


Drepmel—A Multi-Omics Melanoma Drug Repurposing Resource for Prioritizing Drug Combinations and Understanding Tumor Microenvironment

Abstract
1. Introduction
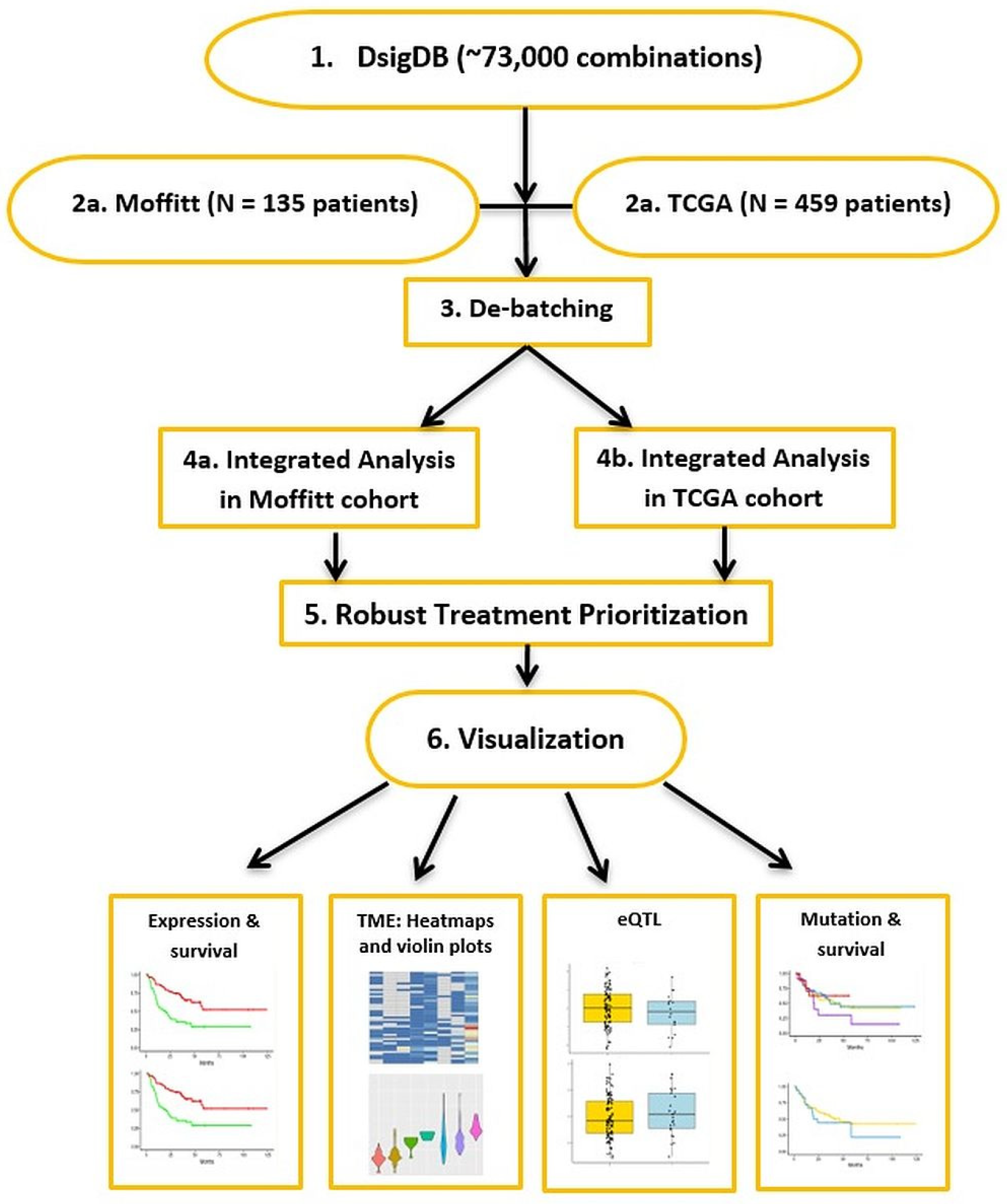
2. Methods
2.1. WES and RNA-Seq Sequence Analyses
2.2. Doublet Combination Therapy Candidates
2.3. Drug Repurposing Models for Doublets
2.4. Potential TME Targeted by the Predicted Doublet Therapies
3. Web Application and Results
- Tables of top doublet combinations summarized the overall results and those for each of the major melanoma genotype groups.
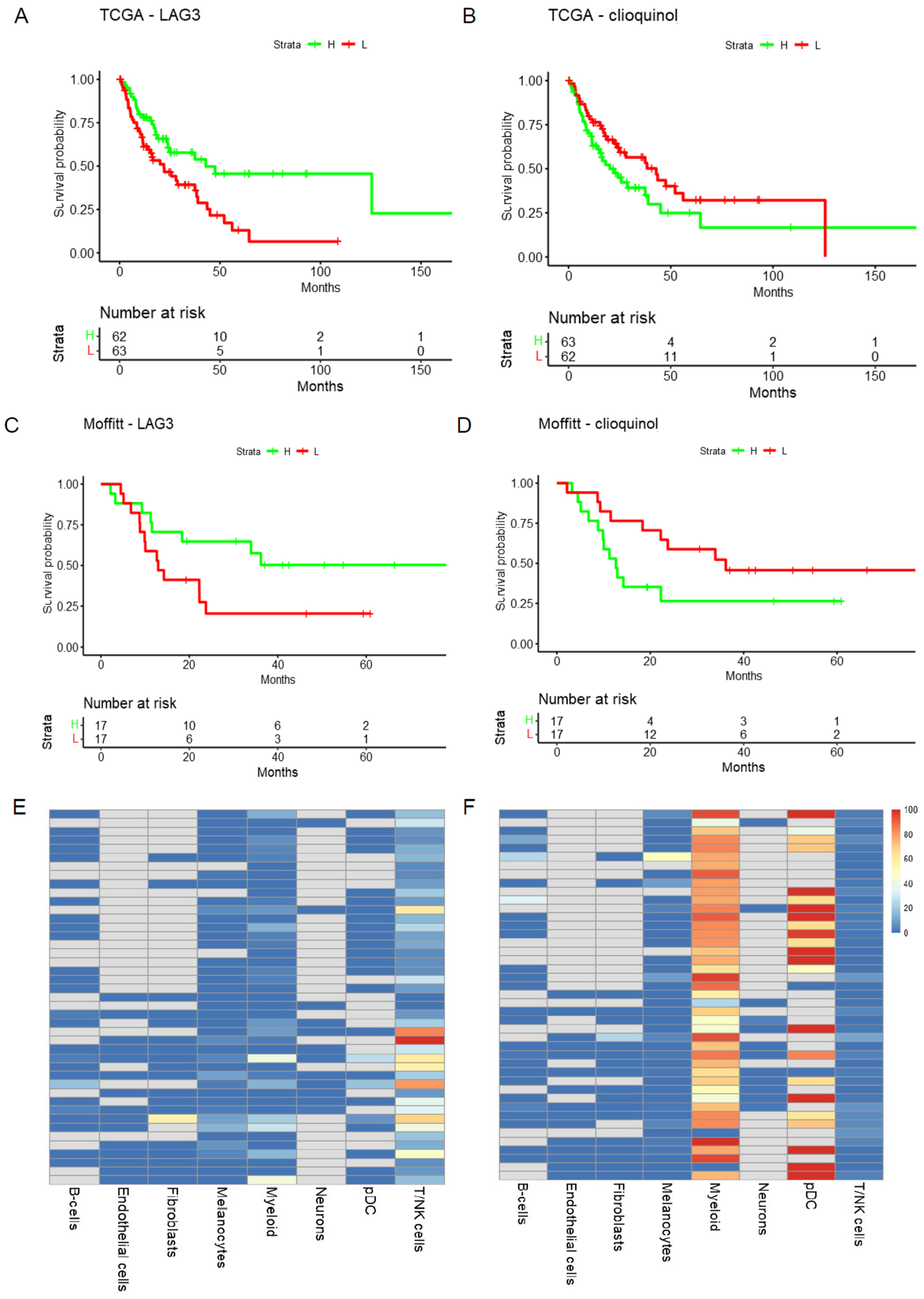
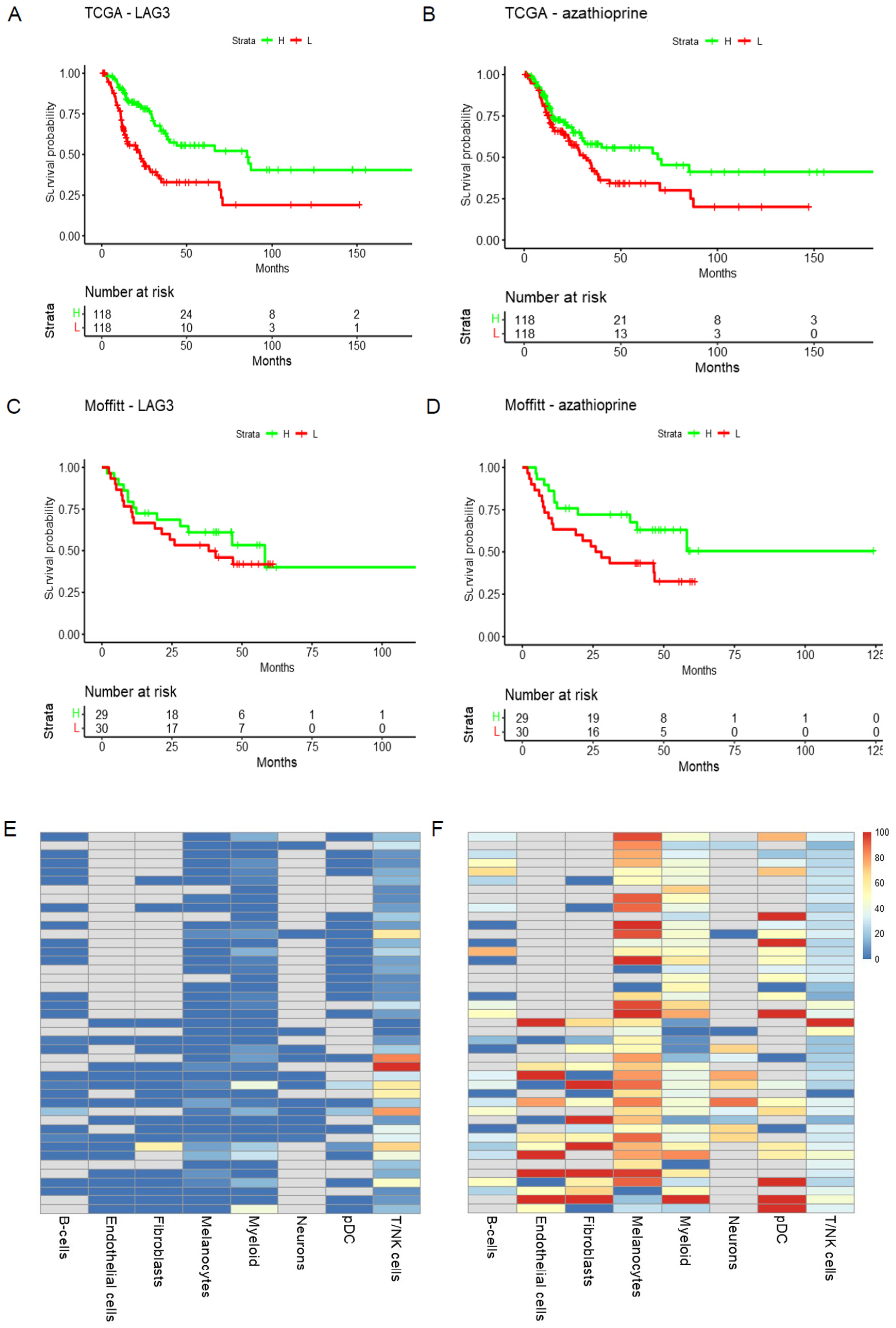
- TME: Heatmap and Violin Plot Highlight Potential Targeted Cells Populations by Each Therapy
- The mutation and survival tab displays Kaplan–Meier plots of overall survival based on mutation status in the target genes of the selected doublets in each cohort.
- The PC1 and survival tab shows the tables of genes and PC1 loadings in the target gene sets of each treatment for each cohort along with the KM plots of PC1 and overall survival for each treatment in both cohorts. The PC1 values are dichotomized at the median.
- The eQTL tab displays the box plots of gene expression in both cohorts by mutation status in the target genes. It also displays the summary statistics of the de-batched expression on a log scale.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsao, H.; Chin, L.; Garraway, L.A.; Fisher, D.E. Melanoma: From mutations to medicine. Genes Dev. 2012, 26, 1131–1155. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Huang, C.; Fu, Y.; Wang, J.; Wu, Z.; Ru, J.; Zheng, C.; Guo, Z.; Chen, X.; Zhou, W.; et al. Large-scale exploration and analysis of drug combinations. Bioinformatics 2015, 31, 2007–2016. [Google Scholar] [CrossRef] [PubMed]
- Wildenhain, J.; Spitzer, M.; Dolma, S.; Jarvik, N.; White, R.; Roy, M.; Griffiths, E.; Bellows, D.S.; Wright, G.D.; Tyers, M. Prediction of Synergism from Chemical-Genetic Interactions by Machine Learning. Cell Syst. 2015, 1, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Preuer, K.; Lewis, R.P.I.; Hochreiter, S.; Bender, A.; Bulusu, K.C.; Klambauer, G. DeepSynergy: Predicting anti-cancer drug synergy with Deep Learning. Bioinformatics 2018, 34, 1538–1546. [Google Scholar] [CrossRef]
- Smalley, I.; Chen, Z.; Phadke, M.; Li, J.; Yu, X.; Wyatt, C.; Evernden, B.; Messina, J.L.; Sarnaik, A.; Sondak, V.K.; et al. Single-Cell Characterization of the Immune Microenvironment of Melanoma Brain and Leptomeningeal Metastases. Clin. Cancer Res. 2021, 27, 4109–4125. [Google Scholar] [CrossRef]
- Wang, X.; Yu, X.; Krauthammer, M.; Hugo, W.; Duan, C.; Kanetsky, P.A.; Teer, J.K.; Thompson, Z.J.; Kalos, D.; Tsai, K.Y.; et al. The Association of MUC16 Mutation with Tumor Mutation Burden and Its Prognostic Implications in Cutaneous Melanoma. Cancer Epidemiol. Biomark. Prev. 2020, 29, 1792–1799. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Rudnicki, C.; Zahavi, I. Antibiotic prophylaxis for the prevention of infective endocarditis. Harefuah 1990, 119, 275–277. [Google Scholar]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.H.; Matilla, A.; Pollack, D.J. A critique and case study of nodular sclerosing Hodgkin’s disease. J. Clin. Pathol. 1976, 29, 923–930. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq–A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef]
- The TCGA SKIN DNAseq Dataset. Available online: https://tcga-dta.nci.nih.gov/tcgafiles/ftp_auth/distro_ftpusers/anonymous/tumor/skcm/gsc/broad.mit.edu/illuminaga_dnaseq_automated/muttions/broad.mit.edu_SKCM.IlluminaGA_DNASeq_automated.Level_2.1.5.0/SKCM_pairs.aggregated.capture.tcga.uuid.automated.somatic.maf (accessed on 1 March 2016).
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Yoo, M.; Shin, J.; Kim, J.; Ryall, K.A.; Lee, K.; Lee, S.; Jeon, M.; Kang, J.; Tan, A.C. DSigDB: Drug signatures database for gene set analysis. Bioinformatics 2015, 31, 3069–3071. [Google Scholar] [CrossRef]
- Khan, R.; Khan, H.; Abdullah, Y.; Dou, Q.P. Feasibility of Repurposing Clioquinol for Cancer Therapy. Recent Pat. Anticancer Drug Discov. 2020, 15, 14–31. [Google Scholar] [CrossRef]
- Treiber, C.; Simons, A.; Strauss, M.; Hafner, M.; Cappai, R.; Bayer, T.A.; Multhaup, G. Clioquinol mediates copper uptake and counteracts copper efflux activities of the amyloid precursor protein of Alzheimer’s disease. J. Biol. Chem. 2004, 279, 51958–51964. [Google Scholar] [CrossRef] [PubMed]
- Phadke, M.S.; Chen, Z.; Li, J.; Mohamed, E.; Davies, M.A.; Smalley, I.; Duckett, D.R.; Palve, V.; Czerniecki, B.J.; Forsyth, P.A.; et al. Targeted Therapy Given after Anti-PD-1 Leads to Prolonged Responses in Mouse Melanoma Models through Sustained Antitumor Immunity. Cancer Immunol. Res. 2021, 9, 554–567. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| TCGA (N = 459) | Moffitt (N = 135) | ||
|---|---|---|---|
| Mutation Cohorts n (%) | |||
| BRAF | 236 (51.4) | 59 (43.7) | |
| NRAS | 125 (27.2) | 34 (25.2) | |
| Triple Wild type | 88 (19.2) | 28 (20.7) | |
| Age mean (sd) | 61.5 (15.0) | 62.7 (15.3) | |
| IPI/NIVO treatment n (%) | 15 (3.6) | 51 (38.8) | |
| BRAF treatment (%) | 5 (1.1) | 29 (21.5) | |
| Gender n (%) | |||
| Female | 175 (38.1) | 49 (36.3) | |
| Male | 284 (61.9) | 86 (63.7) | |
| Transcriptomics | RNA-seq | RNA-seq | |
| DNA mutation | WES | WES | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thompson, Z.J.; Teer, J.K.; Li, J.; Chen, Z.; Welsh, E.A.; Zhang, Y.; Ayoubi, N.; Eroglu, Z.; Tan, A.C.; Smalley, K.S.M.; et al. Drepmel—A Multi-Omics Melanoma Drug Repurposing Resource for Prioritizing Drug Combinations and Understanding Tumor Microenvironment. Cells 2022, 11, 2894. https://doi.org/10.3390/cells11182894
Thompson ZJ, Teer JK, Li J, Chen Z, Welsh EA, Zhang Y, Ayoubi N, Eroglu Z, Tan AC, Smalley KSM, et al. Drepmel—A Multi-Omics Melanoma Drug Repurposing Resource for Prioritizing Drug Combinations and Understanding Tumor Microenvironment. Cells. 2022; 11(18):2894. https://doi.org/10.3390/cells11182894
Chicago/Turabian StyleThompson, Zachary J., Jamie K. Teer, Jiannong Li, Zhihua Chen, Eric A. Welsh, Yonghong Zhang, Noura Ayoubi, Zeynep Eroglu, Aik Choon Tan, Keiran S. M. Smalley, and et al. 2022. "Drepmel—A Multi-Omics Melanoma Drug Repurposing Resource for Prioritizing Drug Combinations and Understanding Tumor Microenvironment" Cells 11, no. 18: 2894. https://doi.org/10.3390/cells11182894
APA StyleThompson, Z. J., Teer, J. K., Li, J., Chen, Z., Welsh, E. A., Zhang, Y., Ayoubi, N., Eroglu, Z., Tan, A. C., Smalley, K. S. M., & Chen, Y. A. (2022). Drepmel—A Multi-Omics Melanoma Drug Repurposing Resource for Prioritizing Drug Combinations and Understanding Tumor Microenvironment. Cells, 11(18), 2894. https://doi.org/10.3390/cells11182894