulfate protamine | Hemodialysis No sulfate protamine |
| Warfarin | |
|---|---|
| Type | Racemic mixture |
| Origin | Synthetic molecule |
| FDA approval | 1954 |
| Target | Vitamin K-dependent coagulation factors II, VII, IX, and X |
| Available administration route | PO |
| Absorption | Stomach and proximal small bowel |
| Bioavailability | 80%–100% |
| Vd | 8–10 L |
| Protein binding | 95%–97% |
| Time to peak concentration (h) | 1–2 |
| Monitoring | INR test |
| Half-life (h) | 20–60 |
| Elimination | Major hepatic metabolism, biotransformation > 90% of inactive metabolites (CYP 2C9) |
| Neutralizing agent | Vitamin K |
| Bivalirudin | Argatroban | |
|---|---|---|
| Type | Small molecule | Small molecule |
| Origin | Synthetic molecule | Synthetic molecule |
| FDA approval | 2000 | 2000 |
| Target | Thrombin | Thrombin |
| Available administration route | IV | IV |
| Absorption | N/A | N/A |
| Bioavailability | 40%–80% | 100% |
| Vd | 0.24 L/Kg | 0.174 L/Kg |
| Protein binding | No plasma proteins (just thrombin) | 55% (20% albumin, 35% alpha-acid glycoprotein) |
| Time to peak activity | 2–4 min | 3–4 h |
| Monitoring | aPTT, ACT, ecarin clotting time | aPTT, ACT |
| Half-life (hours) | 1.0–1.5 | 3–5 |
| Elimination | Renal and proteolytic cleavage | 70% hepatic hydroxylation, 16% renal excretion as unchanged drug, 14% biliary excretion as unchanged drug |
| Neutralizing agent | N/A | N/A |
| Dabigatran | Rivaroxaban | Apixaban | Edoxaban | |
|---|---|---|---|---|
| Type | Small molecule | Small molecule | Small molecule | Small molecule |
| Origin | Synthetic molecule | Synthetic molecule | Synthetic molecule | Synthetic molecule |
| FDA approval | 2010 | 2011 | 2014 | 2014 |
| Target | FIIa | FXa | FXa | FXa |
| Available administration route | PO | PO | PO | PO |
| Absorption | Lower stomach + duodenum | Proximal small bowel + gastric | Proximal small bowel + gastric | Proximal small bowel |
| Bioavailability | 6.5% | 66% | 50% | 60% |
| Vd | 50–70 L | 50 L | 21 L | >107 L |
| Protein binding | 35% | >90% | 87% | 40%–60% |
| Time to peak concentration (h) | 1–2 | 2–4 | 1–4 | 1–2 |
| Monitoring | N/A | N/A | N/A | N/A |
| Half-life (h) | 12–17 | 5–9 | 8–15 | 10–14 |
| Elimination | Up to 20% glucuronidation and then biliary excretion 80% unchanged renal excretion | 66% undergoes metabolic degradation (30% CYP CYP3A4/5, metabolism) 66% renal excretion 28% feces | 30%–35% O-demethylation 15% CYP CYP3A4/5, metabolism 1/3 biliary excretion 1/3 renal excretion | Conjugation, oxidation by CYP 3A4 and hydrolysis 1/2 biliary excretion 1/2 renal excretion |
| Neutralizing agent | Idarucizumab | Andexanet alfa, PCC | Andexanet alfa, PCC | Andexanet alfa (?), PCC |
| Indication | UFH | Enoxaparin | Fondaparinux | Bilvarudin | Argatroban |
|---|---|---|---|---|---|
| Arterial thrombosis | |||||
| Prophylaxis of ischemic complications in the setting of unstable angina or NSTEMI | yes | yes | / | / | / |
| Treatment of atrial fibrillation with embolization | yes | / | / | / | / |
| Prophylaxis of peripheral arterial embolism | yes | / | / | / | / |
| Prophylaxis of clotting in cardiac surgery | yes | / | / | / | / |
| Prophylaxis or treatment of thrombosis in adult patients with HIT, including during PCI procedures | / | / | / | yes | yes |
| Venous thrombosis | |||||
| Extended treatment of symptomatic venous thromboembolism to reduce recurrence in patients | yes | yes | yes | / | / |
| Prophylaxis of postoperative VTE in patients undergoing abdominal surgery, hip replacement surgery, or knee replacement surgery | yes | yes | yes | / | / |
| Prophylaxis of VTE in acutely ill medical patients with severely restricted mobility | / | yes | / | / | / |
| Extended treatment of symptomatic venous thromboembolism to reduce recurrence in patients with cancer | / | yes * | / | / | / |
| Other indications | |||||
| Extracorporeal circulation and dialysis procedures | yes | / | / | / | / |
| Treatment of acute and chronic consumptive coagulopathies | yes | / | / | / | / |
| Indication | Warfarin | Dabigatran | Rivaroxaban | Apixaban | Edoxaban |
|---|---|---|---|---|---|
| Arterial thrombosis | |||||
| Reduced risk of stroke and systemic embolism in patients with NVAF | yes | yes | yes | yes | yes |
| Prophylaxis and treatment of embolic complications associated with atrial fibrillation or cardiac valve replacement | yes | / | / | / | / |
| Reduced risk of death, recurrent myocardial infarction, and stroke or systemic embolism after myocardial infarction | yes | / | / | / | / |
| Venous thrombosis | |||||
| Prophylaxis of DVT and PE in patients who have undergone hip replacement surgery | yes | yes | yes | yes | / |
| Treatment of DVT and PE | / | yes | yes | yes | yes |
| Reduced risk of recurrent DVT and/or PE in patients at continued risk for recurrent of VTE | yes | yes | yes | yes | yes |
| Reduced risk of recurrent DVT and/or PE in patients at continued risk for recurrent of VTE in cancer patients | / | / | yes | yes | yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heestermans, M.; Poenou, G.; Hamzeh-Cognasse, H.; Cognasse, F.; Bertoletti, L. Anticoagulants: A Short History, Their Mechanism of Action, Pharmacology, and Indications. Cells 2022, 11, 3214. https://doi.org/10.3390/cells11203214
Heestermans M, Poenou G, Hamzeh-Cognasse H, Cognasse F, Bertoletti L. Anticoagulants: A Short History, Their Mechanism of Action, Pharmacology, and Indications. Cells. 2022; 11(20):3214. https://doi.org/10.3390/cells11203214
Chicago/Turabian StyleHeestermans, Marco, Géraldine Poenou, Hind Hamzeh-Cognasse, Fabrice Cognasse, and Laurent Bertoletti. 2022. "Anticoagulants: A Short History, Their Mechanism of Action, Pharmacology, and Indications" Cells 11, no. 20: 3214. https://doi.org/10.3390/cells11203214
APA StyleHeestermans, M., Poenou, G., Hamzeh-Cognasse, H., Cognasse, F., & Bertoletti, L. (2022). Anticoagulants: A Short History, Their Mechanism of Action, Pharmacology, and Indications. Cells, 11(20), 3214. https://doi.org/10.3390/cells11203214