
A Defective Viral Particle Approach to COVID-19



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. SARS-CoV-2 DIPs, VLPs and Other Nanoparticles
3. Strategies
3.1. Targeting Macromolecular Assemblies
3.2. Targeting RNA
3.3. Deletions
3.4. Immune Stimulation by TIPs
3.5. Anti-Sense Oligonucleotides, Aptamers, Ribozymes, and Antibodies
3.5.1. Anti-Sense Oligonucleotides
3.5.2. Aptamers
3.5.3. Ribozymes and Antibodies
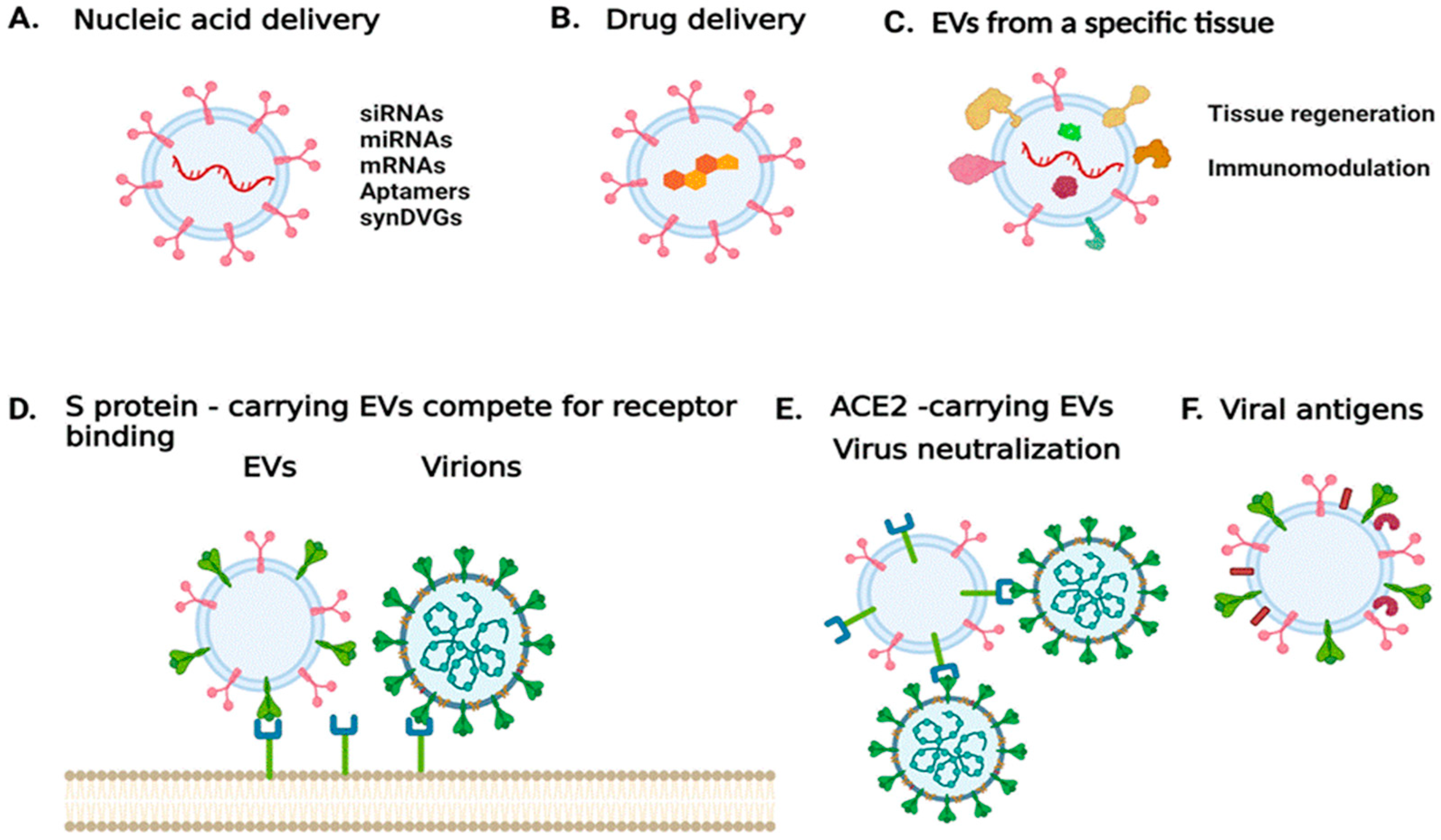
3.6. EVs
3.7. Mimics
4. Problems
4.1. Constructing Large synDVGs
4.2. Obtaining DIPs That Attenuate Rather Than Exacerbate Disease Progress
4.3. Avoiding Loss of EV Activity and Other Complications
4.4. Avoiding Loss of Aptamers
5. Discussion
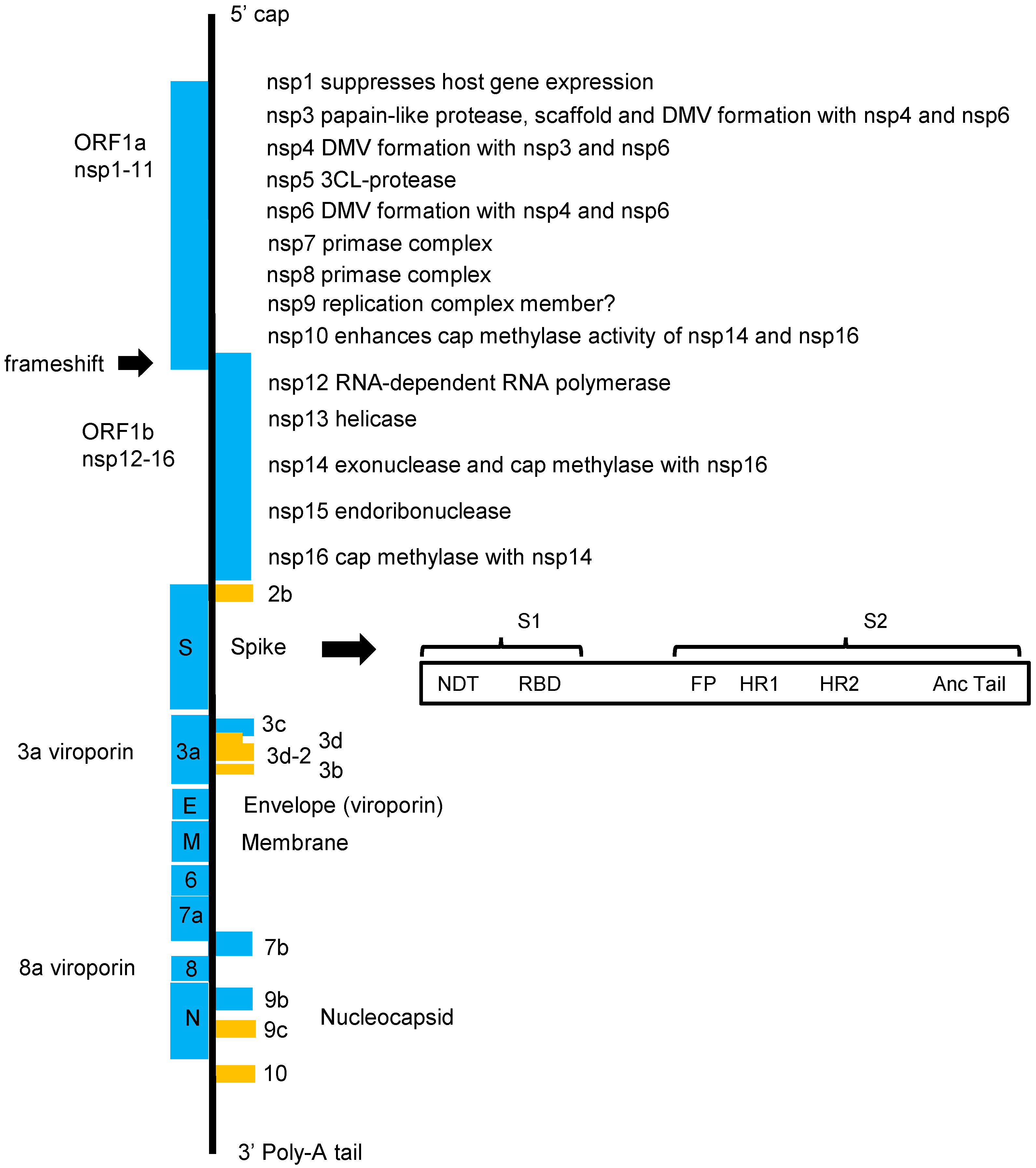
6. SARS-CoV-2 Overview: Viral and Host Targets
6.1. Spike (S) and Viral Entry into the Host Cell
6.2. Nucleocapsid (N)
6.3. Envelope (E)
6.4. Membrane (M)
6.5. RNA-Dependent RNA Polymerase (RdRp) (nsp12) and the Co-Factors nsp7 and nsp8
6.6. Non-Structural Protein 1
6.7. Non-Structural Protein 9
6.8. Viroporins
6.8.1. Open Reading Frame 3a
6.8.2. Open Reading Frame 8
6.9. Viral Proteases
6.9.1. Papain-like Protease (PLP2 or nsp3)
6.9.2. Main Protease (Mpro) or 3C-like Protease (3CLpro) or nsp5
6.10. Viral Endonucleases, Helicase and S-Adenosylmethionine (SAM)-Dependent Methyltransferases (MTases)
6.10.1. Non-Structural Protein 14
6.10.2. Non-Structural Protein 16 and nsp10
6.10.3. Non-Structural Protein 13
6.10.4. Non-Structural Protein 15
6.11. Viral Replication Organelles
6.11.1. Non-Structural Proteins nsp4 and nsp6
6.11.2. Lipid Composition of Viral Replication Organelles
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ganem, D.E. 2000 and Beyond: Confronting the Microbe Menace Lecture One—Microbe Hunters: Tracking Infectious Agents. Available online: https://www.biointeractive.org/sites/default/files/Infectious%2520Diseases%2520Lecture%25201%2520Transcript.pdf (accessed on 9 June 2020).
- Noppornpanth, S.; Smits, S.L.; Lien, T.X.; Poovorawan, Y.; Osterhaus, A.D.; Haagmans, B.L. Characterization of hepatitis c virus deletion mutants circulating in chronically infected patients. J. Virol. 2007, 81, 12496–12503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saira, K.; Lin, X.; DePasse, J.V.; Halpin, R.; Twaddle, A.; Stockwell, T.; Angus, B.; Cozzi-Lepri, A.; Delfino, M.; Dugan, V.; et al. Sequence analysis of in vivo defective interfering-like rna of influenza a h1n1 pandemic virus. J. Virol. 2013, 87, 8064–8074. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Jain, D.; Koziol-White, C.J.; Genoyer, E.; Gilbert, M.; Tapia, K.; Panettieri, R.A., Jr.; Hodinka, R.L.; Lopez, C.B. Immunostimulatory defective viral genomes from respiratory syncytial virus promote a strong innate antiviral response during infection in mice and humans. PLoS Pathog. 2015, 11, e1005122. [Google Scholar] [CrossRef] [Green Version]
- Vignuzzi, M.; Lopez, C.B. Defective viral genomes are key drivers of the virus-host interaction. Nat. Microbiol. 2019, 4, 1075–1087. [Google Scholar] [CrossRef]
- Roux, L.; Simon, A.E.; Holland, J.J. Effects of defective interfering viruses on virus-replication and pathogenesis em in vitro and em in vivo. Adv. Virus Res. 1991, 40, 181–211. [Google Scholar] [PubMed]
- Laske, T.; Heldt, F.S.; Hoffmann, H.; Frensing, T.; Reichl, U. Modeling the intracellular replication of influenza a virus in the presence of defective interfering rnas. Virus Res. 2016, 213, 90–99. [Google Scholar] [CrossRef]
- Duhaut, S.D.; McCauley, J.W. Defective rnas inhibit the assembly of influenza virus genome segments in a segment-specific manner. Virology 1996, 216, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Easton, A.J.; Scott, P.D.; Edworthy, N.L.; Meng, B.; Marriott, A.C.; Dimmock, N.J. A novel broad-spectrum treatment for respiratory virus infections: Influenza-based defective interfering virus provides protection against pneumovirus infection in vivo. Vaccine 2011, 29, 2777–2784. [Google Scholar] [CrossRef] [Green Version]
- Notton, T.; Sardanyes, J.; Weinberger, A.D.; Weinberger, L.S. The case for transmissible antivirals to control population-wide infectious disease. Trends Biotechnol. 2014, 32, 400–405. [Google Scholar] [CrossRef]
- Lai, M.M.; Baric, R.S.; Makino, S.; Keck, J.G.; Egbert, J.; Leibowitz, J.L.; Stohlman, S.A. Recombination between nonsegmented rna genomes of murine coronaviruses. J. Virol. 1985, 56, 449–456. [Google Scholar] [CrossRef] [Green Version]
- Makino, S.; Stohlman, S.A.; Lai, M.M. Leader sequences of murine coronavirus mrnas can be freely reassorted: Evidence for the role of free leader rna in transcription. Proc. Natl. Acad. Sci. USA 1986, 83, 4204–4208. [Google Scholar] [CrossRef] [Green Version]
- Makino, S.; Keck, J.G.; Stohlman, S.A.; Lai, M.M. High-frequency rna recombination of murine coronaviruses. J. Virol. 1986, 57, 729–737. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.M.; Makino, S.; Soe, L.H.; Shieh, C.K.; Keck, J.G.; Fleming, J.O. Coronavirus: A jumping rna transcription. Cold Spring Harb. Symp. Quant. Biol. 1987, 52, 359–365. [Google Scholar] [CrossRef]
- Lai, M.M. Molecular biology of coronavirus 1986. Adv. Exp. Med. Biol. 1987, 218, 7–13. [Google Scholar] [PubMed] [Green Version]
- Keck, J.G.; Soe, L.H.; Makino, S.; Stohlman, S.A.; Lai, M.M. Rna recombination of murine coronaviruses: Recombination between fusion-positive mouse hepatitis virus a59 and fusion-negative mouse hepatitis virus 2. J. Virol. 1988, 62, 1989–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keck, J.G.; Matsushima, G.K.; Makino, S.; Fleming, J.O.; Vannier, D.M.; Stohlman, S.A.; Lai, M.M. In vivo rna-rna recombination of coronavirus in mouse brain. J. Virol. 1988, 62, 1810–1813. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Shi, M.; Li, J.; Song, P.; Li, N. Construction of SARS-CoV-2 virus-like particles by mammalian expression system. Front. Bioeng. Biotechnol. 2020, 8, 862. [Google Scholar] [CrossRef]
- Plescia, C.B.; David, E.A.; Patra, D.; Sengupta, R.; Amiar, S.; Su, Y.; Stahelin, R.V. SARS-CoV-2 viral budding and entry can be modeled using bsl-2 level virus-like particles. J. Biol. Chem. 2021, 296, 100103. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.; Masters, P.S. The small envelope protein e is not essential for murine coronavirus replication. J. Virol. 2003, 77, 4597–4608. [Google Scholar] [CrossRef] [Green Version]
- Ortego, J.; Escors, D.; Laude, H.; Enjuanes, L. Generation of a replication-competent, propagation-deficient virus vector based on the transmissible gastroenteritis coronavirus genome. J. Virol. 2002, 76, 11518–11529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortego, J.; Ceriani, J.E.; Patino, C.; Plana, J.; Enjuanes, L. Absence of e protein arrests transmissible gastroenteritis coronavirus maturation in the secretory pathway. Virology 2007, 368, 296–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeDiego, M.L.; Alvarez, E.; Almazan, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A severe acute respiratory syndrome coronavirus that lacks the e gene is attenuated in vitro and in vivo. J. Virol. 2007, 81, 1701–1713. [Google Scholar] [CrossRef] [Green Version]
- Dediego, M.L.; Pewe, L.; Alvarez, E.; Rejas, M.T.; Perlman, S.; Enjuanes, L. Pathogenicity of severe acute respiratory coronavirus deletion mutants in hace-2 transgenic mice. Virology 2008, 376, 379–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Castano-Rodriguez, C.; Fernandez-Delgado, R.; Usera, F.; Enjuanes, L. Coronavirus virulence genes with main focus on SARS-CoV envelope gene. Virus Res. 2014, 194, 124–137. [Google Scholar] [CrossRef]
- Krishnamachary, B.; Cook, C.; Spikes, L.; Chalise, P.; Dhillon, N.K. The potential role of extracellular vesicles in COVID-19 associated endothelial injury and pro-inflammation. medRxiv 2020. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Hopfield, J.J.; Leibler, S.; Murray, A.W. From molecular to modular cell biology. Nature 1999, 402, C47–C52. [Google Scholar] [CrossRef] [PubMed]
- Schubert, W.; Gieseler, A.; Krusche, A.; Serocka, P.; Hillert, R. Next-generation biomarkers based on 100-parameter functional super-resolution microscopy tis. New Biotechnol. 2012, 29, 599–610. [Google Scholar] [CrossRef]
- V’Kovski, P.; Al-Mulla, H.; Thiel, V.; Neuman, B.W. New insights on the role of paired membrane structures in coronavirus replication. Virus Res. 2015, 202, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Ulasli, M.; Schepers, H.; Mauthe, M.; V’Kovski, P.; Kriegenburg, F.; Thiel, V.; de Haan, C.A.M.; Reggiori, F. Nucleocapsid protein recruitment to replication-transcription complexes plays a crucial role in coronaviral life cycle. J. Virol. 2020, 94, e01925-19. [Google Scholar] [CrossRef] [Green Version]
- Fodor, E.; Mingay, L.J.; Crow, M.; Deng, T.; Brownlee, G.G. A single amino acid mutation in the pa subunit of the influenza virus rna polymerase promotes the generation of defective interfering rnas. J. Virol. 2003, 77, 5017–5020. [Google Scholar] [CrossRef] [Green Version]
- Vasilijevic, J.; Zamarreno, N.; Oliveros, J.C.; Rodriguez-Frandsen, A.; Gomez, G.; Rodriguez, G.; Perez-Ruiz, M.; Rey, S.; Barba, I.; Pozo, F.; et al. Reduced accumulation of defective viral genomes contributes to severe outcome in influenza virus infected patients. PLoS Pathog. 2017, 13, e1006650. [Google Scholar] [CrossRef] [Green Version]
- Poirier, E.Z.; Mounce, B.C.; Rozen-Gagnon, K.; Hooikaas, P.J.; Stapleford, K.A.; Moratorio, G.; Vignuzzi, M. Low-fidelity polymerases of alphaviruses recombine at higher rates to overproduce defective interfering particles. J. Virol. 2015, 90, 2446–2454. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.P.; Gao, Y.T.; Ge, X.Y.; Zhang, Q.; Peng, C.; Yang, X.L.; Tan, B.; Chen, J.; Chmura, A.A.; Daszak, P.; et al. Bat severe acute respiratory syndrome-like coronavirus wiv1 encodes an extra accessory protein, orfx, involved in modulation of the host immune response. J. Virol. 2016, 90, 6573–6582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkina, R.K.; Chambers, T.M.; Nayak, D.P. Expression of defective-interfering influenza virus-specific transcripts and polypeptides in infected cells. J. Virol. 1984, 51, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Manz, B.; Gotz, V.; Wunderlich, K.; Eisel, J.; Kirchmair, J.; Stech, J.; Stech, O.; Chase, G.; Frank, R.; Schwemmle, M. Disruption of the viral polymerase complex assembly as a novel approach to attenuate influenza a virus. J. Biol. Chem. 2011, 286, 8414–8424. [Google Scholar] [CrossRef] [Green Version]
- Lisziewicz, J.; Rappaport, J.; Dhar, R. Tat-regulated production of multimerized tar rna inhibits hiv-1 gene expression. New Biol. 1991, 3, 82–89. [Google Scholar] [PubMed]
- Sola, I.; Almazan, F.; Zuniga, S.; Enjuanes, L. Continuous and discontinuous rna synthesis in coronaviruses. Annu. Rev. Virol. 2015, 2, 265–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Kim, E.J.; Felt, S.A.; Taylor, L.J.; Agarwal, D.; Grant, G.R.; Lopez, C.B. A specific sequence in the genome of respiratory syncytial virus regulates the generation of copy-back defective viral genomes. PLoS Pathog. 2019, 15, e1007707. [Google Scholar]
- Jennings, P.A.; Finch, J.T.; Winter, G.; Robertson, J.S. Does the higher order structure of the influenza virus ribonucleoprotein guide sequence rearrangements in influenza viral rna? Cell 1983, 34, 619–627. [Google Scholar] [CrossRef]
- Yao, S.; Narayanan, A.; Majowicz, S.A.; Jose, J.; Archetti, M. A synthetic defective interfering SARS-CoV-2. PeerJ 2021, 9, e11686. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Vasen, G.; Pablo, M.; Chen, X.; Beutler, N.; Kumar, A.; Tanner, E.; Illouz, S.; Rahgoshay, D.; Burnett, J.; et al. Identification of a therapeutic interfering particle-a single-dose SARS-CoV-2 antiviral intervention with a high barrier to resistance. Cell 2021, 184, 6022–6036.e18. [Google Scholar] [CrossRef]
- Lisziewicz, J.; Zeng, G.; Gratas, C.; Weinstein, J.N.; Lori, F. Combination gene therapy: Synergistic inhibition of human immunodeficiency virus tat and rev functions by a single rna molecule. Hum. Gene 2000, 11, 807–815. [Google Scholar] [CrossRef]
- Somogyi, E.; Xu, J.; Gudics, A.; Toth, J.; Kovacs, A.L.; Lori, F.; Lisziewicz, J. A plasmid DNA immunogen expressing fifteen protein antigens and complex virus-like particles (vlp+) mimicking naturally occurring hiv. Vaccine 2011, 29, 744–753. [Google Scholar] [CrossRef] [PubMed]
- DermaVir. Available online: http://www.geneticimmunity.com/dermavir.html (accessed on 13 January 2022).
- Strahle, L.; Garcin, D.; Kolakofsky, D. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 2006, 351, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Mercado-Lopez, X.; Grier, J.T.; Kim, W.K.; Chun, L.F.; Irvine, E.B.; Del Toro Duany, Y.; Kell, A.; Hur, S.; Gale, M., Jr.; et al. Identification of a natural viral rna motif that optimizes sensing of viral rna by rig-i. mBio 2015, 6, e01265-15. [Google Scholar] [CrossRef] [Green Version]
- Mercado-Lopez, X.; Cotter, C.R.; Kim, W.K.; Sun, Y.; Munoz, L.; Tapia, K.; Lopez, C.B. Highly immunostimulatory rna derived from a sendai virus defective viral genome. Vaccine 2013, 31, 5713–5721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Gil, L.; Goff, P.H.; Hai, R.; Garcia-Sastre, A.; Shaw, M.L.; Palese, P. A sendai virus-derived rna agonist of rig-i as a virus vaccine adjuvant. J. Virol. 2013, 87, 1290–1300. [Google Scholar] [CrossRef] [Green Version]
- Fisher, D.G.; Coppock, G.M.; Lopez, C.B. Virus-derived immunostimulatory rna induces type i ifn-dependent antibodies and t-cell responses during vaccination. Vaccine 2018, 36, 4039–4045. [Google Scholar] [CrossRef]
- Dimmock, N.J.; Rainsford, E.W.; Scott, P.D.; Marriott, A.C. Influenza virus protecting rna: An effective prophylactic and therapeutic antiviral. J. Virol. 2008, 82, 8570–8578. [Google Scholar] [CrossRef] [Green Version]
- Meng, B.; Bentley, K.; Marriott, A.C.; Scott, P.D.; Dimmock, N.J.; Easton, A.J. Unexpected complexity in the interference activity of a cloned influenza defective interfering rna. Virol. J. 2017, 14, 138. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.M.; Scott, P.D.; O’Callaghan, C.; Easton, A.J.; Dimmock, N.J. A defective interfering influenza rna inhibits infectious influenza virus replication in human respiratory tract cells: A potential new human antiviral. Viruses 2016, 8, 237. [Google Scholar] [CrossRef]
- Mantlo, E.; Bukreyeva, N.; Maruyama, J.; Paessler, S.; Huang, C. Antiviral activities of type i interferons to SARS-CoV-2 infection. Antivir. Res 2020, 179, 104811. [Google Scholar] [CrossRef] [PubMed]
- Rand, U.; Kupke, S.Y.; Shkarlet, H.; Hein, M.D.; Hirsch, T.; Marichal-Gallardo, P.; Cicin-Sain, L.; Reichl, U.; Bruder, D. Antiviral activity of influenza a virus defective interfering particles against SARS-CoV-2 replication in vitro through stimulation of innate immunity. Cells 2021, 10, 1756. [Google Scholar] [CrossRef] [PubMed]
- Dogrammatzis, C.; Saleh, S.; Deighan, C.; Kalamvoki, M. Diverse populations of extracellular vesicles with opposite functions during herpes simplex virus 1 infection. J. Virol. 2021, 95, e02357-20. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.L. Coronavirus rna proofreading: Molecular basis and therapeutic targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef]
- Gagliardi, M.; Ashizawa, A.T. The challenges and strategies of antisense oligonucleotide drug delivery. Biomedicines 2021, 9, 433. [Google Scholar] [CrossRef] [PubMed]
- Neuman, B.W.; Stein, D.A.; Kroeker, A.D.; Churchill, M.J.; Kim, A.M.; Kuhn, P.; Dawson, P.; Moulton, H.M.; Bestwick, R.K.; Iversen, P.L.; et al. Inhibition, escape, and attenuated growth of severe acute respiratory syndrome coronavirus treated with antisense morpholino oligomers. J. Virol. 2005, 79, 9665–9676. [Google Scholar] [CrossRef] [Green Version]
- Shimo, T.; Maruyama, R.; Yokota, T. Designing effective antisense oligonucleotides for exon skipping. Methods Mol. Biol. 2018, 1687, 143–155. [Google Scholar]
- Karaki, S.; Benizri, S.; Mejias, R.; Baylot, V.; Branger, N.; Nguyen, T.; Vialet, B.; Oumzil, K.; Barthelemy, P.; Rocchi, P. Lipid-oligonucleotide conjugates improve cellular uptake and efficiency of tctp-antisense in castration-resistant prostate cancer. J. Control Release 2017, 258, 1–9. [Google Scholar] [CrossRef]
- Posocco, P.; Liu, X.; Laurini, E.; Marson, D.; Chen, C.; Liu, C.; Fermeglia, M.; Rocchi, P.; Pricl, S.; Peng, L. Impact of sirna overhangs for dendrimer-mediated sirna delivery and gene silencing. Mol. Pharm. 2013, 10, 3262–3273. [Google Scholar] [CrossRef]
- Luvino, D.; Khiati, S.; Oumzil, K.; Rocchi, P.; Camplo, M.; Barthelemy, P. Efficient delivery of therapeutic small nucleic acids to prostate cancer cells using ketal nucleoside lipid nanoparticles. J. Control Release 2013, 172, 954–961. [Google Scholar] [CrossRef]
- Chan, S.K.; Du, P.; Ignacio, K.; Metha, S.; Newton, I.G.; Steinmetz, N.F. Biomimetic virus-like particles as SARS-CoV-2 positive controls for rt-pcr diagnostics. medRxiv 2021, 15, 1259–1272. [Google Scholar] [CrossRef]
- Karimi, M.; Mirshekari, H.; Moosavi Basri, S.M.; Bahrami, S.; Moghoofei, M.; Hamblin, M.R. Bacteriophages and phage-inspired nanocarriers for targeted delivery of therapeutic cargos. Adv. Drug Deliv. Rev. 2016, 106, 45–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storni, T.; Ruedl, C.; Schwarz, K.; Schwendener, R.A.; Renner, W.A.; Bachmann, M.F. Nonmethylated cg motifs packaged into virus-like particles induce protective cytotoxic t cell responses in the absence of systemic side effects. J. Immunol. 2004, 172, 1777–1785. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.; Tuerk, C.; Gold, L. Selection of high affinity rna ligands to the bacteriophage r17 coat protein. J. Mol. Biol. 1992, 228, 862–869. [Google Scholar] [CrossRef]
- Torres-Chavolla, E.; Alocilja, E.C. Aptasensors for detection of microbial and viral pathogens. Biosens. Bioelectron. 2009, 24, 3175–3182. [Google Scholar] [CrossRef]
- Ku, T.H.; Zhang, T.; Luo, H.; Yen, T.M.; Chen, P.W.; Han, Y.; Lo, Y.H. Nucleic acid aptamers: An emerging tool for biotechnology and biomedical sensing. Sensors 2015, 15, 16281–16313. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, J. Antibodies are challenged. Indian J. Med. Sci. 2010, 64, 144–147. [Google Scholar]
- Szpechcinski, A.; Grzanka, A. Aptamers in clinical diagnostics. Postepy Biochem. 2006, 52, 260–270. [Google Scholar]
- Zou, X.; Wu, J.; Gu, J.; Shen, L.; Mao, L. Application of aptamers in virus detection and antiviral therapy. Front. Microbiol. 2019, 10, 1462. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Zeng, H. Aptamer-based elisa assay for highly specific and sensitive detection of zika ns1 protein. Anal. Chem. 2017, 89, 12743–12748. [Google Scholar] [CrossRef]
- Chen, H.L.; Hsiao, W.H.; Lee, H.C.; Wu, S.C.; Cheng, J.W. Selection and characterization of DNA aptamers targeting all four serotypes of dengue viruses. PLoS ONE 2015, 10, e0131240. [Google Scholar] [CrossRef]
- Gopinath, S.C.; Hayashi, K.; Kumar, P.K. Aptamer that binds to the gd protein of herpes simplex virus 1 and efficiently inhibits viral entry. J. Virol. 2012, 86, 6732–6744. [Google Scholar] [CrossRef] [Green Version]
- Yadavalli, T.; Agelidis, A.; Jaishankar, D.; Mangano, K.; Thakkar, N.; Penmetcha, K.; Shukla, D. Targeting herpes simplex virus-1 gd by a DNA aptamer can be an effective new strategy to curb viral infection. Mol. Nucleic Acids 2017, 9, 365–378. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, D.; Duclair, S.; Datta, S.A.; Ellington, A.; Rein, A.; Prasad, V.R. Rna aptamers directed to human immunodeficiency virus type 1 gag polyprotein bind to the matrix and nucleocapsid domains and inhibit virus production. J. Virol. 2011, 85, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Jang, K.J.; Lee, N.R.; Yeo, W.S.; Jeong, Y.J.; Kim, D.E. Isolation of inhibitory rna aptamers against severe acute respiratory syndrome (sars) coronavirus ntpase/helicase. Biochem. Biophys. Res. Commun. 2008, 366, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Shum, K.T.; Tanner, J.A. Differential inhibitory activities and stabilisation of DNA aptamers against the sars coronavirus helicase. Chembiochem 2008, 9, 3037–3045. [Google Scholar] [CrossRef] [PubMed]
- Bellecave, P.; Cazenave, C.; Rumi, J.; Staedel, C.; Cosnefroy, O.; Andreola, M.L.; Ventura, M.; Tarrago-Litvak, L.; Astier-Gin, T. Inhibition of hepatitis c virus (hcv) rna polymerase by DNA aptamers: Mechanism of inhibition of in vitro rna synthesis and effect on hcv-infected cells. Antimicrob. Agents Chemother. 2008, 52, 2097–2110. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.J.; Woo, H.M.; Kim, K.S.; Oh, J.W.; Jeong, Y.J. Novel system for detecting sars coronavirus nucleocapsid protein using an ssdna aptamer. J. Biosci. Bioeng. 2011, 112, 535–540. [Google Scholar] [CrossRef]
- Chen, Z.; Wu, Q.; Chen, J.; Ni, X.; Dai, J. A DNA aptamer based method for detection of SARS-CoV-2 nucleocapsid protein. Virol. Sin. 2020, 35, 351–354. [Google Scholar] [CrossRef] [PubMed]
- McBride, R.; van Zyl, M.; Fielding, B.C. The coronavirus nucleocapsid is a multifunctional protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Wang, H.; Ji, Y.; Yang, J.; Xu, S.; Huang, X.; Wang, Z.; Qin, L.; Tien, P.; Zhou, X.; et al. The nucleocapsid protein of coronaviruses acts as a viral suppressor of rna silencing in mammalian cells. J. Virol. 2015, 89, 9029–9043. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.L.; McKeague, M. Aptamers in the pursuit of COVID-19 management. Aptamers 2020, 4, 1–2. [Google Scholar]
- Liebich, S. Potent selex aptamer-based therapeutic method for novel SARS-CoV2 virus disease (COVID-19). COVID-19 BabuBio 2020, 53, 101636. [Google Scholar]
- Rangan, R.; Watkins, A.M.; Chacon, J.; Kladwang, W.; Zheludev, I.N.; Townley, J.; Rynge, M.; Thain, G.; Das, R. De novo 3d models of SARS-CoV-2 rna elements and small-molecule-binding rnas to aid drug discovery. bioRxiv 2020, 49, 3092–3108. [Google Scholar] [CrossRef]
- Song, Y.; Song, J.; Wei, X.; Huang, M.; Sun, M.; Zhu, L.; Lin, B.; Shen, H.; Zhu, Z.; Yang, C. Discovery of aptamers targeting the receptor-binding domain of the SARS-CoV-2 spike glycoprotein. Anal. Chem. 2020, 92, 9895–9900. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J.J. Aptamer-targeted rnai for hiv-1 therapy. Methods Mol. Biol. 2011, 721, 355–371. [Google Scholar] [PubMed]
- Matsukura, M.; Zon, G.; Shinozuka, K.; Robert-Guroff, M.; Shimada, T.; Stein, C.A.; Mitsuya, H.; Wong-Staal, F.; Cohen, J.S.; Broder, S. Regulation of viral expression of human immunodeficiency virus in vitro by an antisense phosphorothioate oligodeoxynucleotide against rev (art/trs) in chronically infected cells. Proc. Natl. Acad. Sci. USA 1989, 86, 4244–4248. [Google Scholar] [CrossRef] [Green Version]
- Natsoulis, G.; Boeke, J.D. New antiviral strategy using capsid-nuclease fusion proteins. Nature 1991, 352, 632–635. [Google Scholar] [CrossRef]
- Natsoulis, G.; Seshaiah, P.; Federspiel, M.J.; Rein, A.; Hughes, S.H.; Boeke, J.D. Targeting of a nuclease to murine leukemia virus capsids inhibits viral multiplication. Proc. Natl. Acad. Sci. USA 1995, 92, 364–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Ojwang, J.; Yamada, O.; Hampel, A.; Rapapport, J.; Looney, D.; Wong-Staal, F. A hairpin ribozyme inhibits expression of diverse strains of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 1993, 90, 6340–6344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asha, K.; Kumar, P.; Sanicas, M.; Meseko, C.A.; Khanna, M.; Kumar, B. Advancements in nucleic acid based therapeutics against respiratory viral infections. J. Clin. Med. 2018, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Goguen, R.P.; Malard, C.M.; Scarborough, R.J.; Gatignol, A. Small rnas to treat human immunodeficiency virus type 1 infection by gene therapy. Curr. Opin. Virol. 2019, 38, 10–20. [Google Scholar] [CrossRef]
- Nguyen, P.D.M.; Zheng, J.; Gremminger, T.J.; Qiu, L.; Zhang, D.; Tuske, S.; Lange, M.J.; Griffin, P.R.; Arnold, E.; Chen, S.J.; et al. Binding interface and impact on protease cleavage for an rna aptamer to hiv-1 reverse transcriptase. Nucleic Acids Res. 2020, 48, 2709–2722. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Wang, H.; He, W.; Li, Y.; Mou, H.; Tickner, Z.J.; Tran, M.H.; Ou, T.; Yin, Y.; Diao, H.; et al. A reversible rna on-switch that controls gene expression of aav-delivered therapeutics in vivo. Nat. Biotechnol. 2020, 38, 169–175. [Google Scholar] [CrossRef]
- Nguyen, T.M.; Zhang, Y.; Pandolfi, P.P. Virus against virus: A potential treatment for 2019-ncov (SARS-CoV-2) and other rna viruses. Cell Res. 2020, 30, 189–190. [Google Scholar] [CrossRef] [Green Version]
- Wrapp, D.; De Vlieger, D.; Corbett, K.S.; Torres, G.M.; Wang, N.; Van Breedam, W.; Roose, K.; van Schie, L.; Team, V.-C.C.-R.; Hoffmann, M.; et al. Structural basis for potent neutralization of betacoronaviruses by single-domain camelid antibodies. Cell 2020, 181, 1004–1015.e1015. [Google Scholar] [CrossRef]
- Cappellano, G.; Raineri, D.; Rolla, R.; Giordano, M.; Puricelli, C.; Vilardo, B.; Manfredi, M.; Cantaluppi, V.; Sainaghi, P.P.; Castello, L.; et al. Circulating platelet-derived extracellular vesicles are a hallmark of SARS-CoV-2 infection. Cells 2021, 10, 85. [Google Scholar] [CrossRef]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; et al. Platelets can associate with SARS-CoV-2 rna and are hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418. [Google Scholar] [CrossRef]
- Traby, L.; Kollars, M.; Kussmann, M.; Karer, M.; Sinkovec, H.; Lobmeyr, E.; Hermann, A.; Staudinger, T.; Schellongowski, P.; Rossler, B.; et al. Extracellular vesicles and citrullinated histone h3 in coronavirus disease 2019 patients. Thromb. Haemost. 2021. [Google Scholar]
- Kumar, L.; Verma, S.; Vaidya, B.; Gupta, V. Exosomes: Natural carriers for sirna delivery. Curr. Pharm. Des. 2015, 21, 4556–4565. [Google Scholar] [CrossRef]
- Kumar, S.; Zhi, K.; Mukherji, A.; Gerth, K. Repurposing antiviral protease inhibitors using extracellular vesicles for potential therapy of COVID-19. Viruses 2020, 12, 486. [Google Scholar] [CrossRef]
- Rayamajhi, S.; Nguyen, T.D.T.; Marasini, R.; Aryal, S. Macrophage-derived exosome-mimetic hybrid vesicles for tumor targeted drug delivery. Acta Biomater. 2019, 94, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Vallhov, H.; Gutzeit, C.; Johansson, S.M.; Nagy, N.; Paul, M.; Li, Q.; Friend, S.; George, T.C.; Klein, E.; Scheynius, A.; et al. Exosomes containing glycoprotein 350 released by ebv-transformed b cells selectively target b cells through cd21 and block ebv infection in vitro. J. Immunol. 2011, 186, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Shennawy, L.; Hoffmann, A.D.; Dashzeveg, N.K.; Mehl, P.J.; Yu, Z.; Tokars, V.L.; Nicolaescu, V.; Ostiguin, C.; Jia, Y.; Li, L.; et al. Circulating ace2-expressing exosomes block SARS-CoV-2 infection as an innate antiviral mechanism. bioRxiv 2020. [Google Scholar] [CrossRef]
- Park, K.S.; Bandeira, E.; Shelke, G.V.; Lasser, C.; Lotvall, J. Enhancement of therapeutic potential of mesenchymal stem cell-derived extracellular vesicles. Stem Cell Res. Ther. 2019, 10, 288. [Google Scholar] [CrossRef]
- Shah, T.; Qin, S.; Vashi, M.; Predescu, D.N.; Jeganathan, N.; Bardita, C.; Ganesh, B.; diBartolo, S.; Fogg, L.F.; Balk, R.A.; et al. Alk5/runx1 signaling mediated by extracellular vesicles promotes vascular repair in acute respiratory distress syndrome. Clin. Transl. Med. 2018, 7, 19. [Google Scholar] [CrossRef]
- Inal, J.M. Decoy ace2-expressing extracellular vesicles that competitively bind SARS-CoV-2 as a possible COVID-19 therapy. Clin. Sci. 2020, 134, 1301–1304. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Kang, I.; Yu, K.R. Therapeutic features and updated clinical trials of mesenchymal stem cell (msc)-derived exosomes. J. Clin. Med. 2021, 10, 711. [Google Scholar] [CrossRef]
- Lindenbergh, M.F.S.; Stoorvogel, W. Antigen presentation by extracellular vesicles from professional antigen-presenting cells. Annu. Rev. Immunol. 2018, 36, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Norris, V.; Thierry, A.; Holland, I.B.; Amar, P.; Molina, F. The mimic chain reaction. J. Mol. Microbiol. Biotechnol. 2012, 22, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Deschamps, T.; Kalamvoki, M. Extracellular vesicles released by herpes simplex virus 1-infected cells block virus replication in recipient cells in a sting-dependent manner. J. Virol. 2018, 92, e01102-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogrammatzis, C.; Deschamps, T.; Kalamvoki, M. Biogenesis of extracellular vesicles during herpes simplex virus 1 infection: Role of the cd63 tetraspanin. J. Virol. 2019, 93, e01850-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nkosi, D.; Sun, L.; Duke, L.C.; Patel, N.; Surapaneni, S.K.; Singh, M.; Meckes, D.G., Jr. Epstein-barr virus lmp1 promotes syntenin-1- and hrs-induced extracellular vesicle formation for its own secretion to increase cell proliferation and migration. mBio 2020, 11, e00589-20. [Google Scholar] [CrossRef]
- McNamara, R.P.; Chugh, P.E.; Bailey, A.; Costantini, L.M.; Ma, Z.; Bigi, R.; Cheves, A.; Eason, A.B.; Landis, J.T.; Host, K.M.; et al. Extracellular vesicles from kaposi sarcoma-associated herpesvirus lymphoma induce long-term endothelial cell reprogramming. PLoS Pathog. 2019, 15, e1007536. [Google Scholar] [CrossRef] [Green Version]
- Lakhin, A.V.; Tarantul, V.Z.; Gening, L.V. Aptamers: Problems, solutions and prospects. Acta Nat. 2013, 5, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Norris, V.; Ovadi, J. Role of multifunctional cytoskeletal filaments in coronaviri-dae infections: Therapeutic opportunities for COVID-19 in a nutshell. Cells 2021, 10, 1818. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ace2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ace2 and tmprss2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-ncov) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, S.; Yan, L.; Xu, W.; Agrawal, A.S.; Algaissi, A.; Tseng, C.K.; Wang, Q.; Du, L.; Tan, W.; Wilson, I.A.; et al. A pan-coronavirus fusion inhibitor targeting the hr1 domain of human coronavirus spike. Sci. Adv. 2019, 5, eaav4580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.K.; Lo, S.C.; Wang, Y.S.; Hou, M.H. Recent insights into the development of therapeutics against coronavirus diseases by targeting n protein. Drug Discov. Today 2016, 21, 562–572. [Google Scholar] [CrossRef]
- Wu, C.H.; Chen, P.J.; Yeh, S.H. Nucleocapsid phosphorylation and rna helicase ddx1 recruitment enables coronavirus transition from discontinuous to continuous transcription. Cell Host. Microbe 2014, 16, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Yang, M.; Hong, Z.; Zhang, L.; Huang, Z.; Chen, X.; He, S.; Zhou, Z.; Zhou, Z.; Chen, Q.; et al. Crystal structure of SARS-CoV-2 nucleocapsid protein rna binding domain reveals potential unique drug targeting sites. Acta Pharm. Sin. B 2020, 10, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Sarma, P.; Sekhar, N.; Prajapat, M.; Avti, P.; Kaur, H.; Kumar, S.; Singh, S.; Kumar, H.; Prakash, A.; Dhibar, D.P.; et al. In-silico homology assisted identification of inhibitor of rna binding against 2019-ncov n-protein (n terminal domain). J. Biomol. Struct. Dyn. 2020, 39, 2724–2732. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.P.; Liu, D.X. The missing link in coronavirus assembly. Retention of the avian coronavirus infectious bronchitis virus envelope protein in the pre-golgi compartments and physical interaction between the envelope and membrane proteins. J. Biol. Chem. 2001, 276, 17515–17523. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.P.; Xu, H.Y.; Liu, D.X. Physical interaction between the membrane (m) and envelope (e) proteins of the coronavirus avian infectious bronchitis virus (ibv). Adv. Exp. Med. Biol. 2001, 494, 595–602. [Google Scholar]
- Bos, E.C.; Luytjes, W.; van der Meulen, H.V.; Koerten, H.K.; Spaan, W.J. The production of recombinant infectious di-particles of a murine coronavirus in the absence of helper virus. Virology 1996, 218, 52–60. [Google Scholar] [CrossRef]
- Vennema, H.; Godeke, G.J.; Rossen, J.W.; Voorhout, W.F.; Horzinek, M.C.; Opstelten, D.J.; Rottier, P.J. Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein genes. EMBO J. 1996, 15, 2020–2028. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Verdia-Baguena, C.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Castano-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe acute respiratory syndrome coronavirus e protein transports calcium ions and activates the nlrp3 inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Castano-Rodriguez, C.; Honrubia, J.M.; Gutierrez-Alvarez, J.; DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Verdia-Baguena, C.; Queralt-Martin, M.; et al. Role of severe acute respiratory syndrome coronavirus viroporins e, 3a, and 8a in replication and pathogenesis. mBio 2018, 9, e02325-17. [Google Scholar] [CrossRef] [Green Version]
- Breitinger, U.; Ali, N.K.M.; Sticht, H.; Breitinger, H.-G. Inhibition of SARS Cov envelope protein by flavonoids and classical viroporin inhibitors. Front. Microbiol. 2021, 12, 692423. [Google Scholar] [CrossRef]
- Siu, Y.L.; Teoh, K.T.; Lo, J.; Chan, C.M.; Kien, F.; Escriou, N.; Tsao, S.W.; Nicholls, J.M.; Altmeyer, R.; Peiris, J.S.; et al. The m, e, and n structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. J. Virol. 2008, 82, 11318–11330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.A.; Leibundgut, M.; Thiel, V.; Muhlemann, O.; Ban, N. SARS-CoV-2 nsp1 binds the ribosomal mrna channel to inhibit translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural basis for translational shutdown and immune evasion by the nsp1 protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Frieman, M.; Yount, B.; Agnihothram, S.; Page, C.; Donaldson, E.; Roberts, A.; Vogel, L.; Woodruff, B.; Scorpio, D.; Subbarao, K.; et al. Molecular determinants of severe acute respiratory syndrome coronavirus pathogenesis and virulence in young and aged mouse models of human disease. J. Virol. 2012, 86, 884–897. [Google Scholar] [CrossRef] [Green Version]
- Littler, D.R.; Gully, B.S.; Colson, R.N.; Rossjohn, J. Crystal structure of the SARS-CoV-2 non-structural protein 9, nsp9. iScience 2020, 23, 101258. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, Y.; Li, L.; Yang, Y.; He, J.; Chen, C.; Su, D. Structural basis for the multimerization of nonstructural protein nsp9 from SARS-CoV-2. Mol. Biomed. 2020, 1, 5. [Google Scholar] [CrossRef]
- Siu, K.L.; Yuen, K.S.; Castano-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome coronavirus orf3a protein activates the nlrp3 inflammasome by promoting traf3-dependent ubiquitination of asc. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef] [PubMed]
- Issa, E.; Merhi, G.; Panossian, B.; Salloum, T.; Tokajian, S. SARS-CoV-2 and orf3a: Nonsynonymous mutations, functional domains, and viral pathogenesis. mSystems 2020, 5, e00266-20. [Google Scholar] [CrossRef]
- Mohammad, S.; Bouchama, A.; Mohammad Alharbi, B.; Rashid, M.; Saleem Khatlani, T.; Gaber, N.S.; Malik, S.S. SARS-CoV-2 orf8 and SARS-CoV orf8ab: Genomic divergence and functional convergence. Pathogens 2020, 9, 677. [Google Scholar] [CrossRef]
- Arya, R.; Kumari, S.; Pandey, B.; Mistry, H.; Bihani, S.C.; Das, A.; Prashar, V.; Gupta, G.D.; Panicker, L.; Kumar, M. Structural insights into SARS-CoV-2 proteins. J. Mol. Biol. 2021, 433, 166725. [Google Scholar] [CrossRef]
- Neuman, B.W.; Joseph, J.S.; Saikatendu, K.S.; Serrano, P.; Chatterjee, A.; Johnson, M.A.; Liao, L.; Klaus, J.P.; Yates, J.R., 3rd; Wuthrich, K.; et al. Proteomics analysis unravels the functional repertoire of coronavirus nonstructural protein 3. J. Virol. 2008, 82, 5279–5294. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Yu, X. Adp-ribosyltransferases and poly adp-ribosylation. Curr. Protein Pept. Sci. 2015, 16, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Harcourt, B.H.; Jukneliene, D.; Kanjanahaluethai, A.; Bechill, J.; Severson, K.M.; Smith, C.M.; Rota, P.A.; Baker, S.C. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 2004, 78, 13600–13612. [Google Scholar] [CrossRef] [Green Version]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes. Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Wolff, G.; Limpens, R.; Zevenhoven-Dobbe, J.C.; Laugks, U.; Zheng, S.; de Jong, A.W.M.; Koning, R.I.; Agard, D.A.; Grunewald, K.; Koster, A.J.; et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science 2020, 369, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Stobart, C.C.; Sexton, N.R.; Munjal, H.; Lu, X.; Molland, K.L.; Tomar, S.; Mesecar, A.D.; Denison, M.R. Chimeric exchange of coronavirus nsp5 proteases (3clpro) identifies common and divergent regulatory determinants of protease activity. J. Virol. 2013, 87, 12611–12618. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; O’Meara, M.J.; Guo, J.Z.; Swaney, D.L.; Tummino, T.A.; Huttenhain, R.; et al. A SARS-CoV-2-human protein-protein interaction map reveals drug targets and potential drug-repurposing. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Moustaqil, M.; Ollivier, E.; Chiu, H.-P.; Van Tol, S.; Rudolffi-Soto, P.; Stevens, C.; Bhumkar, A.; Hunter, D.J.B.; Freiberg, A.; Jacques, D.; et al. SARS-CoV-2 proteases cleave irf3 and critical modulators of inflammatory pathways (nlrp12 and tab1): Implications for disease presentation across species and the search for reservoir hosts. bioRxiv 2020, 31, 392–403. [Google Scholar] [CrossRef]
- Snijder, E.J.; Bredenbeek, P.J.; Dobbe, J.C.; Thiel, V.; Ziebuhr, J.; Poon, L.L.; Guan, Y.; Rozanov, M.; Spaan, W.J.; Gorbalenya, A.E. Unique and conserved features of genome and proteome of sars-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003, 331, 991–1004. [Google Scholar] [CrossRef]
- Gribble, J.; Pruijssers, A.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Stevens, L.J.; Routh, A.L.; Denison, M.R. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. PLoS Pathog. 2021, 17, e1009226. [Google Scholar] [CrossRef]
- Case, J.B.; Ashbrook, A.W.; Dermody, T.S.; Denison, M.R. Mutagenesis of s-adenosyl-l-methionine-binding residues in coronavirus nsp14 n7-methyltransferase demonstrates differing requirements for genome translation and resistance to innate immunity. J. Virol. 2016, 90, 7248–7256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, A.; Le, N.T.; Selisko, B.; Eydoux, C.; Alvarez, K.; Guillemot, J.C.; Decroly, E.; Peersen, O.; Ferron, F.; Canard, B. Remdesivir and SARS-CoV-2: Structural requirements at both nsp12 rdrp and nsp14 exonuclease active-sites. Antivir. Res 2020, 178, 104793. [Google Scholar] [CrossRef] [PubMed]
- Eckerle, L.D.; Lu, X.; Sperry, S.M.; Choi, L.; Denison, M.R. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 2007, 81, 12135–12144. [Google Scholar] [CrossRef] [Green Version]
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus susceptibility to the antiviral remdesivir (gs-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio 2018, 9, e00221-18. [Google Scholar] [CrossRef] [Green Version]
- Ferron, F.; Subissi, L.; Silveira De Morais, A.T.; Le, N.T.T.; Sevajol, M.; Gluais, L.; Decroly, E.; Vonrhein, C.; Bricogne, G.; Canard, B.; et al. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus rna. Proc. Natl. Acad. Sci. USA 2018, 115, E162–E171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Sun, Y.; Wu, A.; Xu, S.; Pan, R.; Zeng, C.; Jin, X.; Ge, X.; Shi, Z.; Ahola, T.; et al. Coronavirus nsp10/nsp16 methyltransferase can be targeted by nsp10-derived peptide in vitro and in vivo to reduce replication and pathogenesis. J. Virol. 2015, 89, 8416–8427. [Google Scholar] [CrossRef] [Green Version]
- Krafcikova, P.; Silhan, J.; Nencka, R.; Boura, E. Structural analysis of the SARS-CoV-2 methyltransferase complex involved in rna cap creation bound to sinefungin. Nat. Commun. 2020, 11, 3717. [Google Scholar] [CrossRef]
- Hyde, J.L.; Diamond, M.S. Innate immune restriction and antagonism of viral rna lacking 2-o methylation. Virology 2015, 479–480, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Z.; Yan, L.; Ren, Z.; Wu, L.; Wang, J.; Guo, J.; Zheng, L.; Ming, Z.; Zhang, L.; Lou, Z.; et al. Delicate structural coordination of the severe acute respiratory syndrome coronavirus nsp13 upon atp hydrolysis. Nucleic Acids Res. 2019, 47, 6538–6550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, J.A.; Watt, R.M.; Chai, Y.B.; Lu, L.Y.; Lin, M.C.; Peiris, J.S.; Poon, L.L.; Kung, H.F.; Huang, J.D. The severe acute respiratory syndrome (sars) coronavirus ntpase/helicase belongs to a distinct class of 5′ to 3′ viral helicases. J. Biol. Chem. 2003, 278, 39578–39582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, K.A.; Ziebuhr, J. Human coronavirus 229e nonstructural protein 13: Characterization of duplex-unwinding, nucleoside triphosphatase, and rna 5′-triphosphatase activities. J. Virol. 2004, 78, 7833–7838. [Google Scholar] [CrossRef] [Green Version]
- Habtemariam, S.; Nabavi, S.F.; Banach, M.; Berindan-Neagoe, I.; Sarkar, K.; Sil, P.C.; Nabavi, S.M. Should we try SARS-CoV-2 helicase inhibitors for COVID-19 therapy? Arch. Med. Res. 2020, 52, 733–735. [Google Scholar] [CrossRef]
- Deng, X.; Baker, S.C. An “Old” Protein with a new story: Coronavirus endoribonuclease is important for evading host antiviral defenses. Virology 2018, 517, 157–163. [Google Scholar] [CrossRef]
- Kim, Y.; Jedrzejczak, R.; Maltseva, N.I.; Endres, M.; Godzik, A.; Michalska, K.; Joachimiak, A. Crystal structure of nsp15 endoribonuclease nendou from SARS-CoV-2. Protein Sci. 2020, 29, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Hackbart, M.; Deng, X.; Baker, S.C. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc. Natl. Acad. Sci. USA 2020, 117, 8094–8103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 2013, 4, e00524-13. [Google Scholar] [CrossRef] [Green Version]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottam, E.M.; Whelband, M.C.; Wileman, T. Coronavirus nsp6 restricts autophagosome expansion. Autophagy 2014, 10, 1426–1441. [Google Scholar] [CrossRef] [Green Version]
- Benvenuto, D.; Angeletti, S.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evolutionary analysis of SARS-CoV-2: How mutation of non-structural protein 6 (nsp6) could affect viral autophagy. J. Infect. 2020, 81, e24–e27. [Google Scholar] [CrossRef]
- Chazal, N.; Gerlier, D. Virus entry, assembly, budding, and membrane rafts. Microbiol. Mol. Biol. Rev. 2003, 67, 226–237. [Google Scholar] [CrossRef] [Green Version]
- Hsu, N.Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for rna replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, P.D.; Strating, J.R.; van Kuppeveld, F.J. Building viral replication organelles: Close encounters of the membrane types. PLoS Pathog. 2016, 12, e1005912. [Google Scholar] [CrossRef]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis c virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010, 6, e1000719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoops, K.; Kikkert, M.; Worm, S.H.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. Sars-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Hardt, M.; Schwudke, D.; Neuman, B.W.; Pleschka, S.; Ziebuhr, J. Inhibition of cytosolic phospholipase a2alpha impairs an early step of coronavirus replication in cell culture. J. Virol. 2018, 92, e01463-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, B.; Chu, H.; Yang, D.; Sze, K.H.; Lai, P.M.; Yuan, S.; Shuai, H.; Wang, Y.; Kao, R.Y.; Chan, J.F.; et al. Characterization of the lipidomic profile of human coronavirus-infected cells: Implications for lipid metabolism remodeling upon coronavirus replication. Viruses 2019, 11, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Chu, H.; Chan, J.F.; Ye, Z.W.; Wen, L.; Yan, B.; Lai, P.M.; Tee, K.M.; Huang, J.; Chen, D.; et al. Srebp-dependent lipidomic reprogramming as a broad-spectrum antiviral target. Nat. Commun. 2019, 10, 120. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalamvoki, M.; Norris, V. A Defective Viral Particle Approach to COVID-19. Cells 2022, 11, 302. https://doi.org/10.3390/cells11020302
Kalamvoki M, Norris V. A Defective Viral Particle Approach to COVID-19. Cells. 2022; 11(2):302. https://doi.org/10.3390/cells11020302
Chicago/Turabian StyleKalamvoki, Maria, and Vic Norris. 2022. "A Defective Viral Particle Approach to COVID-19" Cells 11, no. 2: 302. https://doi.org/10.3390/cells11020302
APA StyleKalamvoki, M., & Norris, V. (2022). A Defective Viral Particle Approach to COVID-19. Cells, 11(2), 302. https://doi.org/10.3390/cells11020302