

Cigarette Smoke and Morphine Promote Treg Plasticity to Th17 via Enhancing Trained Immunity

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Statistical Analysis of RNA-Seq Data
2.2. Ingenuity Pathway Analysis
2.3. Metascape Analysis
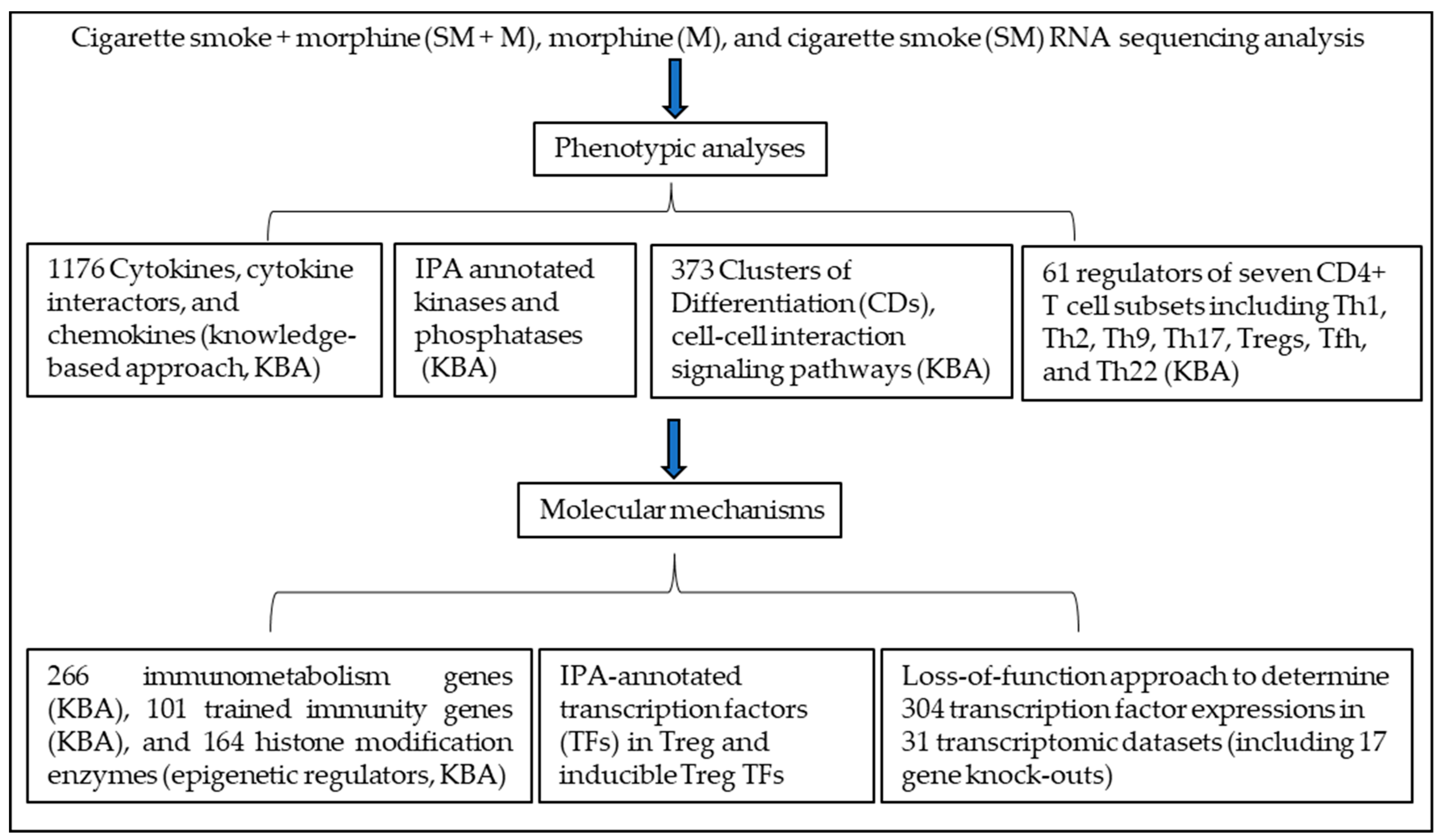
3. Results
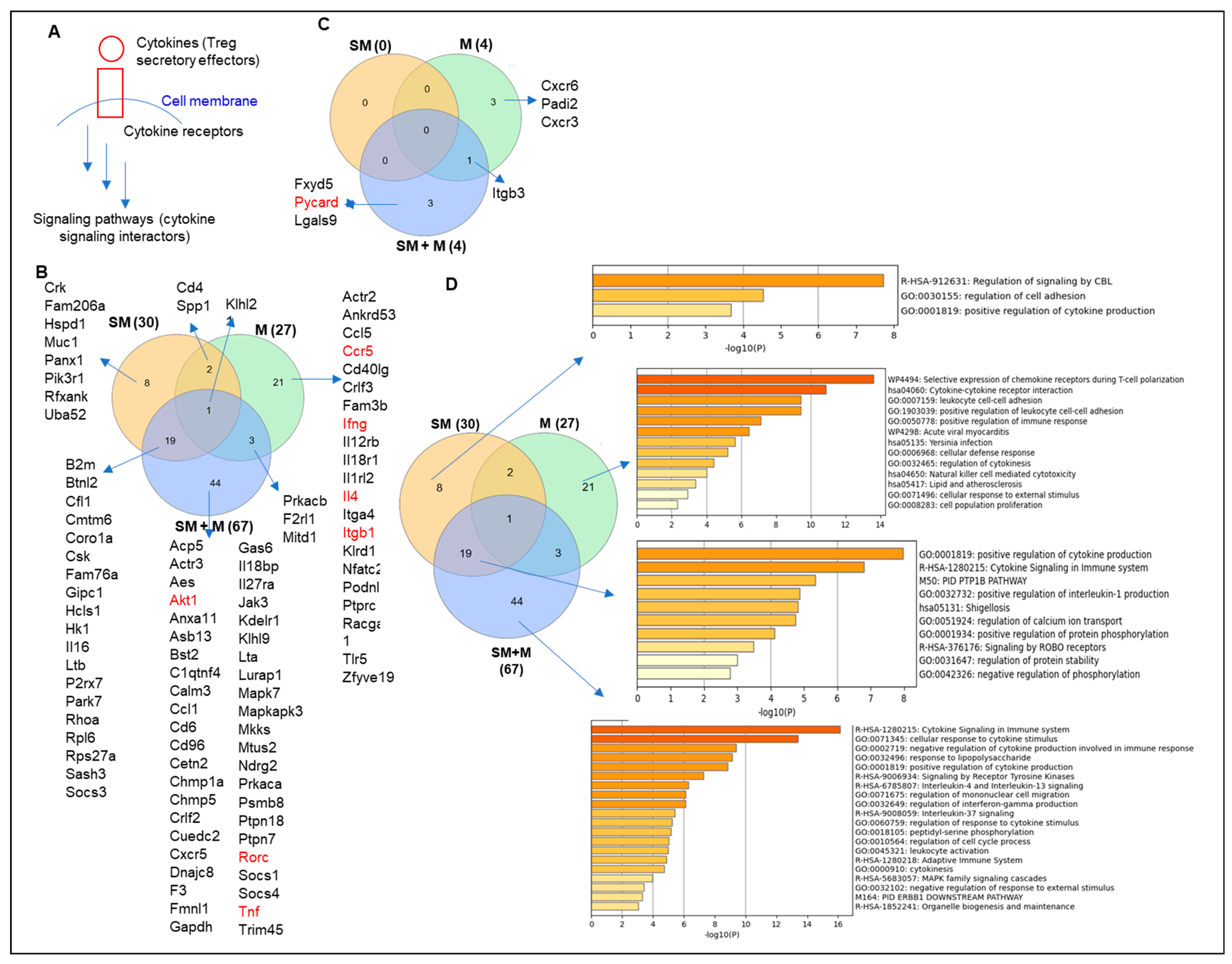
3.1. Cigarette Smoke Plus Morphine, Morphine, and Cigarette Smoke Upregulate the Transcripts of 44, 21, and 8 Cytokines and Their Interactors in Tregs, Respectively, to Promote Plastic/Dysfunctional Tregs
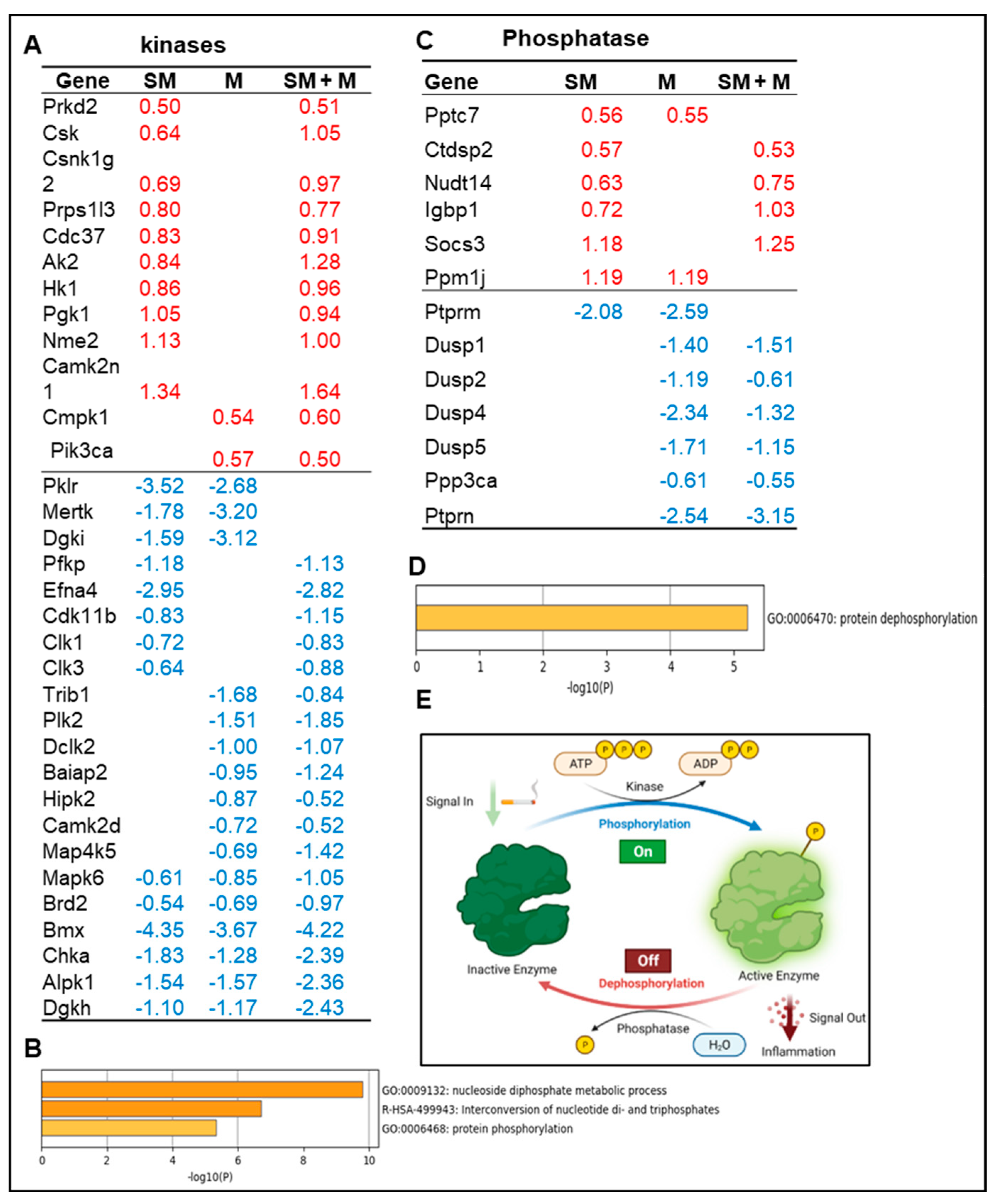
3.2. Cigarette Smoke Plus Morphine, Morphine, and Cigarette Smoke Upregulate the Transcripts of 12, 2, and 10 and Downregulate the Transcripts 18, 16, and 14 Kinases in Splenic Tregs, Respectively; Also, Upregulate the Transcripts of 4, 2, and 6 and Downregulate the Transcripts of 6, 7, and 1 Phosphatases in Splenic Tregs, Respectively
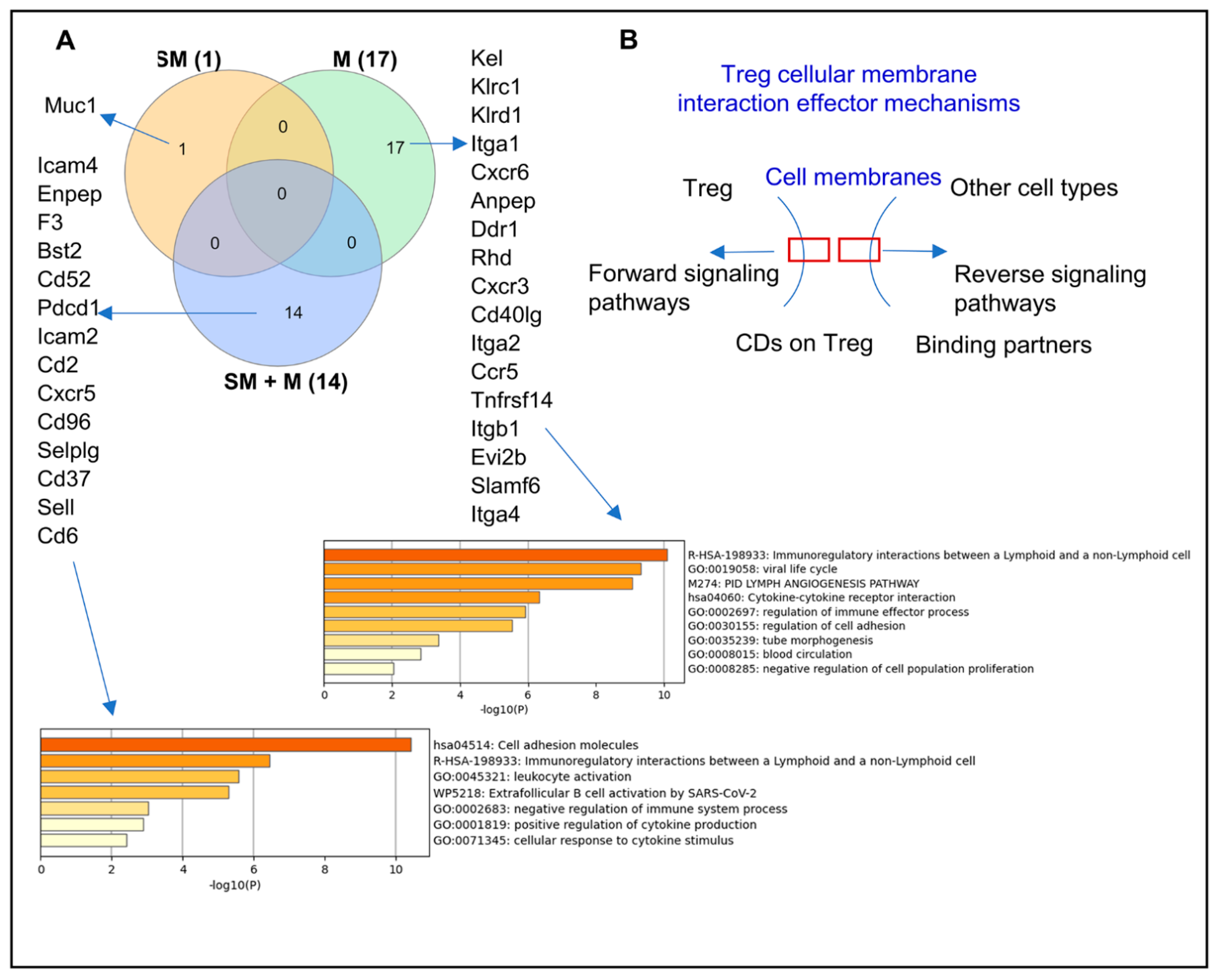
3.3. Cigarette Smoke Plus Morphine, Morphine, and Cigarette Smoke Upregulate the Transcripts of 14, 17, and 1 Clusters of Differentiation (CDs) in Tregs, Respectively, to Promote Weakened, Plastic/Dysfunctional Tregs
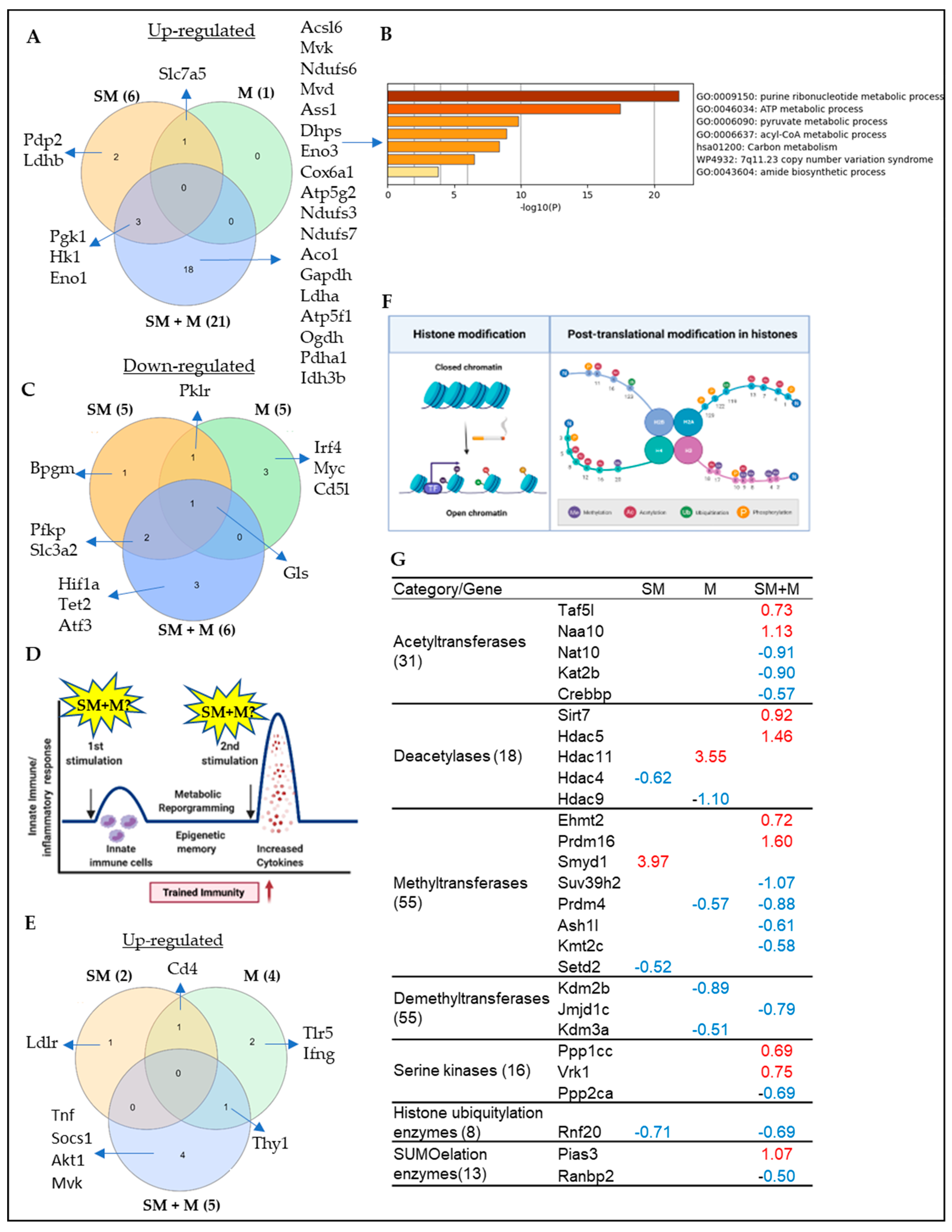
3.4. Cigarette Smoke Plus Morphine, Morphine, and Cigarette Smoke Upregulate the Transcripts of 21, 1, and 6 Immunometabolism Genes; 5, 4, and 2 Trained Immunity Genes, Respectively; Also, SM + M, M, and SM Upregulate the Transcripts of 9, 1, and 1 and Downregulate the Transcripts of 11, 4, and 3 Histone Modification Enzymes in Splenic Tregs, Respectively
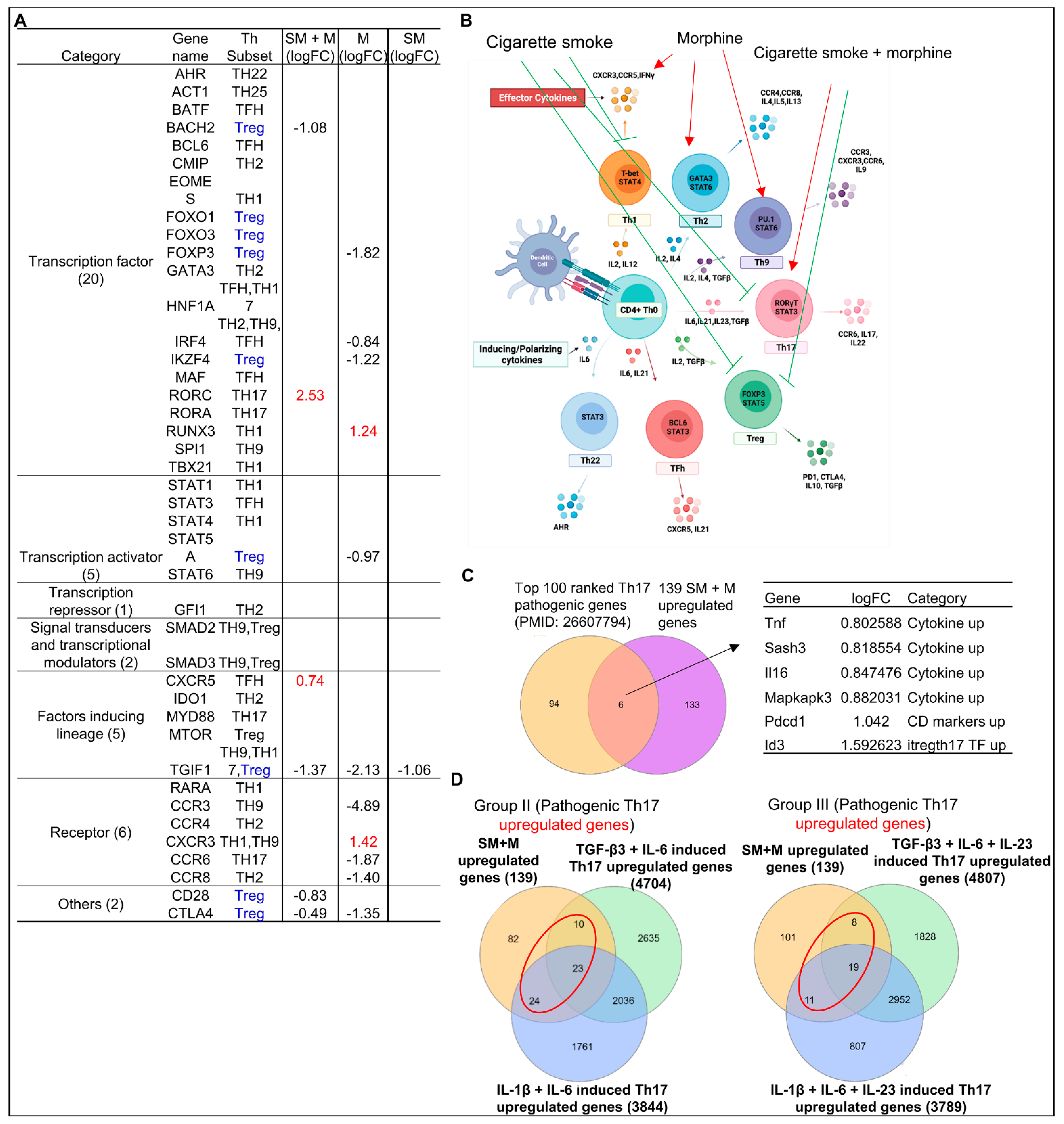
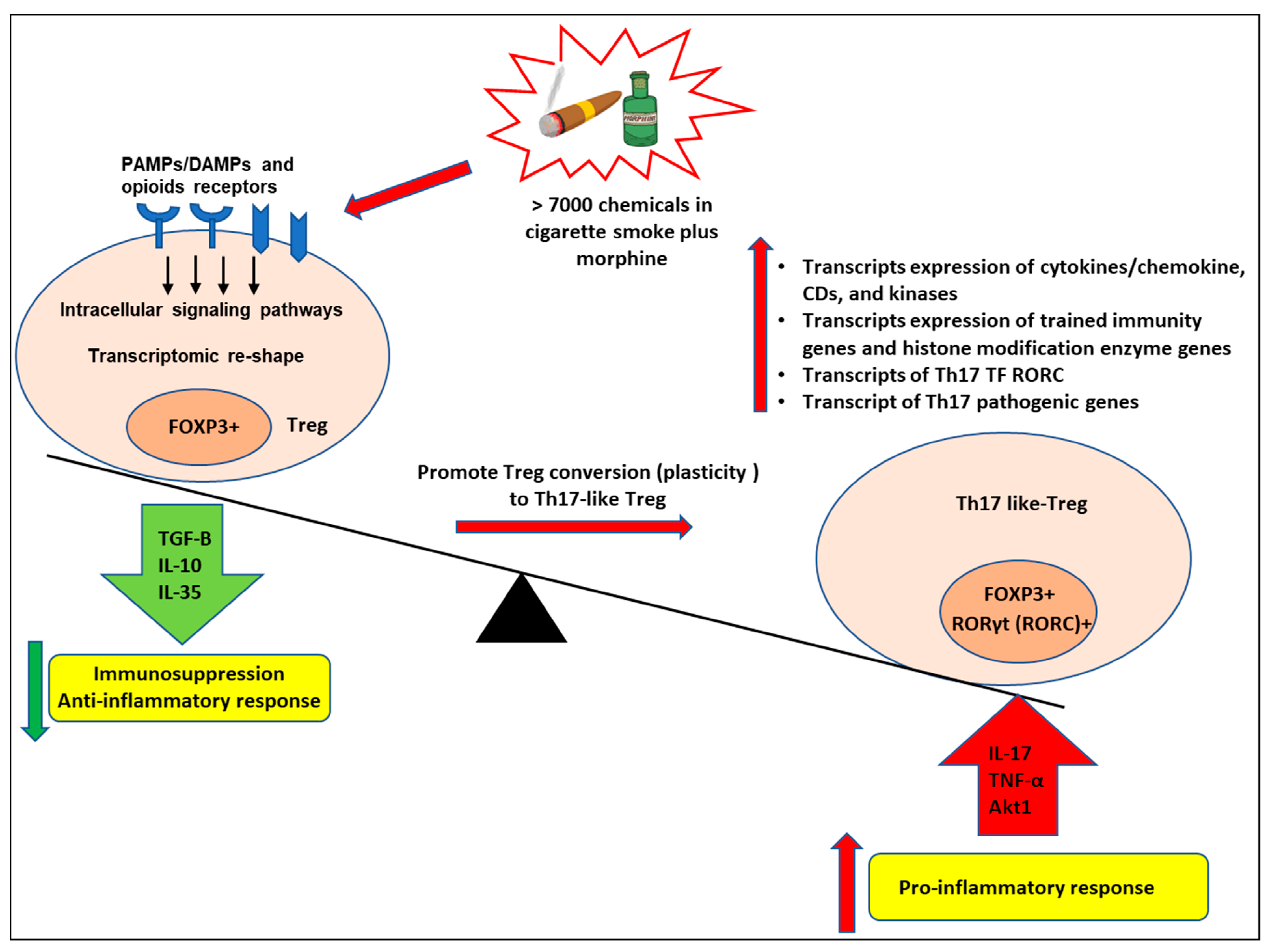
3.5. Cigarette Smoke Plus Morphine Increase the Transcripts of Th17 Transcription Factor (TF) RORC and CXCR5; Morphine Decreases Treg TF FOXP3, and IKZF4 and Increases Th1 TF RUNX3; and SM Inhibits Th1, Th17 and Treg Linage Marker TGIF1 in Tregs; and Six out of 139 SM + M Upregulated Genes Were Matched with Th17 Pathogenic Genes and 57, 38 out of 139 SM + M Upregulated Genes Were Matched with Group II and Group III Th17 Pathogenic Genes, Respectively
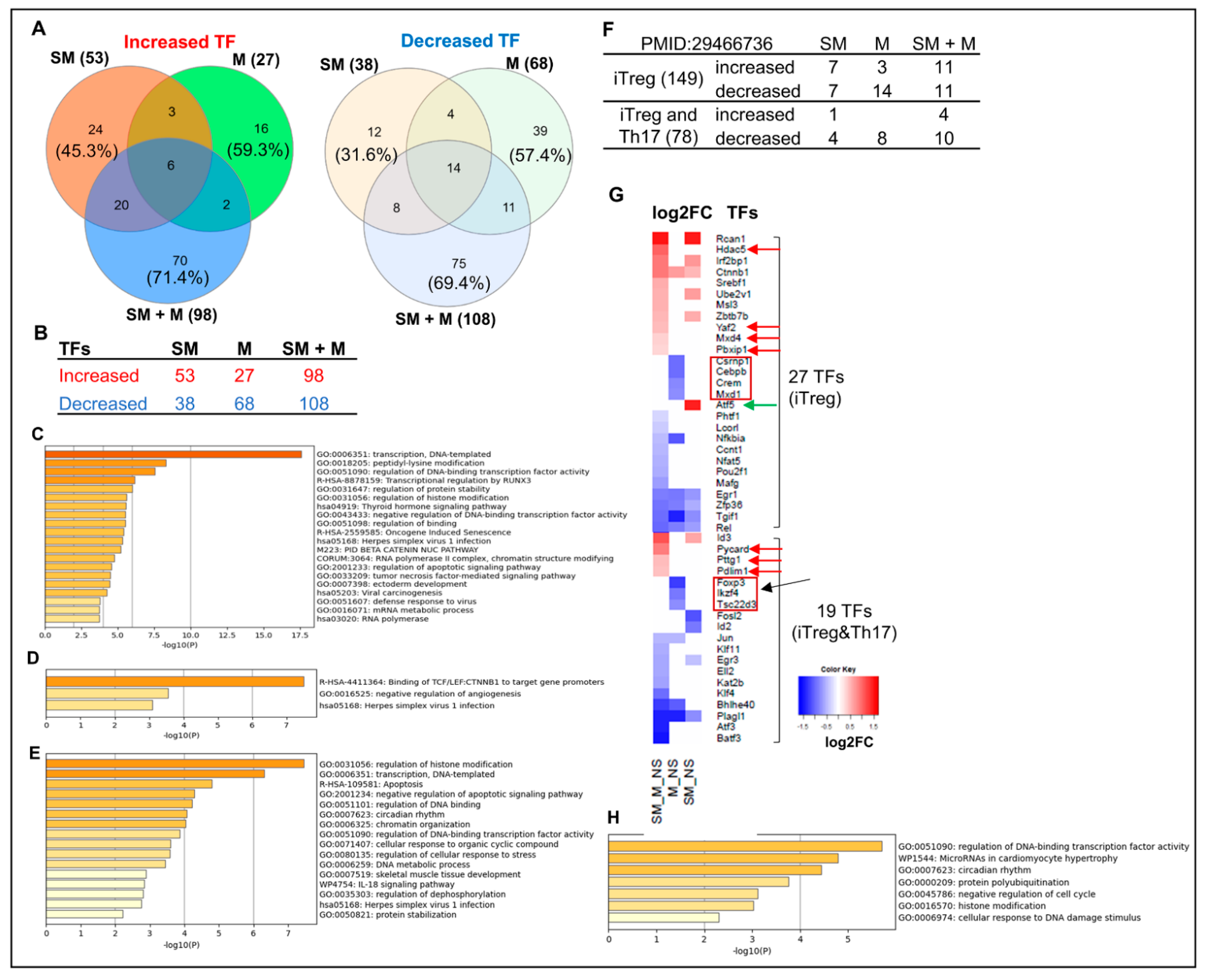
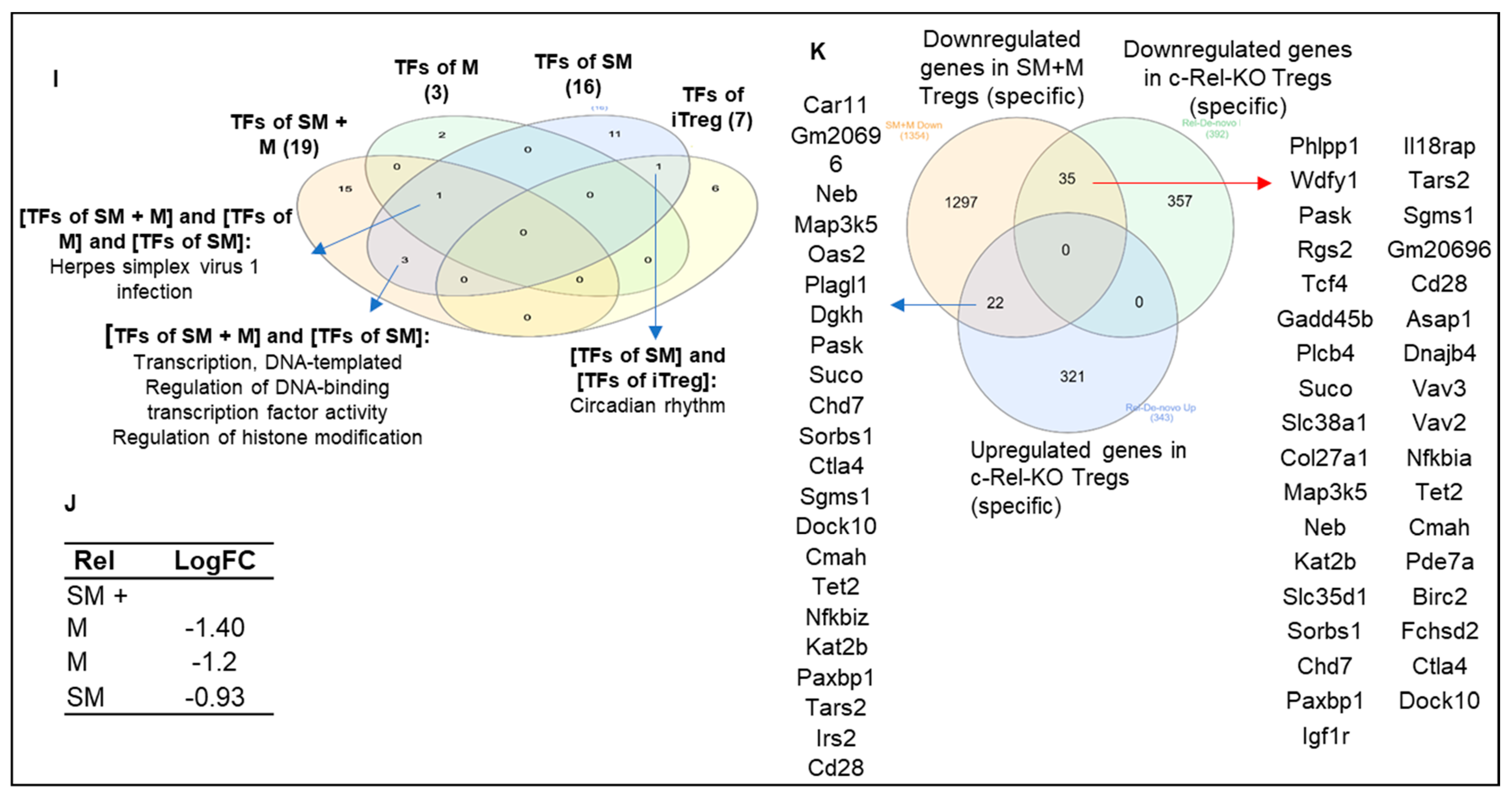
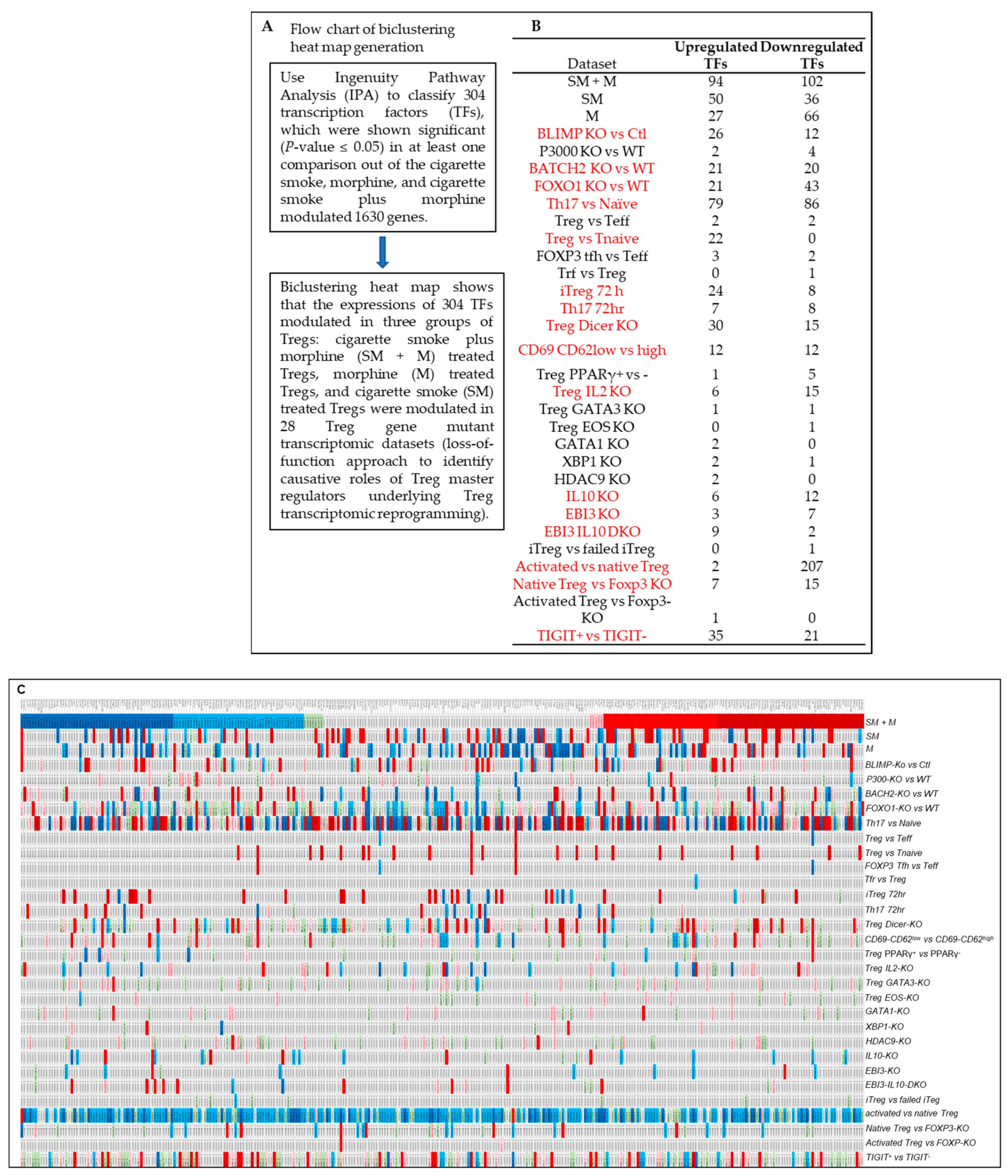
3.6. Cigarette Smoke Plus Morphine, Morphine, and Cigarette Smoke Upregulate the Transcripts of 70, 16, and 24 and Downregulate the Transcripts of 75, 39, and 12 IPA-Annotated Transcription Factors (TFs) in Tregs, Respectively; and SM + M, M, and SM Modulate the Transcripts of 22, 17, and 14 out of 149 Inducible Treg (iTreg) Specific TFs and 14, 8, and 5 out of 78 iTregs and Th17 Shared TFs in Mouse Splenic Tregs, Respectively
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Benowitz, N.L. Nicotine addiction. N. Engl. J. Med. 2010, 362, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Plumb, J.; Smyth, L.J.; Adams, H.R.; Vestbo, J.; Bentley, A.; Singh, S.D. Increased T-regulatory cells within lymphocyte follicles in moderate COPD. Eur. Respir. J. 2009, 34, 89–94. [Google Scholar] [CrossRef]
- Silva, L.E.F.; Lourenco, J.D.; Silva, K.R.; Santana, F.P.R.; Kohler, J.B.; Moreira, A.R.; Velosa, A.P.P.; Prado, C.M.; Vieira, R.P.; Aun, M.V.; et al. Th17/Treg imbalance in COPD development: Suppressors of cytokine signaling and signal transducers and activators of transcription proteins. Sci. Rep. 2020, 10, 15287. [Google Scholar] [CrossRef]
- Pawankar, R.; Hayashi, M.; Yamanishi, S.; Igarashi, T. The paradigm of cytokine networks in allergic airway inflammation. Curr. Opin. Allergy Clin. Immunol. 2015, 15, 41–48. [Google Scholar] [CrossRef] [PubMed]
- DuPage, M.; Bluestone, J.A. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat. Reviews. Immunol. 2016, 16, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.Y.; Shao, Y.; Lopez-Pastrana, J.; Mai, J.; Wang, H.; Yang, X.F. Pathological conditions re-shape physiological Tregs into pathological Tregs. Burn. Trauma 2015, 3, 1. [Google Scholar] [CrossRef]
- Ke, X.; Wang, J.; Li, L.; Chen, I.H.; Wang, H.; Yang, X.F. Roles of CD4+CD25(high) FOXP3+ Tregs in lymphomas and tumors are complex. Front. Biosci. 2008, 13, 3986–4001. [Google Scholar] [CrossRef][Green Version]
- Gowthaman, U.; Chen, J.S.; Zhang, B.; Flynn, W.F.; Lu, Y.; Song, W.; Joseph, J.; Gertie, J.A.; Xu, L.; Collet, M.A.; et al. Identification of a T follicular helper cell subset that drives anaphylactic IgE. Science 2019, 365, eaaw6433. [Google Scholar] [CrossRef]
- Webb, L.M.C.; Linterman, M.A. Signals that drive T follicular helper cell formation. Immunology 2017, 152, 185–194. [Google Scholar] [CrossRef]
- Tuzlak, S.; Dejean, A.S.; Iannacone, M.; Quintana, F.J.; Waisman, A.; Ginhoux, F.; Korn, T.; Becher, B. Repositioning TH cell polarization from single cytokines to complex help. Nat. Immunol. 2021, 22, 1210–1217. [Google Scholar] [CrossRef]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 2004, 22, 531–562. [Google Scholar] [CrossRef] [PubMed]
- Salvany-Celades, M.; van der Zwan, A.; Benner, M.; Setrajcic-Dragos, V.; Bougleux Gomes, H.A.; Iyer, V.; Norwitz, E.R.; Strominger, J.L.; Tilburgs, T. Three Types of Functional Regulatory T Cells Control T Cell Responses at the Human Maternal-Fetal Interface. Cell Rep. 2019, 27, 2537–2547.e2535. [Google Scholar] [CrossRef] [PubMed]
- Zemmour, D.; Zilionis, R.; Kiner, E.; Klein, A.M.; Mathis, D.; Benoist, C. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat. Immunol. 2018, 19, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xu, K.; Shao, Y.; Sun, Y.; Saredy, J.; Cutler, E.; Yao, T.; Liu, M.; Liu, L.; Drummer Iv, C.; et al. Tissue Treg Secretomes and Transcription Factors Shared With Stem Cells Contribute to a Treg Niche to Maintain Treg-Ness With 80% Innate Immune Pathways, and Functions of Immunosuppression and Tissue Repair. Front. Immunol. 2020, 11, 632239. [Google Scholar] [CrossRef]
- Miragaia, R.J.; Gomes, T.; Chomka, A.; Jardine, L.; Riedel, A.; Hegazy, A.N.; Whibley, N.; Tucci, A.; Chen, X.; Lindeman, I.; et al. Single-Cell Transcriptomics of Regulatory T Cells Reveals Trajectories of Tissue Adaptation. Immunity 2019, 50, 493–504.e497. [Google Scholar] [CrossRef]
- Mason, G.M.; Lowe, K.; Melchiotti, R.; Ellis, R.; de Rinaldis, E.; Peakman, M.; Heck, S.; Lombardi, G.; Tree, T.I. Phenotypic Complexity of the Human Regulatory T Cell Compartment Revealed by Mass Cytometry. J. Immunol. 2015, 195, 2030–2037. [Google Scholar] [CrossRef]
- Shevyrev, D.; Tereshchenko, V. Treg Heterogeneity, Function, and Homeostasis. Front. Immunol. 2019, 10, 3100. [Google Scholar] [CrossRef]
- Xu, K.; Yang, W.Y.; Nanayakkara, G.K.; Shao, Y.; Yang, F.; Hu, W.; Choi, E.T.; Wang, H.; Yang, X. gaTa3, hDac6, and Bcl6 regulate FOXP3+ Treg Plasticity and Determine Treg conversion into either novel antigen-Presenting cell-like Treg or Th1-Treg. Front. Immunol. 2018, 9, 45. [Google Scholar] [CrossRef]
- Rubtsov, Y.P.; Niec, R.E.; Josefowicz, S.; Li, L.; Darce, J.; Mathis, D.; Benoist, C.; Rudensky, A.Y. Stability of the regulatory T cell lineage in vivo. Science 2010, 329, 1667–1671. [Google Scholar] [CrossRef]
- Li, J.; McArdle, S.; Gholami, A.; Kimura, T.; Wolf, D.; Gerhardt, T.; Miller, J.; Weber, C.; Ley, K. CCR5+T-bet+FoxP3+ Effector CD4 T Cells Drive Atherosclerosis. Circ. Res. 2016, 118, 1540–1552. [Google Scholar] [CrossRef] [PubMed]
- Butcher, M.J.; Filipowicz, A.R.; Waseem, T.C.; McGary, C.M.; Crow, K.J.; Magilnick, N.; Boldin, M.; Lundberg, P.S.; Galkina, E.V. Atherosclerosis-Driven Treg Plasticity Results in Formation of a Dysfunctional Subset of Plastic IFNγ+ Th1/Tregs. Circ. Res. 2016, 119, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Kitz, A.; de Marcken, M.; Gautron, A.S.; Mitrovic, M.; Hafler, D.A.; Dominguez-Villar, M. AKT isoforms modulate Th1-like Treg generation and function in human autoimmune disease. EMBO Rep. 2016, 17, 1169–1183. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Gerhardt, T.; Winkels, H.; Michel, N.A.; Pramod, A.B.; Ghosheh, Y.; Brunel, S.; Buscher, K.; Miller, J.; McArdle, S.; et al. Pathogenic Autoimmunity in Atherosclerosis Evolves From Initially Protective Apolipoprotein B(100)-Reactive CD4(+) T-Regulatory Cells. Circulation 2020, 142, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- Lai, B.; Wang, J.; Fagenson, A.; Sun, Y.; Saredy, J.; Lu, Y.; Nanayakkara, G.; Yang, W.Y.; Yu, D.; Shao, Y.; et al. Twenty Novel Disease Group-Specific and 12 New Shared Macrophage Pathways in Eight Groups of 34 Diseases Including 24 Inflammatory Organ Diseases and 10 Types of Tumors. Front. Immunol. 2019, 10, 2612. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.J.; Yang, X.F.; McLaughlin, S.; Neuberg, D.; Canning, C.; Stein, B.; Alyea, E.P.; Soiffer, R.J.; Dranoff, G.; Ritz, J. Detection of a potent humoral response associated with immune-induced remission of chronic myelogenous leukemia. J. Clin. Investig. 2000, 106, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.F.; Wu, C.J.; McLaughlin, S.; Chillemi, A.; Wang, K.S.; Canning, C.; Alyea, E.P.; Kantoff, P.; Soiffer, R.J.; Dranoff, G.; et al. CML66, a broadly immunogenic tumor antigen, elicits a humoral immune response associated with remission of chronic myelogenous leukemia. Proc. Natl. Acad. Sci. USA 2001, 98, 7492–7497. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.F.; Wu, C.J.; Chen, L.; Alyea, E.P.; Canning, C.; Kantoff, P.; Soiffer, R.J.; Dranoff, G.; Ritz, J. CML28 is a broadly immunogenic antigen, which is overexpressed in tumor cells. Cancer Res. 2002, 62, 5517–5522. [Google Scholar] [PubMed]
- Yang, F.; Yang, X.F. New concepts in tumor antigens: Their significance in future immunotherapies for tumors. Cell Mol. Immunol. 2005, 2, 331–341. [Google Scholar] [PubMed]
- Yan, Y.; Phan, L.; Yang, F.; Talpaz, M.; Yang, Y.; Xiong, Z.; Ng, B.; Timchenko, N.A.; Wu, C.J.; Ritz, J.; et al. A novel mechanism of alternative promoter and splicing regulates the epitope generation of tumor antigen CML66-L. J. Immunol. 2004, 172, 651–660. [Google Scholar] [CrossRef]
- Ng, B.; Yang, F.; Huston, D.P.; Yan, Y.; Yang, Y.; Xiong, Z.; Peterson, L.E.; Wang, H.; Yang, X.F. Increased noncanonical splicing of autoantigen transcripts provides the structural basis for expression of untolerized epitopes. J. Allergy Clin. Immunol. 2004, 114, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Liu, E.; Yan, Y.; Silver, R.T.; Yang, F.; Chen, I.H.; Chen, Y.; Verstovsek, S.; Wang, H.; Prchal, J.; et al. An unconventional antigen translated by a novel internal ribosome entry site elicits antitumor humoral immune reactions. J. Immunol. 2006, 177, 4907–4916. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.Y.; Wang, J.; Yang, X.F. [New principles in tumor antigens and their significance in future immunotherapies for lymphomas and other malignancies--editorial]. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2006, 14, 419–426. [Google Scholar] [PubMed]
- Yang, F.; Chen, I.H.; Xiong, Z.; Yan, Y.; Wang, H.; Yang, X.F. Model of stimulation-responsive splicing and strategies in identification of immunogenic isoforms of tumor antigens and autoantigens. Clin. Immunol. 2006, 121, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Yan, Y.; Liu, E.; Silver, R.T.; Verstovsek, S.; Yang, F.; Wang, H.; Prchal, J.; Yang, X.F. Novel tumor antigens elicit anti-tumor humoral immune reactions in a subset of patients with polycythemia vera. Clin. Immunol. 2007, 122, 279–287. [Google Scholar] [CrossRef]
- Xiong, Z.; Liu, E.; Yan, Y.; Silver, R.T.; Yang, F.; Chen, I.H.; Hodge, I.; Verstovsek, S.; Segura, F.J.; Wang, H.; et al. A novel unconventional antigen MPD5 elicits anti-tumor humoral immune responses in a subset of patients with polycythemia vera. Int. J. Immunopathol. Pharmacol. 2007, 20, 373–380. [Google Scholar] [CrossRef]
- Wu, C.J.; Biernacki, M.; Kutok, J.L.; Rogers, S.; Chen, L.; Yang, X.F.; Soiffer, R.J.; Ritz, J. Graft-versus-leukemia target antigens in chronic myelogenous leukemia are expressed on myeloid progenitor cells. Clin. Cancer Res. 2005, 11, 4504–4511. [Google Scholar] [CrossRef]
- Yan, Y.; Chen, Y.; Yang, F.; Chen, I.H.; Xiong, Z.; Wang, J.; Lachman, L.B.; Wang, H.; Yang, X.F. HLA-A2.1-restricted T cells react to SEREX-defined tumor antigen CML66L and are suppressed by CD4+CD25+ regulatory T cells. Int. J. Immunopathol. Pharmacol. 2007, 20, 75–89. [Google Scholar] [CrossRef]
- Yang, X.F.; Weber, G.F.; Cantor, H. A novel Bcl-x isoform connected to the T cell receptor regulates apoptosis in T cells. Immunity 1997, 7, 629–639. [Google Scholar] [CrossRef]
- Shao, Y.; Yang, W.Y.; Saaoud, F.; Drummer, C.t.; Sun, Y.; Xu, K.; Lu, Y.; Shan, H.; Shevach, E.M.; Jiang, X.; et al. IL-35 promotes CD4+Foxp3+ Tregs and inhibits atherosclerosis via maintaining CCR5-amplified Treg-suppressive mechanisms. JCI Insight 2021, 6, e152511. [Google Scholar] [CrossRef]
- Shen, H.; Wu, N.; Nanayakkara, G.; Fu, H.; Yang, Q.; Yang, W.Y.; Li, A.; Sun, Y.; Drummer Iv, C.; Johnson, C.; et al. Co-signaling receptors regulate T-cell plasticity and immune tolerance. Front. Biosci. 2019, 24, 96–132. [Google Scholar]
- Holt, P.G. Immune and inflammatory function in cigarette smokers. Thorax 1987, 42, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Sopori, M.L.; Kozak, W. Immunomodulatory effects of cigarette smoke. J. Neuroimmunol. 1998, 83, 148–156. [Google Scholar] [CrossRef]
- Saaoud, F.; Shao, Y.; Cornwell, W.; Wang, H.; Rogers, T.; Yang, X. Cigarette smoke modulates inflammation and immunity via ROS-regulated trained immunity and trained tolerance mechanisms. Antioxid Redox Signal. 2022. [Google Scholar] [CrossRef]
- Siasos, G.; Tsigkou, V.; Kokkou, E.; Oikonomou, E.; Vavuranakis, M.; Vlachopoulos, C.; Verveniotis, A.; Limperi, M.; Genimata, V.; Papavassiliou, A.G.; et al. Smoking and atherosclerosis: Mechanisms of disease and new therapeutic approaches. Curr. Med. Chem. 2014, 21, 3936–3948. [Google Scholar] [CrossRef]
- Barua, R.S.; Sharma, M.; Dileepan, K.N. Cigarette Smoke Amplifies Inflammatory Response and Atherosclerosis Progression Through Activation of the H1R-TLR2/4-COX2 Axis. Front. Immunol. 2015, 6, 572. [Google Scholar] [CrossRef]
- Plein, L.M.; Rittner, H.L. Opioids and the immune system-friend or foe. Br. J. Pharmacol. 2018, 175, 2717–2725. [Google Scholar] [CrossRef]
- Steele, A.D.; Henderson, E.E.; Rogers, T.J. Mu-opioid modulation of HIV-1 coreceptor expression and HIV-1 replication. Virology 2003, 309, 99–107. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Sun, Y.; Shao, Y.; Yang, W.Y.; Jiang, X.; Wang, H.; Yang, X. Anti-inflammatory cytokines IL-35 and IL-10 block atherogenic lysophosphatidylcholine-induced, mitochondrial ROS-mediated innate immune activation, but spare innate immune memory signature in endothelial cells. Redox Biol. 2020, 28, 101373. [Google Scholar] [CrossRef]
- Mai, J.; Nanayakkara, G.; Lopez-Pastrana, J.; Li, X.; Li, Y.F.; Wang, X.; Song, A.; Virtue, A.; Shao, Y.; Shan, H.; et al. Interleukin-17A Promotes Aortic Endothelial Cell Activation via Transcriptionally and Post-translationally Activating p38 Mitogen-activated Protein Kinase (MAPK) Pathway. J. Biol. Chem. 2016, 291, 4939–4954. [Google Scholar] [CrossRef]
- Abo-Elnazar, S.; Moaaz, M.; Ghoneim, H.; Molokhia, T.; El-Korany, W. Th17/Treg imbalance in opioids and cannabinoids addiction: Relationship to NF-kappaB activation in CD4+ T cells. Egypt J. Immunol. 2014, 21, 33–47. [Google Scholar] [PubMed]
- Shao, Y.; Cornwell, W.; Xu, K.; Kirchhoff, A.; Saasoud, F.; Lu, Y.; Jiang, X.; Criner, G.J.; Wang, H.; Rogers, T.J.; et al. Chronic Exposure to the Combination of Cigarette Smoke and Morphine Decreases CD4(+) Regulatory T Cell Numbers by Reprogramming the Treg Cell Transcriptome. Front. Immunol. 2022, 13, 887681. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, L.; Fang, P.; Sun, Y.; Jiang, X.; Wang, H.; Yang, X.-F. Lysophospholipids induce innate immune transdifferentiation of endothelial cells, resulting in prolonged endothelial activation. J. Biol. Chem. 2018, 293, 11033–11045. [Google Scholar] [CrossRef] [PubMed]
- Beiting, D.P.; Hidano, S.; Baggs, J.E.; Geskes, J.M.; Fang, Q.; Wherry, E.J.; Hunter, C.A.; Roos, D.S.; Cherry, S. The Orphan Nuclear Receptor TLX Is an Enhancer of STAT1-Mediated Transcription and Immunity to Toxoplasma gondii. PLoS Biol. 2015, 13, e1002200. [Google Scholar] [CrossRef]
- Wang, L.; Fu, H.; Nanayakkara, G.; Li, Y.; Shao, Y.; Johnson, C.; Cheng, J.; Yang, W.Y.; Yang, F.; Lavallee, M.; et al. Novel extracellular and nuclear caspase-1 and inflammasomes propagate inflammation and regulate gene expression: A comprehensive database mining study. J. Hematol. Oncol. 2016, 9, 122. [Google Scholar] [CrossRef]
- Li, Y.F.; Nanayakkara, G.; Sun, Y.; Li, X.; Wang, L.; Cueto, R.; Shao, Y.; Fu, H.; Johnson, C.; Cheng, J.; et al. Analyses of caspase-1-regulated transcriptomes in various tissues lead to identification of novel IL-1beta-, IL-18- and sirtuin-1-independent pathways. J. Hematol. Oncol. 2017, 10, 40. [Google Scholar] [CrossRef]
- Zhang, R.; Saredy, J.; Shao, Y.; Yao, T.; Liu, L.; Saaoud, F.; Yang, W.Y.; Sun, Y.; Johnson, C.; Drummer, C.; et al. End-stage renal disease is different from chronic kidney disease in upregulating ROS-modulated proinflammatory secretome in PBMCs-A novel multiple-hit model for disease progression. REDOX Biol. 2020, 34, 101460. [Google Scholar] [CrossRef]
- Pompura, S.L.; Dominguez-Villar, M. The PI3K/AKT signaling pathway in regulatory T-cell development, stability, and function. J. Leukoc. Biol. 2018, 103, 1065–1076. [Google Scholar] [CrossRef]
- Drummer, C.t.; Saaoud, F.; Shao, Y.; Sun, Y.; Xu, K.; Lu, Y.; Ni, D.; Atar, D.; Jiang, X.; Wang, H.; et al. Trained Immunity and Reactivity of Macrophages and Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1032–1046. [Google Scholar] [CrossRef]
- Wajant, H.; Beilhack, A. Targeting Regulatory T Cells by Addressing Tumor Necrosis Factor and Its Receptors in Allogeneic Hematopoietic Cell Transplantation and Cancer. Front. Immunol. 2019, 10, 2040. [Google Scholar] [CrossRef]
- Yin, Y.; Pastrana, J.L.; Li, X.; Huang, X.; Mallilankaraman, K.; Choi, E.T.; Madesh, M.; Wang, H.; Yang, X.F. Inflammasomes: Sensors of metabolic stresses for vascular inflammation. Front. Biosci. 2013, 18, 638–649. [Google Scholar]
- Hou, X.; He, S.; Zhang, D.; Yang, C.; Shi, Y.; Zhang, K. Expression and Clinical Significance of CMTM6 in Nonsmall Cell Lung Cancer. DNA Cell Biol. 2020, 39, 2265–2271. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Zhang, S.W.; Chao, X.; Wang, C.H.; Yang, X.; Zhang, X.K.; Wen, Y.L.; Yun, J.P.; Luo, R.Z. Coexpression of CMTM6 and PD-L1 as a predictor of poor prognosis in macrotrabecular-massive hepatocellular carcinoma. Cancer Immunol. Immunother. 2020, 70, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Shao, Y.; Tian, Y.; Ouyang, C.; Wang, X. Nuclear Alarmin Cytokines in Inflammation. J. Immunol. Res. 2020, 2020, 7206451. [Google Scholar] [CrossRef]
- Luo, S.; Zhu, R.; Yu, T.; Fan, H.; Hu, Y.; Mohanta, S.K.; Hu, D. Chronic Inflammation: A Common Promoter in Tertiary Lymphoid Organ Neogenesis. Front. Immunol. 2019, 10, 2938. [Google Scholar] [CrossRef]
- Moorman, H.R.; Poschel, D.; Klement, J.D.; Lu, C.; Redd, P.S.; Liu, K. Osteopontin: A Key Regulator of Tumor Progression and Immunomodulation. Cancers 2020, 12, 3379. [Google Scholar] [CrossRef]
- Vianello, E.; Kalousova, M.; Dozio, E.; Tacchini, L.; Zima, T.; Corsi Romanelli, M.M. Osteopontin: The Molecular Bridge between Fat and Cardiac-Renal Disorders. Int. J. Mol. Sci. 2020, 21, 5568. [Google Scholar] [CrossRef]
- Alberts, R.; Chen, H.; Pommerenke, C.; Smit, A.B.; Spijker, S.; Williams, R.W.; Geffers, R.; Bruder, D.; Schughart, K. Expression QTL mapping in regulatory and helper T cells from the BXD family of strains reveals novel cell-specific genes, gene-gene interactions and candidate genes for auto-immune disease. BMC Genom. 2011, 12, 610. [Google Scholar] [CrossRef]
- Mei, Z.Z.; Chen, X.Y.; Hu, S.W.; Wang, N.; Ou, X.L.; Wang, J.; Luo, H.H.; Liu, J.; Jiang, Y. Kelch-like Protein 21 (KLHL21) Targets IkappaB Kinase-beta to Regulate Nuclear Factor kappa-Light Chain Enhancer of Activated B Cells (NF-kappaB) Signaling Negatively. J. Biol. Chem. 2016, 291, 18176–18189. [Google Scholar] [CrossRef]
- Yin, Y.; Li, X.; Sha, X.; Xi, H.; Li, Y.F.; Shao, Y.; Mai, J.; Virtue, A.; Lopez-Pastrana, J.; Meng, S.; et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arter. Thromb. Vasc. Biol. 2015, 35, 804–816. [Google Scholar] [CrossRef]
- Birrell, M.A.; Wong, S.; Catley, M.C.; Belvisi, M.G. Impact of tobacco-smoke on key signaling pathways in the innate immune response in lung macrophages. J. Cell Physiol. 2008, 214, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, F.; Xiong, Z.; Yan, Y.; Wang, X.; Nishino, M.; Mirkovic, D.; Nguyen, J.; Wang, H.; Yang, X.F. An N-terminal region of translationally controlled tumor protein is required for its antiapoptotic activity. Oncogene 2005, 24, 4778–4788. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xiong, Z.; Zhang, S.; Yan, Y.; Nguyen, J.; Ng, B.; Lu, H.; Brendese, J.; Yang, F.; Wang, H.; et al. Bcl-xL inhibits T-cell apoptosis induced by expression of SARS coronavirus E protein in the absence of growth factors. Biochem. J. 2005, 392, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Xiong, Z.; Zhang, S.; Song, J.; Huang, Y.; Thornton, A.M.; Wang, H.; Yang, X.F. CD25high T cells with a prolonged survival inhibit development of diabetes. Int. J. Immunopathol. Pharmacol. 2008, 21, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Song, J.; Yan, Y.; Huang, Y.; Cowan, A.; Wang, H.; Yang, X.F. Higher expression of Bax in regulatory T cells increases vascular inflammation. Front. Biosci. 2008, 13, 7143–7155. [Google Scholar] [CrossRef]
- Xiong, Z.; Yan, Y.; Song, J.; Fang, P.; Yin, Y.; Yang, Y.; Cowan, A.; Wang, H.; Yang, X.F. Expression of TCTP antisense in CD25(high) regulatory T cells aggravates cuff-injured vascular inflammation. Atherosclerosis 2009, 203, 401–408. [Google Scholar] [CrossRef]
- Rakocevic, J.; Orlic, D.; Mitrovic-Ajtic, O.; Tomasevic, M.; Dobric, M.; Zlatic, N.; Milasinovic, D.; Stankovic, G.; Ostojic, M.; Labudovic-Borovic, M. Endothelial cell markers from clinician’s perspective. Exp. Mol. Pathol. 2017, 102, 303–313. [Google Scholar] [CrossRef]
- Woelfinger, P.; Epp, K.; Schaefer, L.; Kriege, D.; Theobald, M.; Bopp, T.; Wagner-Drouet, E.M. CD52-negative T cells predict acute graft-versus-host disease after an alemtuzumab-based conditioning regimen. Br. J. Haematol. 2020, 191, 253–262. [Google Scholar] [CrossRef]
- Vasir, B.; Zarwan, C.; Ahmad, R.; Crawford, K.D.; Rajabi, H.; Matsuoka, K.; Rosenblatt, J.; Wu, Z.; Mills, H.; Kufe, D.; et al. Induction of antitumor immunity ex vivo using dendritic cells transduced with fowl pox vector expressing MUC1, CEA, and a triad of costimulatory molecules (rF-PANVAC). J. Immunother. 2012, 35, 555–569. [Google Scholar] [CrossRef]
- Lu, Y.; Sun, Y.; Drummer, C.t.; Nanayakkara, G.K.; Shao, Y.; Saaoud, F.; Johnson, C.; Zhang, R.; Yu, D.; Li, X.; et al. Increased acetylation of H3K14 in the genomic regions that encode trained immunity enzymes in lysophosphatidylcholine-activated human aortic endothelial cells-Novel qualification markers for chronic disease risk factors and conditional DAMPs. Redox Biol. 2019, 24, 101221. [Google Scholar] [CrossRef]
- Netea, M.G.; Dominguez-Andres, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Riksen, N.P. Trained immunity and atherosclerotic cardiovascular disease. Curr. Opin. Lipidol. 2019, 30, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Schnack, L.; Sohrabi, Y.; Lagache, S.M.M.; Kahles, F.; Bruemmer, D.; Waltenberger, J.; Findeisen, H.M. Mechanisms of Trained Innate Immunity in oxLDL Primed Human Coronary Smooth Muscle Cells. Front. Immunol. 2019, 10, 13. [Google Scholar] [CrossRef]
- Drummer, C.I.V.; Saaoud, F.; Sun, Y.; Atar, D.; Xu, K.; Lu, Y.; Shao, Y.; Johnson, C.; Liu, L.; Shen, H.; et al. Hyperlipidemia May Synergize with Hypomethylation in Establishing Trained Immunity and Promoting Inflammation in NASH and NAFLD. J. Immunol. Res. 2021, 2021, 3928323. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Sun, Y.; Xu, K.; Saaoud, F.; Shao, Y.; Drummer, C.t.; Wu, S.; Hu, W.; Yu, J.; Kunapuli, S.P.; et al. Aorta in Pathologies May Function as an Immune Organ by Upregulating Secretomes for Immune and Vascular Cell Activation, Differentiation and Trans-Differentiation-Early Secretomes may Serve as Drivers for Trained Immunity. Front. Immunol. 2022, 13, 858256. [Google Scholar] [CrossRef]
- Bekkering, S.; Blok, B.A.; Joosten, L.A.; Riksen, N.P.; van Crevel, R.; Netea, M.G. In Vitro Experimental Model of Trained Innate Immunity in Human Primary Monocytes. Clin. Vaccine Immunol. 2016, 23, 926–933. [Google Scholar] [CrossRef]
- Bekkering, S.; Quintin, J.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Riksen, N.P. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arter. Thromb. Vasc. Biol. 2014, 34, 1731–1738. [Google Scholar] [CrossRef]
- Kleinnijenhuis, J.; Quintin, J.; Preijers, F.; Joosten, L.A.; Jacobs, C.; Xavier, R.J.; van der Meer, J.W.; van Crevel, R.; Netea, M.G. BCG-induced trained immunity in NK cells: Role for non-specific protection to infection. Clin. Immunol. 2014, 155, 213–219. [Google Scholar] [CrossRef]
- Christ, A.; Günther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baßler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175.e114. [Google Scholar] [CrossRef]
- Cavalli, G.; Tengesdal, I.W.; Gresnigt, M.; Nemkov, T.; Arts, R.J.W.; Domínguez-Andrés, J.; Molteni, R.; Stefanoni, D.; Cantoni, E.; Cassina, L.; et al. The anti-inflammatory cytokine interleukin-37 is an inhibitor of trained immunity. Cell Rep. 2021, 35, 108955. [Google Scholar] [CrossRef]
- Van der Heijden, C.; Noz, M.P.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P.; Keating, S.T. Epigenetics and Trained Immunity. Antioxid Redox Signal. 2018, 29, 1023–1040. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Korn, T.; Oukka, M.; Kuchroo, V.K. Induction and effector functions of T(H)17 cells. Nature 2008, 453, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Arts, R.J.W.; Novakovic, B.; Kourtzelis, I.; van der Heijden, C.; Li, Y.; Popa, C.D.; Ter Horst, R.; van Tuijl, J.; Netea-Maier, R.T.; et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 2018, 172, 135–146.e139. [Google Scholar] [CrossRef]
- Shao, Y.; Saredy, J.; Yang, W.Y.; Sun, Y.; Lu, Y.; Saaoud, F.; Drummer, C.t.; Johnson, C.; Xu, K.; Jiang, X.; et al. Vascular Endothelial Cells and Innate Immunity. Arter. Thromb. Vasc. Biol. 2020, 40, e138–e152. [Google Scholar] [CrossRef]
- Li, X.; Shao, Y.; Sha, X.; Fang, P.; Kuo, Y.M.; Andrews, A.J.; Li, Y.; Yang, W.Y.; Maddaloni, M.; Pascual, D.W.; et al. IL-35 (Interleukin-35) Suppresses Endothelial Cell Activation by Inhibiting Mitochondrial Reactive Oxygen Species-Mediated Site-Specific Acetylation of H3K14 (Histone 3 Lysine 14). Arter. Thromb. Vasc. Biol. 2018, 38, 599–609. [Google Scholar] [CrossRef]
- Zhong, C.; Yang, X.; Feng, Y.; Yu, J. Trained Immunity: An Underlying Driver of Inflammatory Atherosclerosis. Front. Immunol. 2020, 11, 284. [Google Scholar] [CrossRef]
- Shao, Y.; Chernaya, V.; Johnson, C.; Yang, W.Y.; Cueto, R.; Sha, X.; Zhang, Y.; Qin, X.; Sun, J.; Choi, E.T.; et al. Metabolic Diseases Downregulate the Majority of Histone Modification Enzymes, Making a Few Upregulated Enzymes Novel Therapeutic Targets--"Sand Out and Gold Stays". J. Cardiovasc. Transl. Res. 2016, 9, 49–66. [Google Scholar] [CrossRef]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef]
- Capone, A.; Volpe, E. Transcriptional Regulators of T Helper 17 Cell Differentiation in Health and Autoimmune Diseases. Front. Immunol. 2020, 11, 348. [Google Scholar] [CrossRef]
- Volpe, E.; Servant, N.; Zollinger, R.; Bogiatzi, S.I.; Hupé, P.; Barillot, E.; Soumelis, V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat. Immunol. 2008, 9, 650–657. [Google Scholar] [CrossRef]
- Manel, N.; Unutmaz, D.; Littman, D.R. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat. Immunol. 2008, 9, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Gaublomme, J.T.; Yosef, N.; Lee, Y.; Gertner, R.S.; Yang, L.V.; Wu, C.; Pandolfi, P.P.; Mak, T.; Satija, R.; Shalek, A.K.; et al. Single-Cell Genomics Unveils Critical Regulators of Th17 Cell Pathogenicity. Cell 2015, 163, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Awasthi, A.; Yosef, N.; Quintana, F.J.; Xiao, S.; Peters, A.; Wu, C.; Kleinewietfeld, M.; Kunder, S.; Hafler, D.A.; et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 2012, 13, 991–999. [Google Scholar] [CrossRef]
- Ubaid, U.; Andrabi, S.B.A.; Tripathi, S.K.; Dirasantha, O.; Kanduri, K.; Rautio, S.; Gross, C.C.; Lehtimaki, S.; Bala, K.; Tuomisto, J.; et al. Transcriptional Repressor HIC1 Contributes to Suppressive Function of Human Induced Regulatory T Cells. Cell Rep. 2018, 22, 2094–2106. [Google Scholar] [CrossRef] [PubMed]
- Fulford, T.S.; Grumont, R.; Wirasinha, R.C.; Ellis, D.; Barugahare, A.; Turner, S.J.; Naeem, H.; Powell, D.; Lyons, P.A.; Smith, K.G.C.; et al. c-Rel employs multiple mechanisms to promote the thymic development and peripheral function of regulatory T cells in mice. Eur. J. Immunol. 2021, 51, 2006–2026. [Google Scholar] [CrossRef]
- Cretney, E.; Xin, A.; Shi, W.; Minnich, M.; Masson, F.; Miasari, M.; Belz, G.T.; Smyth, G.K.; Busslinger, M.; Nutt, S.L.; et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat. Immunol. 2011, 12, 304–311. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Predina, J.; Han, R.; Beier, U.H.; Wang, L.C.; Kapoor, V.; Bhatti, T.R.; Akimova, T.; Singhal, S.; et al. Inhibition of p300 impairs Foxp3+ T regulatory cell function and promotes antitumor immunity. Nat. Med. 2013, 19, 1173–1177. [Google Scholar] [CrossRef]
- Roychoudhuri, R.; Hirahara, K.; Mousavi, K.; Clever, D.; Klebanoff, C.A.; Bonelli, M.; Sciumè, G.; Zare, H.; Vahedi, G.; Dema, B.; et al. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis. Nature 2013, 498, 506–510. [Google Scholar] [CrossRef]
- Ouyang, W.; Liao, W.; Luo, C.T.; Yin, N.; Huse, M.; Kim, M.V.; Peng, M.; Chan, P.; Ma, Q.; Mo, Y.; et al. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature 2012, 491, 554–559. [Google Scholar] [CrossRef]
- Luo, C.T.; Liao, W.; Dadi, S.; Toure, A.; Li, M.O. Graded Foxo1 activity in Treg cells differentiates tumour immunity from spontaneous autoimmunity. Nature 2016, 529, 532–536. [Google Scholar] [CrossRef]
- Linterman, M.A.; Pierson, W.; Lee, S.K.; Kallies, A.; Kawamoto, S.; Rayner, T.F.; Srivastava, M.; Divekar, D.P.; Beaton, L.; Hogan, J.J.; et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 2011, 17, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Jeker, L.T.; Fife, B.T.; Zhu, S.; Anderson, M.S.; McManus, M.T.; Bluestone, J.A. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J. Exp. Med. 2008, 205, 1983–1991. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Yuan, X.; Tsai, M.S.; Podack, E.R.; Yu, A.; Malek, T.R. IL-2 receptor signaling is essential for the development of Klrg1+ terminally differentiated T regulatory cells. J. Immunol. 2012, 189, 1780–1791. [Google Scholar] [CrossRef]
- Cipolletta, D.; Cohen, P.; Spiegelman, B.M.; Benoist, C.; Mathis, D. Appearance and disappearance of the mRNA signature characteristic of Treg cells in visceral adipose tissue: Age, diet, and PPARγ effects. Proc. Natl. Acad. Sci. USA 2015, 112, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Zhu, L.; Altman, N.H.; Malek, T.R. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity 2009, 30, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Rudra, D.; deRoos, P.; Chaudhry, A.; Niec, R.E.; Arvey, A.; Samstein, R.M.; Leslie, C.; Shaffer, S.A.; Goodlett, D.R.; Rudensky, A.Y. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat. Immunol. 2012, 13, 1010–1019. [Google Scholar] [CrossRef]
- Fu, W.; Ergun, A.; Lu, T.; Hill, J.A.; Haxhinasto, S.; Fassett, M.S.; Gazit, R.; Adoro, S.; Glimcher, L.; Chan, S.; et al. A multiply redundant genetic switch ’locks in’ the transcriptional signature of regulatory T cells. Nat. Immunol. 2012, 13, 972–980. [Google Scholar] [CrossRef]
- Beier, U.H.; Wang, L.; Han, R.; Akimova, T.; Liu, Y.; Hancock, W.W. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci. Signal. 2012, 5, ra45. [Google Scholar] [CrossRef] [PubMed]
- Pillai, M.R.; Collison, L.W.; Wang, X.; Finkelstein, D.; Rehg, J.E.; Boyd, K.; Szymczak-Workman, A.L.; Doggett, T.; Griffith, T.S.; Ferguson, T.A.; et al. The plasticity of regulatory T cell function. J. Immunol. 2011, 187, 4987–4997. [Google Scholar] [CrossRef]
- Haribhai, D.; Lin, W.; Edwards, B.; Ziegelbauer, J.; Salzman, N.H.; Carlson, M.R.; Li, S.H.; Simpson, P.M.; Chatila, T.A.; Williams, C.B. A central role for induced regulatory T cells in tolerance induction in experimental colitis. J. Immunol. 2009, 182, 3461–3468. [Google Scholar] [CrossRef]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan, T.G.; Sefik, E.; Yajnik, V.; et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014, 40, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Plaisier, C.L.; O’Brien, S.; Bernard, B.; Reynolds, S.; Simon, Z.; Toledo, C.M.; Ding, Y.; Reiss, D.J.; Paddison, P.J.; Baliga, N.S. Causal Mechanistic Regulatory Network for Glioblastoma Deciphered Using Systems Genetics Network Analysis. Cell Syst. 2016, 3, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J. T Helper Cell Differentiation, Heterogeneity, and Plasticity. Cold Spring Harb. Perspect. Biol. 2018, 10, a030338. [Google Scholar] [CrossRef]
- Meng, X.; Yang, J.; Dong, M.; Zhang, K.; Tu, E.; Gao, Q.; Chen, W.; Zhang, C.; Zhang, Y. Regulatory T cells in cardiovascular diseases. Nat. Rev. Cardiol. 2016, 13, 167–179. [Google Scholar] [CrossRef]
- Yang, X.F.; Yin, Y.; Wang, H. VASCULAR INFLAMMATION AND ATHEROGENESIS ARE ACTIVATED VIA RECEPTORS FOR PAMPs AND SUPPRESSED BY REGULATORY T CELLS. Drug Discov. Today Ther. Strateg. 2008, 5, 125–142. [Google Scholar] [CrossRef]
- Lopez-Pastrana, J.; Shao, Y.; Chernaya, V.; Wang, H.; Yang, X.F. Epigenetic enzymes are the therapeutic targets for CD4(+)CD25(+/high)Foxp3(+) regulatory T cells. Transl. Res. 2015, 165, 221–240. [Google Scholar] [CrossRef]
- Pastrana, J.L.; Sha, X.; Virtue, A.; Mai, J.; Cueto, R.; Lee, I.A.; Wang, H.; Yang, X.F. Regulatory T cells and Atherosclerosis. J. Clin. Exp. Cardiolog. 2012, 2012, 2. [Google Scholar] [CrossRef]
- Osorio, F.; LeibundGut-Landmann, S.; Lochner, M.; Lahl, K.; Sparwasser, T.; Eberl, G.; Reis e Sousa, C. DC activated via dectin-1 convert Treg into IL-17 producers. Eur. J. Immunol. 2008, 38, 3274–3281. [Google Scholar] [CrossRef]
- Li, L.; Patsoukis, N.; Petkova, V.; Boussiotis, V.A. Runx1 and Runx3 are involved in the generation and function of highly suppressive IL-17-producing T regulatory cells. PLoS ONE 2012, 7, e45115. [Google Scholar] [CrossRef]
- Sharabi, A.; Tsokos, M.G.; Ding, Y.; Malek, T.R.; Klatzmann, D.; Tsokos, G.C. Regulatory T cells in the treatment of disease. Nat. Rev. Drug Discov. 2018, 17, 823–844. [Google Scholar] [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression-implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Ni, D.; Tang, T.; Lu, Y.; Xu, K.; Shao, Y.; Saaoud, F.; Saredy, J.; Liu, L.; Drummer, C.t.; Sun, Y.; et al. Canonical Secretomes, Innate Immune Caspase-1-, 4/11-Gasdermin D Non-Canonical Secretomes and Exosomes May Contribute to Maintain Treg-Ness for Treg Immunosuppression, Tissue Repair and Modulate Anti-Tumor Immunity via ROS Pathways. Front. Immunol. 2021, 12, 678201. [Google Scholar] [CrossRef]
- Romano, M.; Tung, S.L.; Smyth, L.A.; Lombardi, G. Treg therapy in transplantation: A general overview. Transpl. Int. 2017, 30, 745–753. [Google Scholar] [CrossRef]
- Sacerdote, P. Opioids and the immune system. Palliat Med. 2006, 20 (Suppl. 1), 9–15. [Google Scholar] [CrossRef]
- Hirahara, K.; Poholek, A.; Vahedi, G.; Laurence, A.; Kanno, Y.; Milner, J.D.; O’Shea, J.J. Mechanisms underlying helper T-cell plasticity: Implications for immune-mediated disease. J. Allergy Clin. Immunol. 2013, 131, 1276–1287. [Google Scholar] [CrossRef]
- Xu, L.; Kitani, A.; Fuss, I.; Strober, W. Cutting edge: Regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J. Immunol. 2007, 178, 6725–6729. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Z.; Cao, W.; Wu, Q.; Yuan, Y.; Zhang, X. Regulatory T cells in COVID-19. Aging Dis. 2021, 12, 1545–1553. [Google Scholar] [CrossRef]
- D’Alessio, F.R.; Tsushima, K.; Aggarwal, N.R.; West, E.E.; Willett, M.H.; Britos, M.F.; Pipeling, M.R.; Brower, R.G.; Tuder, R.M.; McDyer, J.F.; et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J. Clin. Investig. 2009, 119, 2898–2913. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e1039. [Google Scholar] [CrossRef]
- Hoffmann, A.D.; Weinberg, S.E.; Swaminathan, S.; Chaudhuri, S.; Mubarak, H.F.; Schipma, M.J.; Mao, C.; Wang, X.; El-Shennawy, L.; Dashzeveg, N.K.; et al. Unique molecular signatures sustained in circulating monocytes and regulatory T cells in Convalescent COVID-19 patients. bioRxiv 2022. [Google Scholar] [CrossRef]
- Alsalman, A.; Al-Mterin, M.A.; Elkord, E. Role of T Regulatory Cells and Myeloid-Derived Suppressor Cells in COVID-19. J. Immunol. Res. 2022, 2022, 5545319. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, Y.; Saaoud, F.; Cornwell, W.; Xu, K.; Kirchhoff, A.; Lu, Y.; Jiang, X.; Wang, H.; Rogers, T.J.; Yang, X. Cigarette Smoke and Morphine Promote Treg Plasticity to Th17 via Enhancing Trained Immunity. Cells 2022, 11, 2810. https://doi.org/10.3390/cells11182810
Shao Y, Saaoud F, Cornwell W, Xu K, Kirchhoff A, Lu Y, Jiang X, Wang H, Rogers TJ, Yang X. Cigarette Smoke and Morphine Promote Treg Plasticity to Th17 via Enhancing Trained Immunity. Cells. 2022; 11(18):2810. https://doi.org/10.3390/cells11182810
Chicago/Turabian StyleShao, Ying, Fatma Saaoud, William Cornwell, Keman Xu, Aaron Kirchhoff, Yifan Lu, Xiaohua Jiang, Hong Wang, Thomas J. Rogers, and Xiaofeng Yang. 2022. "Cigarette Smoke and Morphine Promote Treg Plasticity to Th17 via Enhancing Trained Immunity" Cells 11, no. 18: 2810. https://doi.org/10.3390/cells11182810
APA StyleShao, Y., Saaoud, F., Cornwell, W., Xu, K., Kirchhoff, A., Lu, Y., Jiang, X., Wang, H., Rogers, T. J., & Yang, X. (2022). Cigarette Smoke and Morphine Promote Treg Plasticity to Th17 via Enhancing Trained Immunity. Cells, 11(18), 2810. https://doi.org/10.3390/cells11182810