

Depletion of VGLL4 Causes Perinatal Lethality without Affecting Myocardial Development


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Gene Expression
2.3. Statistics
3. Results
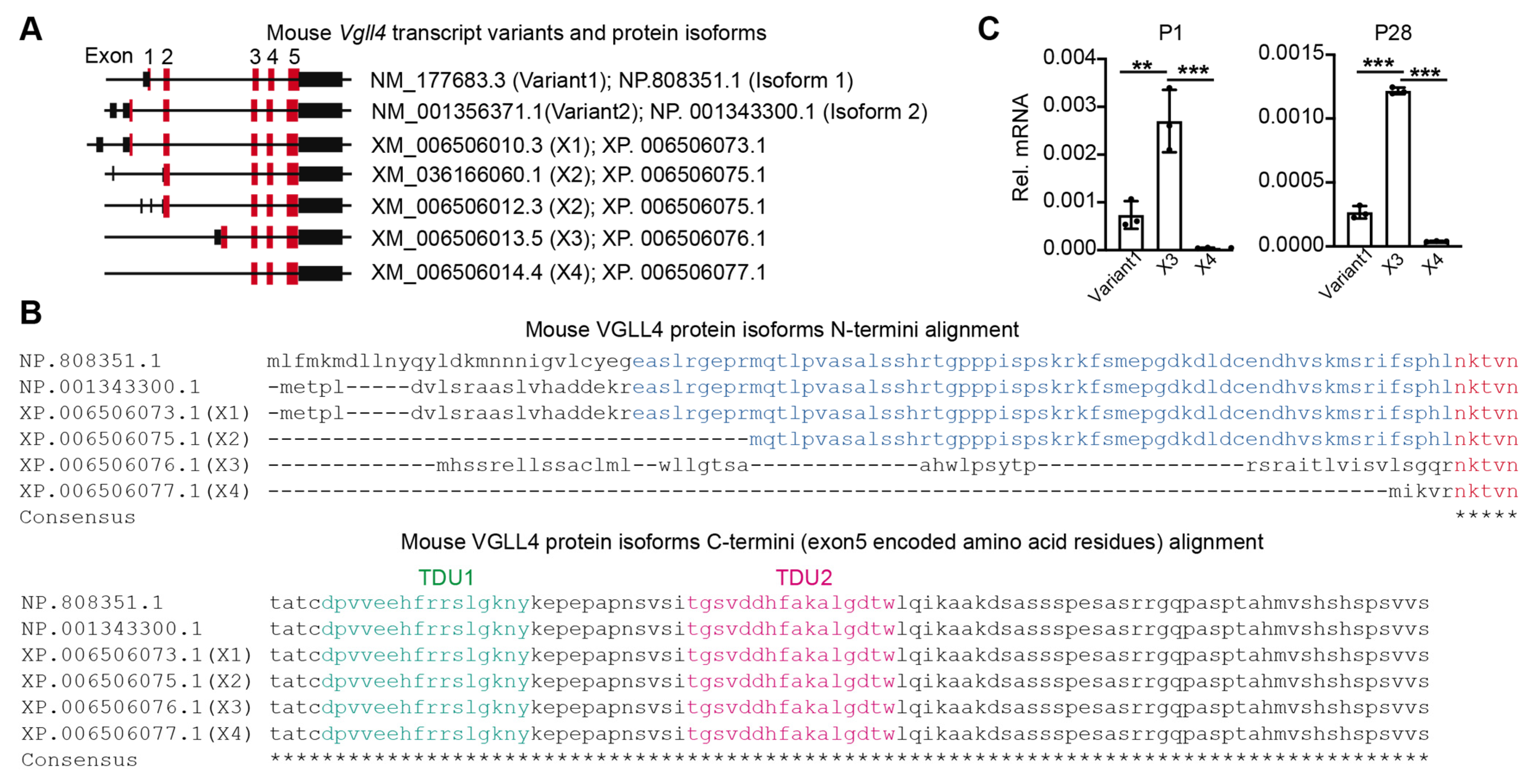
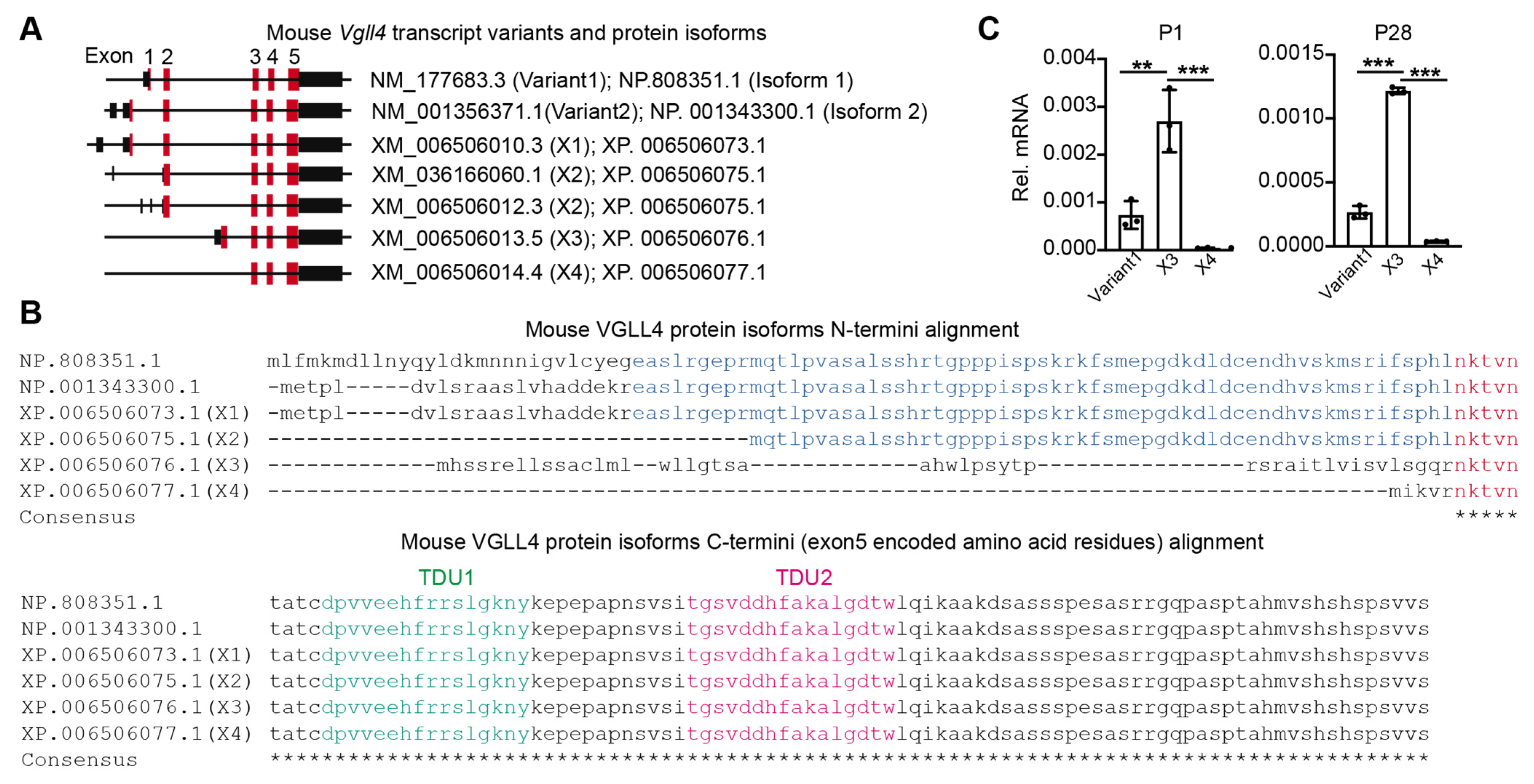
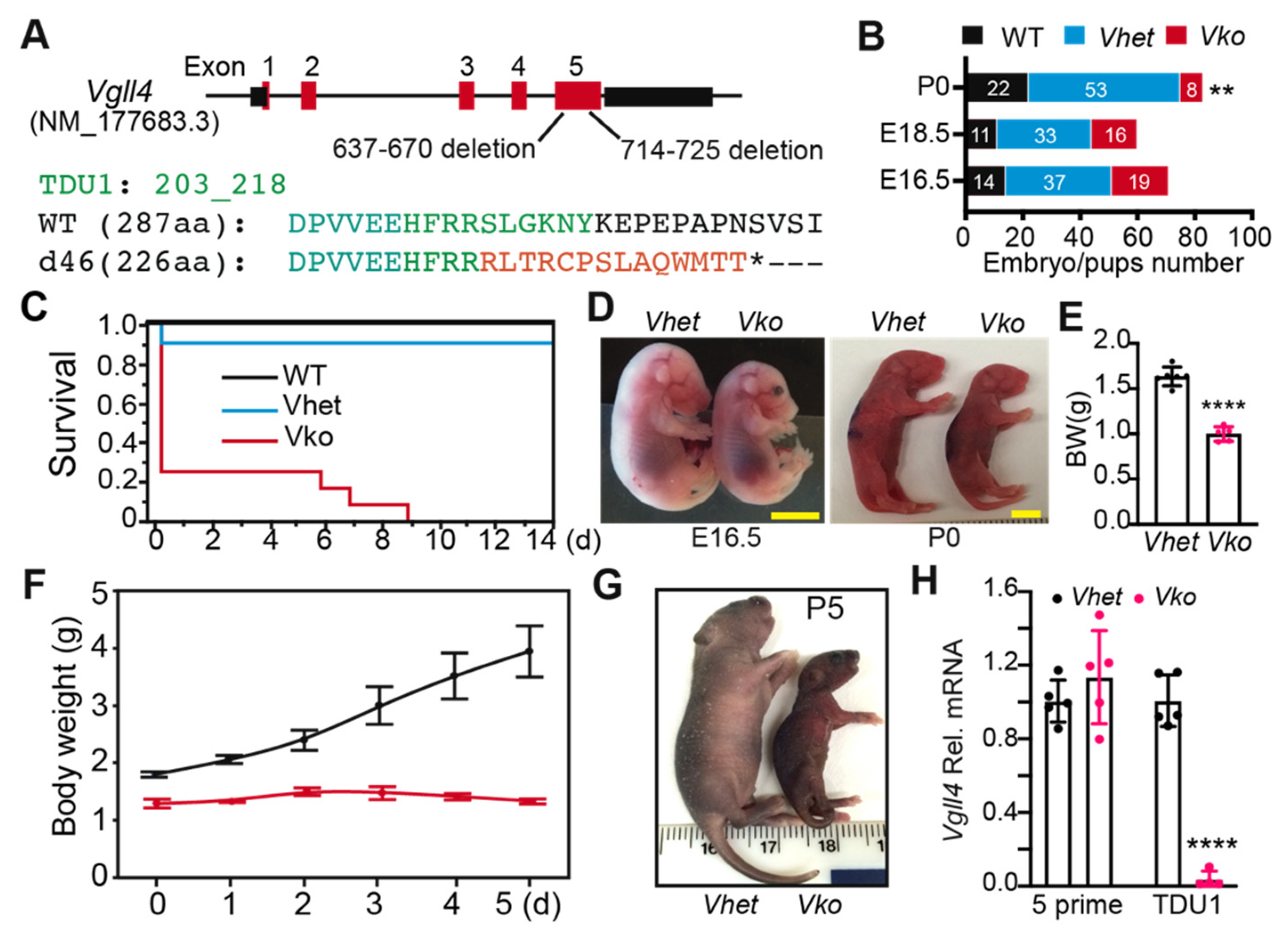
3.1. Vgll4 Gene Has Different Transcript Variants
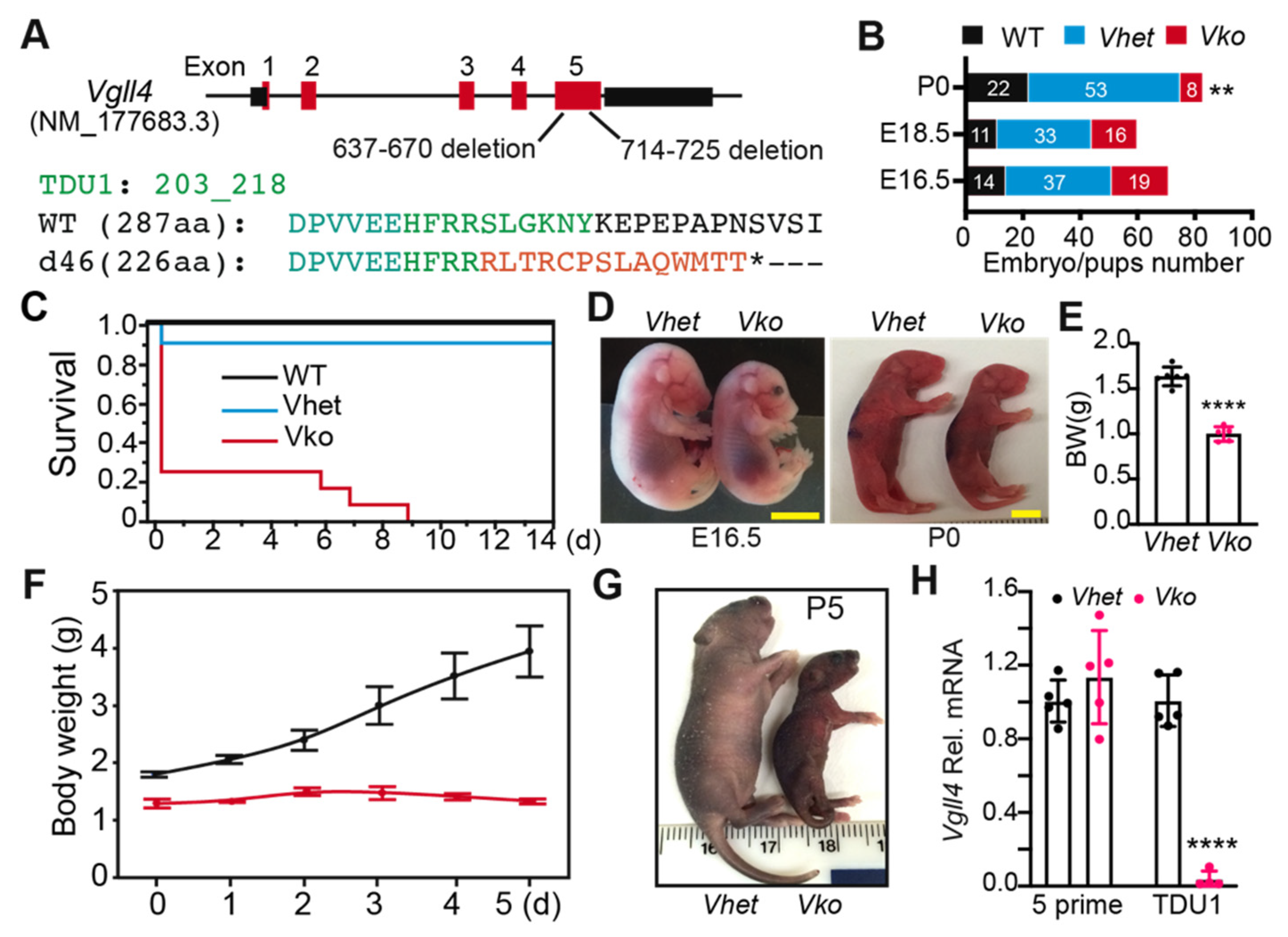
3.2. Whole-Body Deletion of VGLL4 Domains Results in Perinatal Lethality
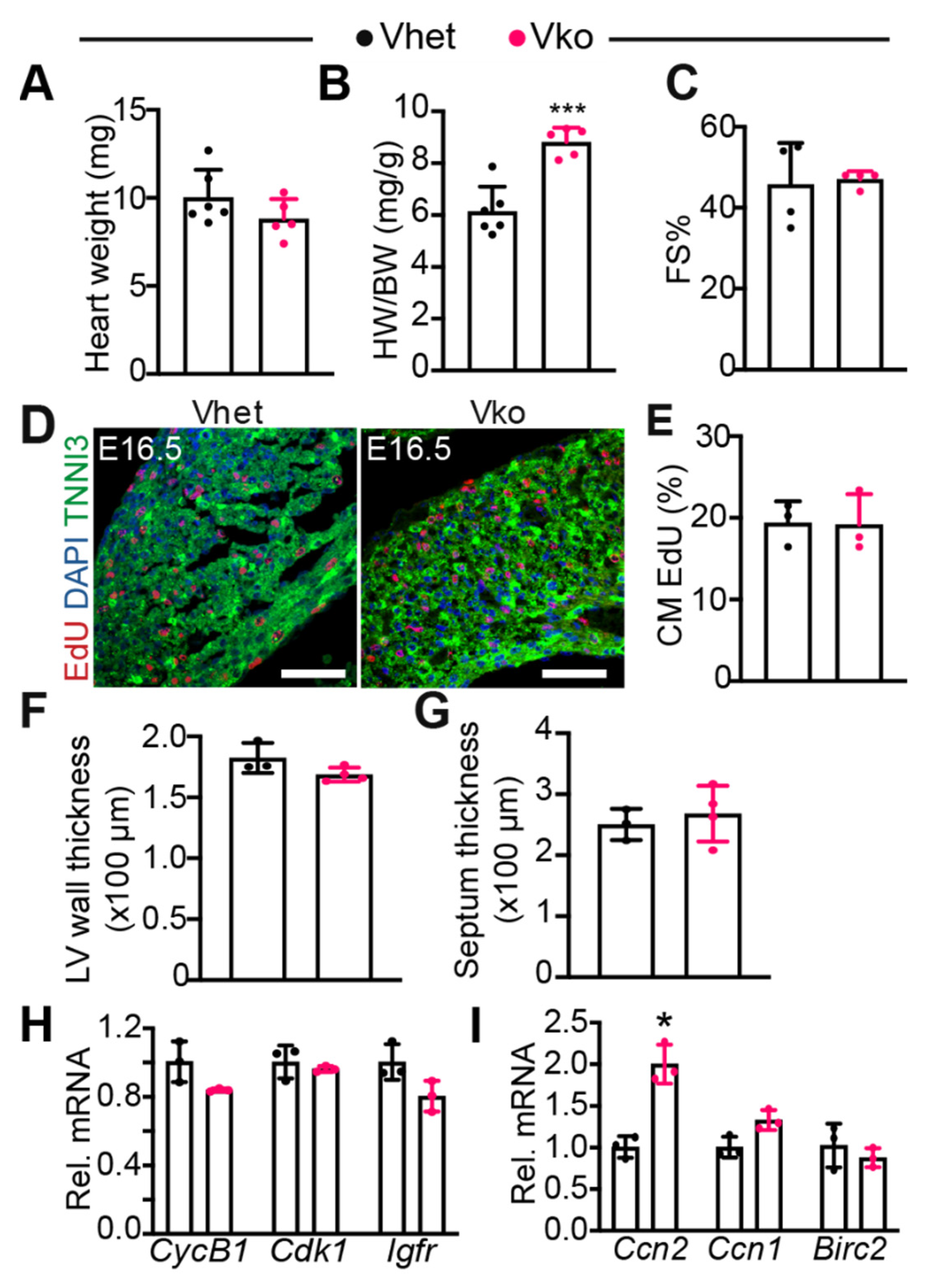
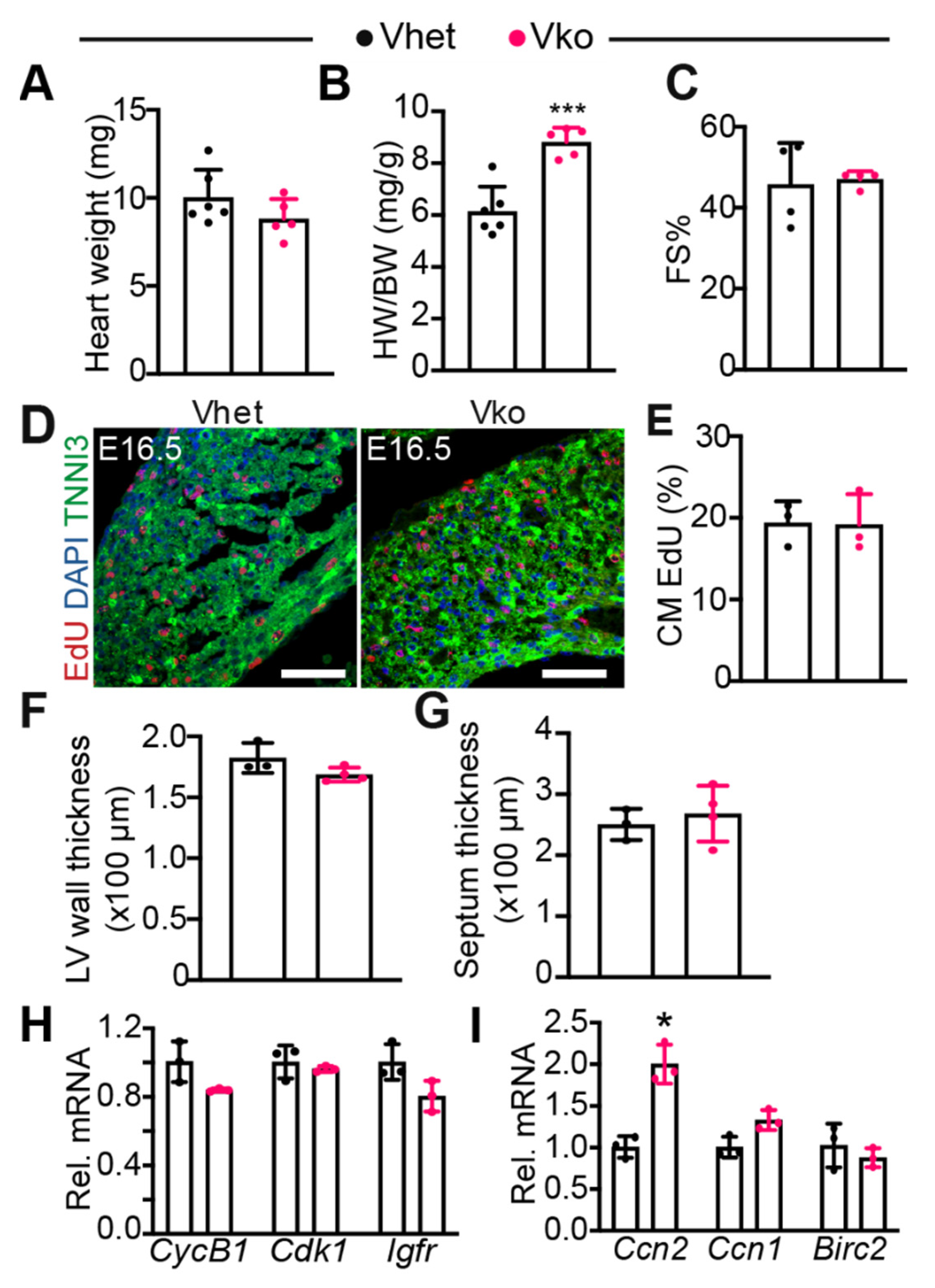
3.3. Whole-Body Deletion of VGLL4 Does Not Affect Myocardial Development and Heart Function
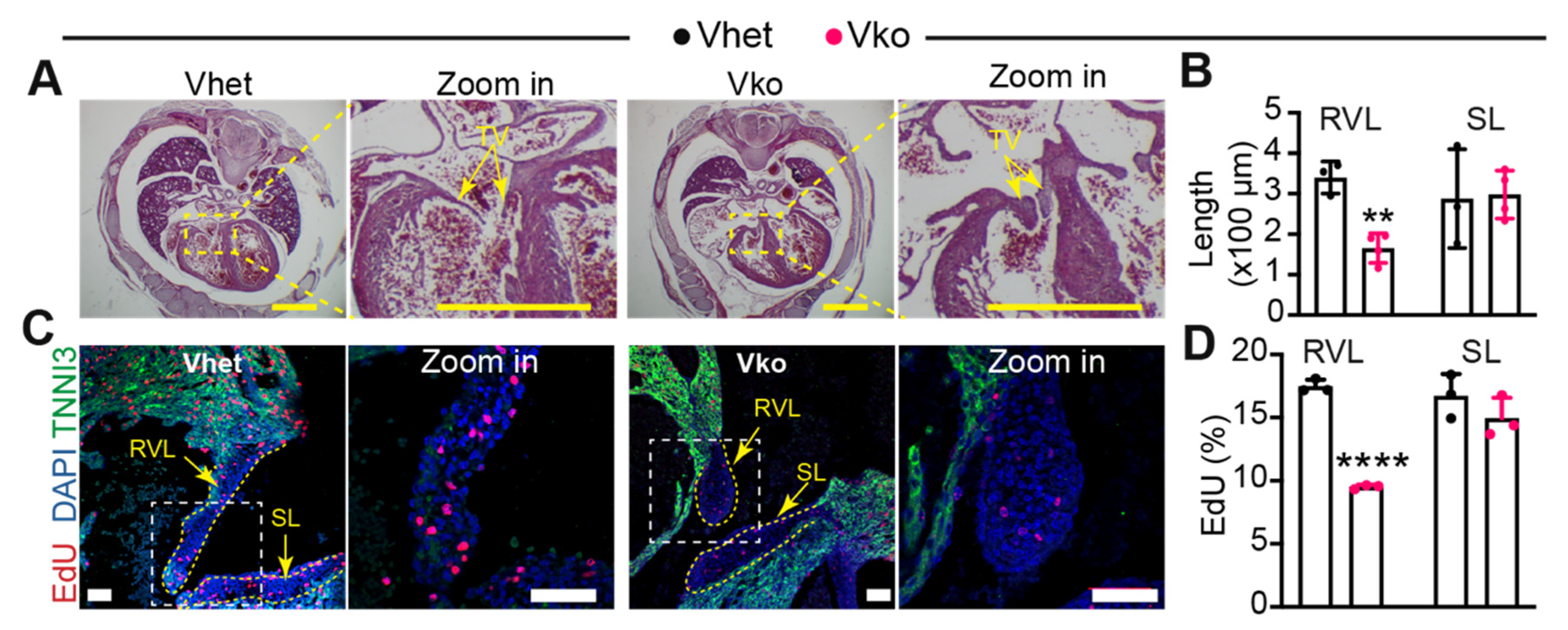
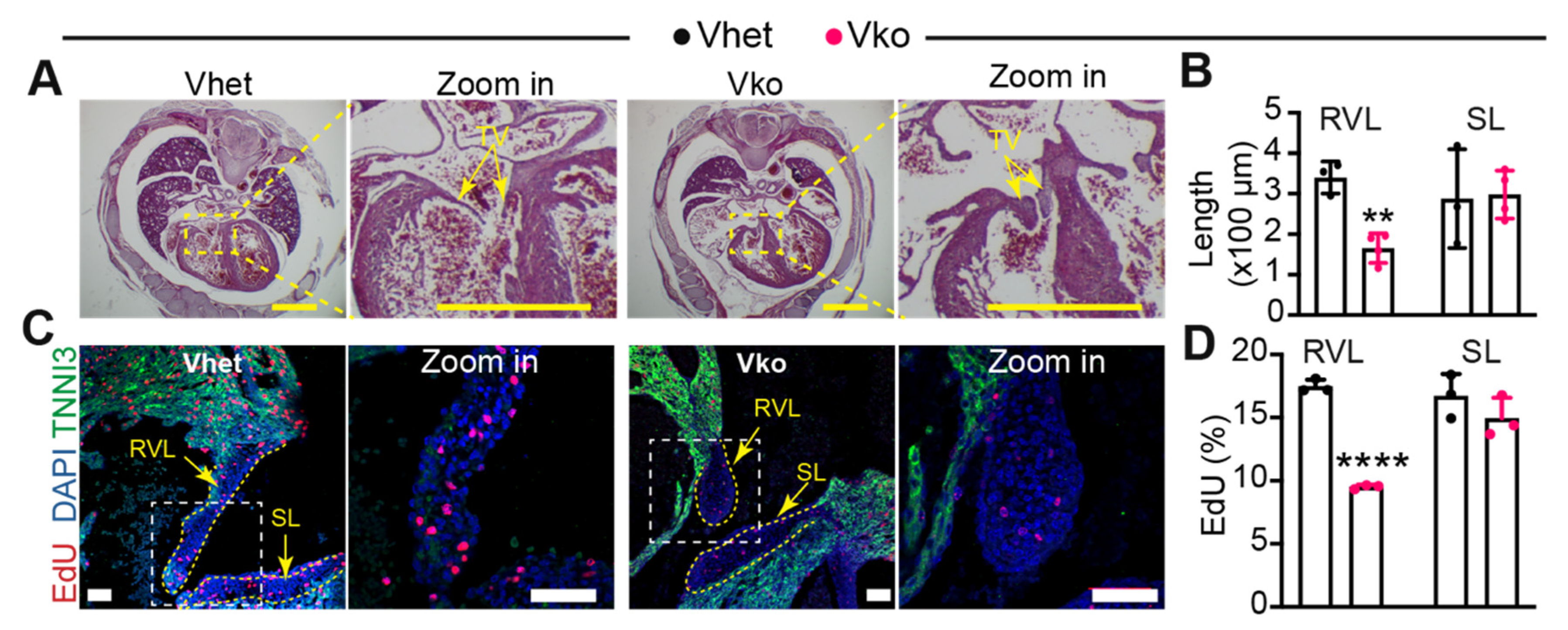
3.4. Whole-Body Deletion of VGLL4 Causes Tricuspid Valve Malformation
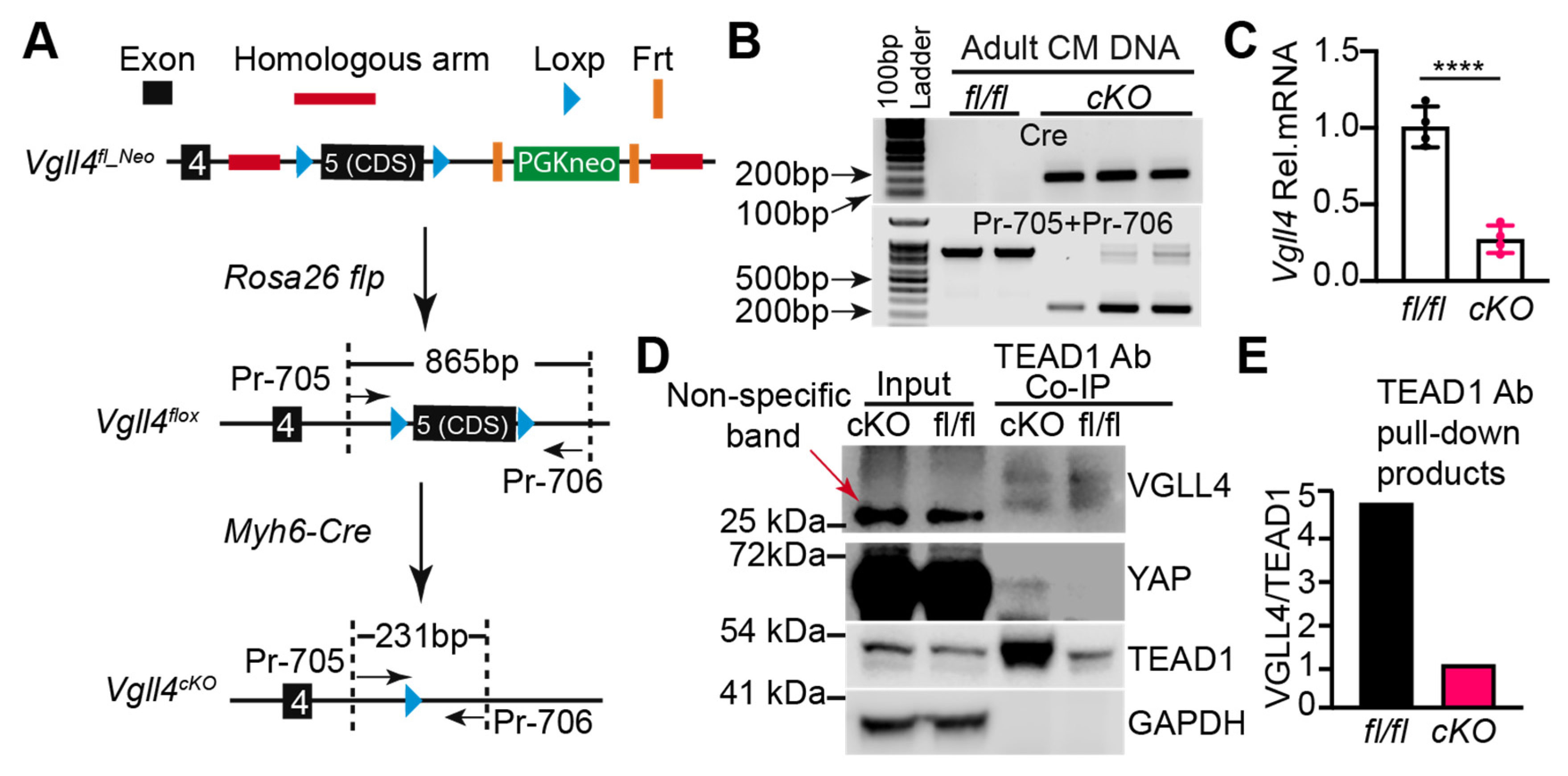
3.5. Generation of a Vgll4 Flox Allele
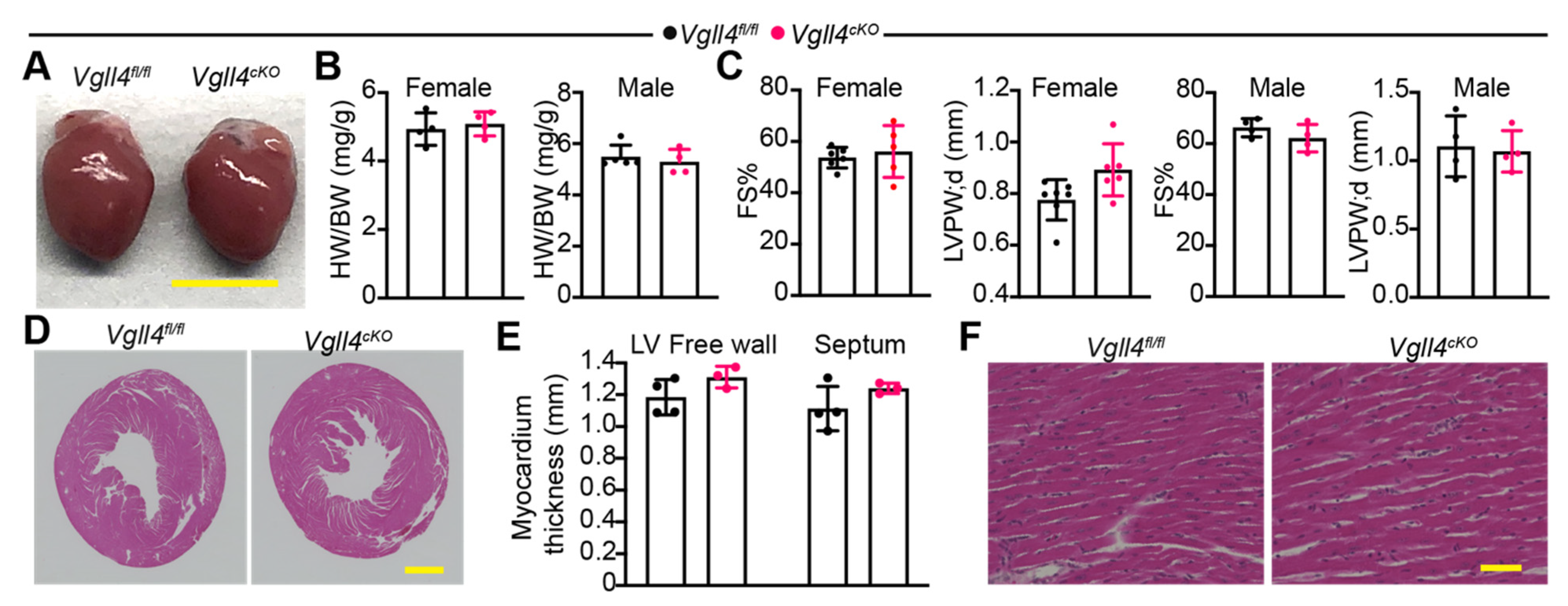
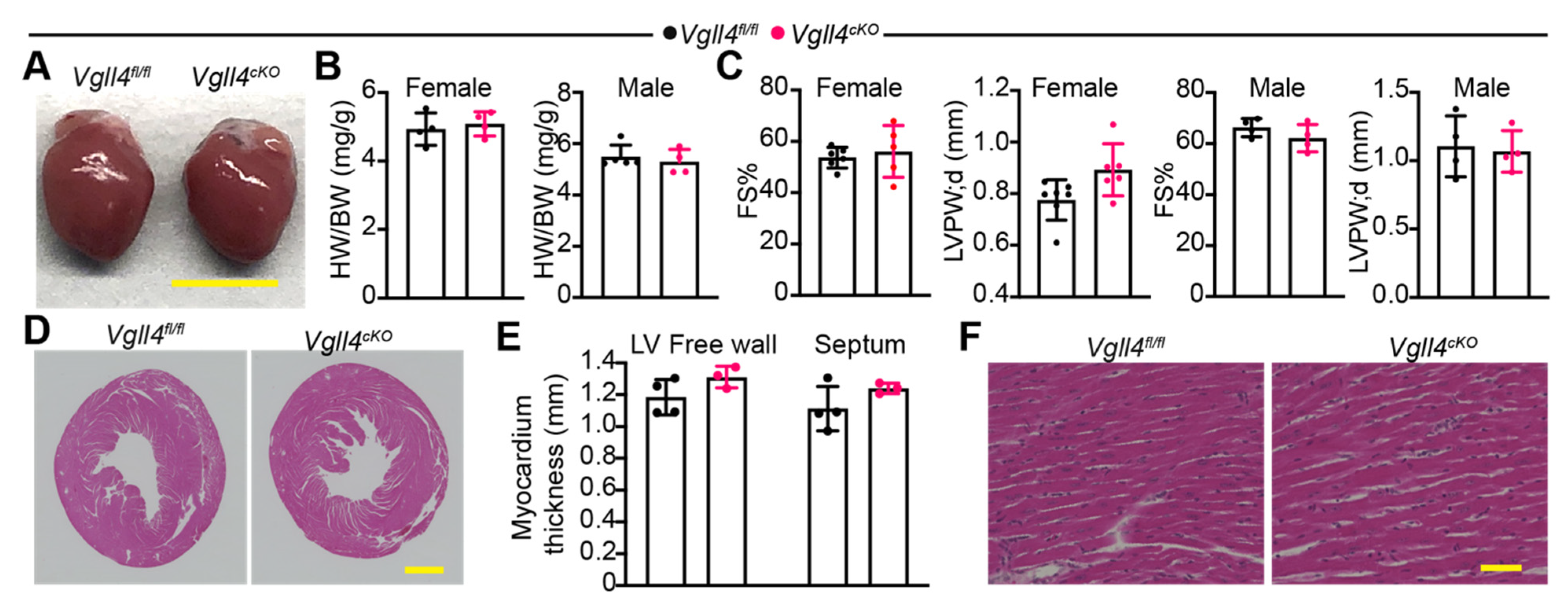
3.6. CM-Specific Depletion of VGLL4 Does Not Affect Heart Growth and Function
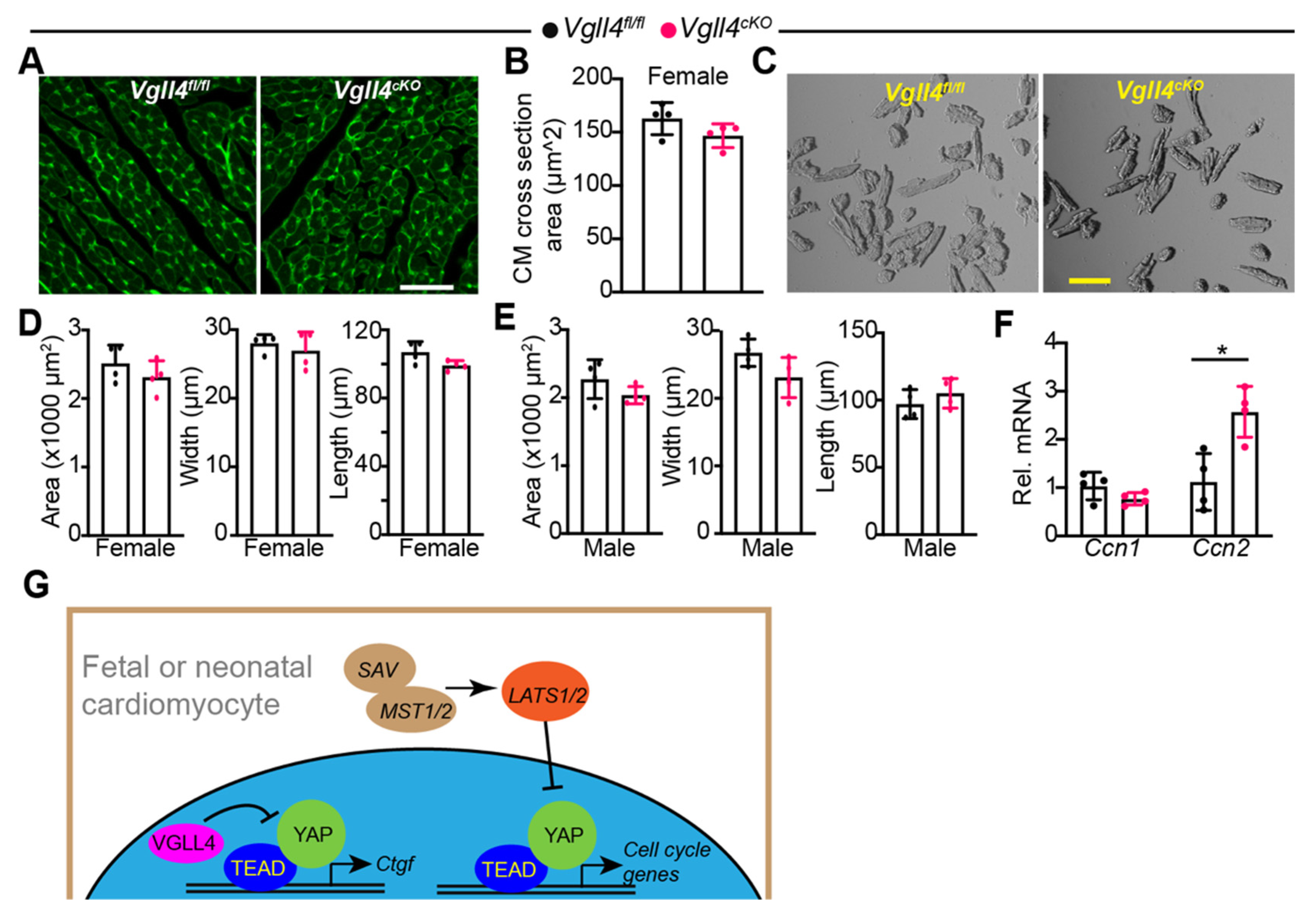
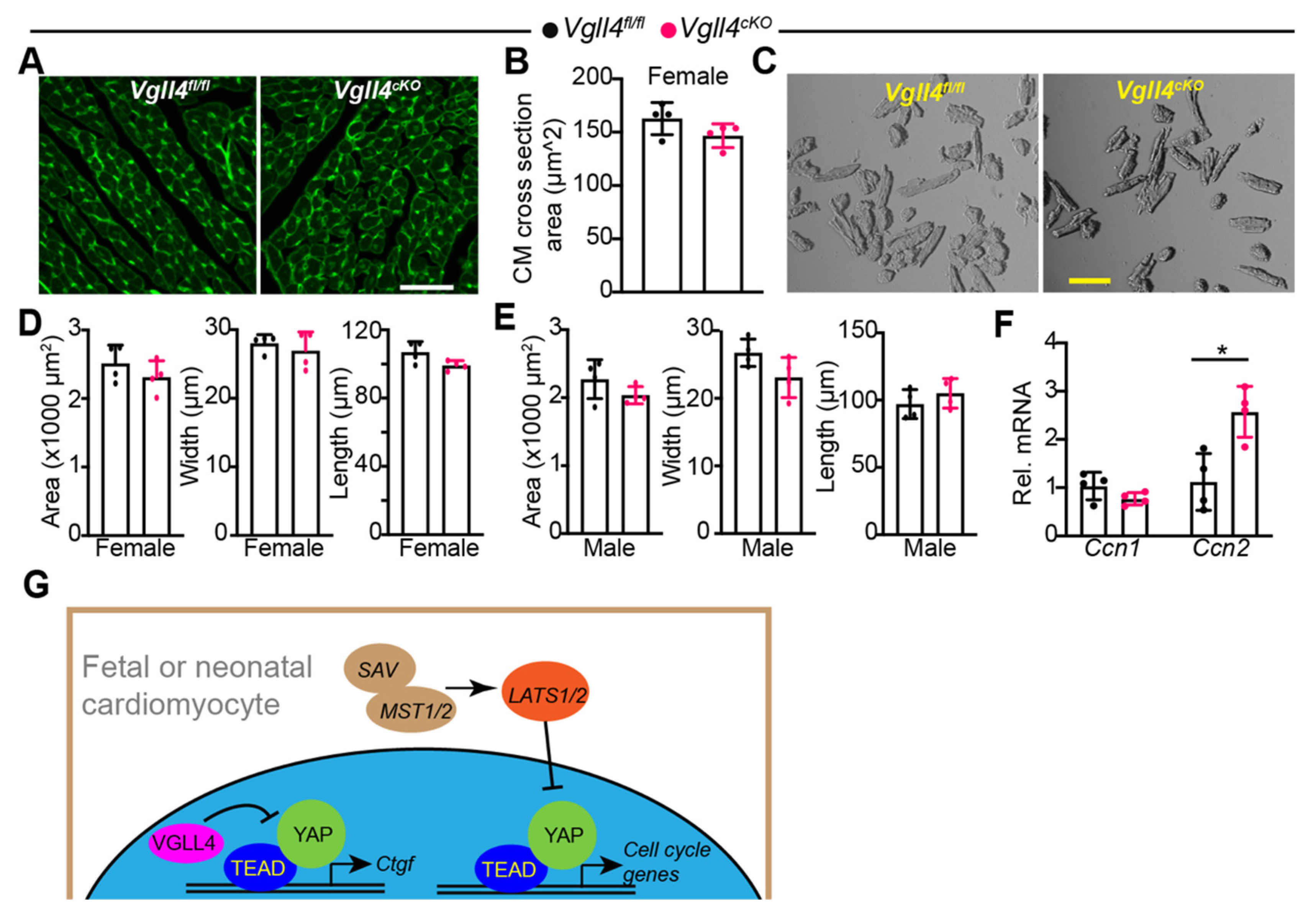
3.7. CM-Specific Depletion of VGLL4 Does Not Affect CM Size
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Triedman, J.K.; Newburger, J.W. Trends in Congenital Heart Disease: The Next Decade. Circulation 2016, 133, 2716–2733. [Google Scholar] [CrossRef] [PubMed]
- Rossano, J.W. Congenital heart disease: A global public health concern. Lancet Child Adolesc. Health 2020, 4, 168–169. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Yu, W.; Zhou, B. Hippo signaling pathway in cardiovascular development and diseases. Hereditas 2017, 39, 576–587. [Google Scholar]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A. The Hippo Pathway Is Activated and Is a Causal Mechanism for Adipogenesis in Arrhythmogenic Cardiomyopathy. Circ. Res. 2014, 114, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, Y.; Heallen, T.; Leach, J.; Xiao, Y.; Martin, J.F. Dystrophin–glycoprotein complex sequesters Yap to inhibit cardiomyocyte proliferation. Nature 2017, 547, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Vita, G.L.; Polito, F.; Oteri, R.; Arrigo, R.; Ciranni, A.M.; Musumeci, O.; Messina, S.; Rodolico, C.; Di Giorgio, R.M.; Vita, G.; et al. Hippo signaling pathway is altered in Duchenne muscular dystrophy. PLoS ONE 2018, 13, e0205514. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The Hippo Signaling Pathway in Development and Cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a Universal Size-Control Mechanism in Drosophila and Mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.-Y.; Guan, K.-L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCFβ-TRCP. Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo Pathway Inhibits Wnt Signaling to Restrain Cardiomyocyte Proliferation and Heart Size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef]
- Heallen, T.; Morikawa, Y.; Leach, J.; Tao, G.; Willerson, J.T.; Johnson, R.L.; Martin, J.F. Hippo signaling impedes adult heart regeneration. Development 2013, 140, 4683–4690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Gise, A.; Lin, Z.; Schlegelmilch, K.; Honor, L.B.; Pan, G.M.; Buck, J.N.; Ma, Q.; Ishiwata, T.; Zhou, B.; Camargo, F.D.; et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc. Natl. Acad. Sci. USA 2012, 109, 2394–2399. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Qi, X.; McAnally, J.; Schwartz, R.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Regulation of Insulin-Like Growth Factor Signaling by Yap Governs Cardiomyocyte Proliferation and Embryonic Heart Size. Sci. Signal. 2011, 4, ra70. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Jagannathan, R.; Li, F.; Lee, J.; Balasubramanyam, N.; Kim, B.S.; Yang, P.; Yechoor, V.K.; Moulik, M. Tead1 is required for perinatal cardiomyocyte proliferation. PLoS ONE 2019, 14, e0212017. [Google Scholar] [CrossRef]
- Zhang, H.; von Gise, A.; Liu, Q.; Hu, T.; Tian, X.; He, L.; Pu, W.; Huang, X.; He, L.; Cai, C.-L.; et al. Yap1 Is Required for Endothelial to Mesenchymal Transition of the Atrioventricular Cushion. J. Biol. Chem. 2014, 289, 18681–18692. [Google Scholar] [CrossRef]
- Deng, X.; Fang, L. VGLL4 is a transcriptional cofactor acting as a novel tumor suppressor via interacting with TEADs. Am. J. Cancer Res. 2018, 8, 932–943. [Google Scholar]
- Guo, T.; Lu, Y.; Li, P.; Yin, M.-X.; Lv, D.; Zhang, W.; Wang, H.; Zhou, Z.; Ji, H.; Zhao, Y.; et al. A novel partner of Scalloped regulates Hippo signaling via antagonizing Scalloped-Yorkie activity. Cell Res. 2013, 23, 1201–1214. [Google Scholar] [CrossRef]
- Koontz, L.M.; Liu-Chittenden, Y.; Yin, F.; Zheng, Y.; Yu, J.; Huang, B.; Chen, Q.; Wu, S.; Pan, D. The Hippo Effector Yorkie Controls Normal Tissue Growth by Antagonizing Scalloped-Mediated Default Repression. Dev. Cell 2013, 25, 388–401. [Google Scholar] [CrossRef]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A Peptide Mimicking VGLL4 Function Acts as a YAP Antagonist Therapy against Gastric Cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef]
- Jiao, S.; Li, C.; Hao, Q.; Miao, H.; Zhang, L.; Li, L.; Zhou, Z. VGLL4 targets a TCF4–TEAD4 complex to coregulate Wnt and Hippo signalling in colorectal cancer. Nat. Commun. 2017, 8, 14058. [Google Scholar] [CrossRef]
- Lin, Z.; Guo, H.; Cao, Y.; Zohrabian, S.; Zhou, P.; Ma, Q.; VanDusen, N.; Guo, Y.; Zhang, J.; Stevens, S.M.; et al. Acetylation of VGLL4 Regulates Hippo-YAP Signaling and Postnatal Cardiac Growth. Dev. Cell 2016, 39, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Ma, X.; Xu, J.; Heumüller, A.W.; Fei, Z.; Feng, X.; Wang, X.; Liu, K.; Li, J.; Cui, G.; et al. VGLL4 plays a critical role in heart valve development and homeostasis. PLoS Genet. 2019, 15, e1007977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 Knockin Mice for Genome Editing and Cancer Modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.-Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pasolli, H.A.; Fuchs, E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc. Natl. Acad. Sci. USA 2011, 108, 2270–2275. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.T.N.; MacIvor, D.M.; Hug, B.A.; Heusel, J.W.; Ley, T.J. Long-range disruption of gene expression by a selectable marker cassette. Proc. Natl. Acad. Sci. USA 1996, 93, 13090–13095. [Google Scholar] [CrossRef] [PubMed]
- Farley, F.W.; Soriano, P.; Steffen, L.S.; Dymecki, S.M. Widespread recombinase expression using FLPeR (flipper) mice. Genesis 2000, 28, 106–110. [Google Scholar] [CrossRef]
- Agah, R.; A Frenkel, P.; A French, B.; Michael, L.H.; Overbeek, P.; Schneider, M.D. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J. Clin. Investig. 1997, 100, 169–179. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Murakami, M.; Qi, X.; McAnally, J.; Porrello, E.R.; Mahmoud, A.I.; Tan, W.; Shelton, J.M.; et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13839–13844. [Google Scholar] [CrossRef]
- Del Re, D.P.; Yang, Y.; Nakano, N.; Cho, J.; Zhai, P.; Yamamoto, T.; Zhang, N.; Yabuta, N.; Nojima, H.; Pan, D.; et al. Yes-associated Protein Isoform 1 (Yap1) Promotes Cardiomyocyte Survival and Growth to Protect against Myocardial Ischemic Injury. J. Biol. Chem. 2013, 288, 3977–3988. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.; Del Re, D.P.; Zhai, P.; Ikeda, S.; Shirakabe, A.; Mizushima, W.; Miyamoto, S.; Brown, J.H.; Sadoshima, J. Yes-associated protein (YAP) mediates adaptive cardiac hypertrophy in response to pressure overload. J. Biol. Chem. 2019, 294, 3603–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monroe, T.; Hill, M.; Morikawa, Y.; Leach, J.; Heallen, T.; Cao, S.; Krijger, P.; de Laat, W.; Wehrens, X.; Rodney, G.; et al. YAP Partially Reprograms Chromatin Accessibility to Directly Induce Adult Cardiogenesis In Vivo. Dev. Cell 2019, 48, 765–779.e7. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gao, E.; Vite, A.; Yi, R.; Gomez, L.; Goossens, S.; van Roy, F.; Radice, G.L. Alpha-Catenins Control Cardiomyocyte Proliferation by Regulating Yap Activity. Circ. Res. 2015, 116, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Mao, B.; Luo, W.; Wei, B.; Jiang, W.; Liu, D.; Song, L.; Ji, G.; Yang, Z.; Lai, Y.-Q.; et al. The alteration of Hippo/YAP signaling in the development of hypertrophic cardiomyopathy. Basic Res. Cardiol. 2014, 109, 1–11. [Google Scholar] [CrossRef]
- Chen, J.; Ma, Q.; King, J.S.; Sun, Y.; Xu, B.; Zhang, X.; Zohrabian, S.; Guo, H.; Cai, W.; Li, G.; et al. aYAP modRNA reduces cardiac inflammation and hypertrophy in a murine ischemia-reperfusion model. Life Sci. Alliance 2019, 3, e201900424. [Google Scholar] [CrossRef]
- Lin, Z.; Von Gise, A.; Zhou, P.; Gu, F.; Ma, Q.; Jiang, J.; Yau, A.L.; Buck, J.N.; Gouin, K.; Van Gorp, P.R.; et al. Cardiac-Specific YAP Activation Improves Cardiac Function and Survival in an Experimental Murine MI Model. Circ. Res. 2014, 115, 354–363. [Google Scholar] [CrossRef]
- Leach, J.; Heallen, T.; Zhang, M.; Rahmani, M.; Morikawa, Y.; Hill, M.; Segura, A.; Willerson, J.T.; Martin, J.F. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 2017, 550, 260–264. [Google Scholar] [CrossRef]
- Feng, X.; Wang, Z.; Wang, F.; Lu, T.; Xu, J.; Ma, X.; Li, J.; He, L.; Zhang, W.; Li, S.; et al. Dual function of VGLL 4 in muscle regeneration. EMBO J. 2019, 38, e101051. [Google Scholar] [CrossRef]
- O’Connell, T.D.; Rodrigo, M.C.; Simpson, P.C. Isolation and culture of adult mouse cardiac myocytes. Methods Mol. Biol. 2007, 357, 271–296. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheldon, C.; Farley, A.; Ma, Q.; Pu, W.T.; Lin, Z. Depletion of VGLL4 Causes Perinatal Lethality without Affecting Myocardial Development. Cells 2022, 11, 2832. https://doi.org/10.3390/cells11182832
Sheldon C, Farley A, Ma Q, Pu WT, Lin Z. Depletion of VGLL4 Causes Perinatal Lethality without Affecting Myocardial Development. Cells. 2022; 11(18):2832. https://doi.org/10.3390/cells11182832
Chicago/Turabian StyleSheldon, Caroline, Aaron Farley, Qing Ma, William T. Pu, and Zhiqiang Lin. 2022. "Depletion of VGLL4 Causes Perinatal Lethality without Affecting Myocardial Development" Cells 11, no. 18: 2832. https://doi.org/10.3390/cells11182832
APA StyleSheldon, C., Farley, A., Ma, Q., Pu, W. T., & Lin, Z. (2022). Depletion of VGLL4 Causes Perinatal Lethality without Affecting Myocardial Development. Cells, 11(18), 2832. https://doi.org/10.3390/cells11182832